
Abstract
Mounting evidence suggests that immunological mechanisms play a fundamental role in the pathogenesis of diabetic retinopathy (DR) and diabetic macular edema (DME). Upregulation of cytokines and other proinflammatory mediators leading to persistent low-grade inflammation is believed to actively contribute to the DR-associated damage to the retinal vasculature, inducing breakdown of the blood-retinal barrier, subsequent macular edema formation, and promotion of retinal neovascularization. This review summarizes the current knowledge of the biological processes providing an inflammatory basis for DR and DME. In addition, emerging therapeutic approaches targeting inflammation are discussed, including blockade of angiopoietin 2 and other molecular targets such as interleukin (IL)-6, IL-1β, plasma kallikrein, and integrins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic eye disease
Diabetes mellitus (DM) constitutes an unrelenting global epidemic affecting more than 422 million people worldwide and its prevalence is expected to grow up to 592 million by 2035 (World Health Organization, www.who.int/diabetes/global-report). Diabetic eye disease (DED) encompasses a variety of ocular conditions that affect people with DM, including diabetic retinopathy (DR), diabetic macular edema (DME), cataract, and glaucoma (Fig. 1). DR refers to the pathological and functional changes observed in the retinal vasculature as a result of chronic hyperglycemia and is recognized as the most frequent microvascular complication of DM [1, 2]. It affects approximately one third of type 1 and type 2 diabetic patients, with increasing risk associated with longer disease duration and higher hemoglobin A1C values [3]. DR is the principal cause of visual impairment in the working-age population [4] and its prevalence continues to rise worldwide, accounting for 15–17% of total blindness [5, 6]. In the USA, studies estimate that 28.5–40.3% of diabetic patients have DR [7] and a third of those are afflicted with vision-threatening diabetic retinopathy (VTDR) [8]. DR is categorized into multiple clinical stages (Table 1) based upon the severity of the disease [9, 10] including mild, moderate, and severe nonproliferative retinopathy (NPDR), and proliferative retinopathy (PDR) (Fig. 2). Of note, at any stage of DR, patients may develop DME) (Fig. 3), which refers to the accumulation of intra and extracellular fluid within the macular retina and represents the most common cause of central vision loss in patients with DM [11]. Even though DME can occur at any time during the course of the disease, the risk increases with the severity of DR [12].

Schematic of diabetic eye disease and ocular anatomy, including the blood-retinal barriers

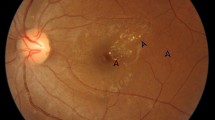
Clinical features of proliferative diabetic retinopathy (PDR). Color fundus photograph (left) and fundus fluorescein angiogram (right). Retinal hemorrhages (white asterisks), retinal neovascularization (black arrow heads), preretinal hemorrhage (white stars), fibrovascular lesions (black stars), areas of retinal ischemia (white arrows), and vascular leakage from retinal new vessels (white arrow heads)

Color fundus photograph (a), fluorescein angiography (b), and spectral domain optical coherence tomography (SD-OCT) (c) of a patient with nonproliferative diabetic retinopathy (NPDR) and diabetic macular edema (DME). a Hard exudates (top left, white arrow heads), microaneurysms and retinal hemorrhages (white asterisks). b Multiple hyperfluorescent spots predominantly from areas with microaneurysmatic changes and vascular leakage. c SD-OCT displaying multiple intraretinal cysts (white asterisk) and subfoveal neuroretinal detachment (white arrow head)
Despite major scientific advances being made over the last decades, the pathogenesis of DED—and specifically DR—remains to be fully elucidated. Current knowledge propones that long-standing hyperglycemic noxa induces retinal microvascular disease, inflammation, and retinal neurodegeneration, causing significant structural changes in the diabetic retina which may ultimately lead to vision loss [12]. Furthermore, an accumulating body of evidence has emerged highlighting the prominent role of immunological mechanisms in the pathogenesis of DED [13, 14]. Indeed, inflammation may be a key player in DR and DME by mediating deleterious effects in the neuronal and vascular components of the retina. Chronic retinal inflammation has been found from early phases to the sight-threatening advanced stages of DR [15]. There is a complex milieu of dysregulated pro-inflammatory factors in the diabetic retina such as interleukin (IL)-6, IL-1β, IL-8, monocyte chemoattractant protein (MCP)-1, and tumor necrosis factor (TNF)-α [13, 16] and increased levels of these cytokines have been found in ocular fluids of diabetic patients compared to controls [17].
In this review, we will discuss the preclinical and clinical evidence supporting the preponderance of inflammation in the pathogenesis of DR and DME, as well as the translational relevance of potential new targets for drug development.
Structure of the eye
The eye is a highly organized and complex organ with unique immunological properties [18]. The anatomy of the eye is structured in an outer layer, composed by cornea and sclera; a middle layer, predominantly vascular, named uvea (which comprises the iris, ciliary body, and choroid); and the innermost layer, the retina. From another point of view, the eye is separated in several compartments: the anterior chamber (localized between the iris and the cornea and filled with aqueous humor), the posterior chamber (between the lens and the iris), and the vitreous cavity, which contains the vitreous humor (a type II/XI collagenous avascular extracellular matrix which forms a gel). Those intraocular compartments ensure a sterile environment separated from the immune system by the blood-ocular barriers, which in homeostatic conditions prevent free trafficking of cells and large molecules into and from the eye. The two blood-ocular barriers are the blood-aqueous barrier (BAB) and the blood-retinal barrier (BRB) [19]. The BRB is composed by the inner barrier (iBRB), formed by the tight junction proteins (TJ) located between endothelial cells of the intraretinal capillaries, together with pericytes, Müller cells, and astrocytes, and the outer barrier (oBRB), which is formed by TJ between retinal pigment epithelium (RPE) cells that separate the choroid from the retina [20]. The BRB plays a fundamental role in the structural and functional integrity of the retina, regulating the flux of molecules within the eye and protecting the retinal tissue from pathogens [18, 19]. The TJs not only strictly regulate the permeability of the BRB and contribute to its high level of trans-endothelial/epithelial electrical resistance (TEER) but they also control the paracellular flux of molecules across the barrier [21]. Breakdown of the BRB can thus occur either at the level of retinal vessels or at the RPE layer. Understanding of the complex cellular and molecular processes involved in BRB leakage has not been fully clarified and appropriate animal models for studying macular edema are lacking [22].
Throughout lifetime, the retina is challenged by a variety of noxious insults including oxidative stress, hypoxia, or hyperglycemia. To deal with those challenges, the retina is equipped with a highly sensitive innate immune system [18, 23]. This immune system includes surveilling microglia cells, which migrate to the site of damage and phagocytose apoptotic material [24, 25], activation of the complement system to opsonize cellular debris [26, 27], and inflammasome assembly in the RPE [28]. In the event of a transient imbalance in retinal physiology, rapid activation of the immune response will induce restoration of tissue homeostasis and function [18]. However, upon persistent insult as in the case of DM, chronic over-activation of the inflammatory response can lead to devastating tissue remodeling and loss of functionality [29]. The resulting low-grade, chronic inflammatory response known as para-inflammation [30] is associated with a decline in RPE function and structure, breach of the BRB, new vessel formation, and recruitment of choroidal macrophages [29]. Therefore, inhibiting this sustained pathologic inflammation represents a plausible therapeutic target to treat DR.
Pathophysiology of diabetic eye disease
Metabolic changes in the diabetic retina
DR and DME are thought to result from the chronic effects of multiple systemic factors, the most important of which are hyperglycemia and dyslipidemia (Fig. 4) [31]. In particular, chronic hyperglycemia is associated with increased risk of retinopathy [32]. Excess flux of glucose through ectopically activated cellular biochemical and metabolic pathways alters the delicate balance between glucose-metabolizing enzymes, their intermediates, and end products. Under hyperglycemic conditions, stimulation of the polyol pathway, hexosamine pathway [33] and enhanced activity of an increasingly dysfunctional mitochondrial electron transport chain result in glucose auto-oxidation and the production of free radicals [34]. Over time, these free radicals overpower the existing anti-oxidant defense mechanisms and produce oxidative stress [35]. Oxidative modification of cellular proteins, lipids, and DNA promotes further mitochondrial damage, inflammation, apoptosis, and hence tissue damage. In parallel with altered intracellular processing of glucose, hyperglycemia also promotes the glycation of cellular, tissue, and serum proteins creating highly stable advanced glycation end products (AGE)-adducts [36,37,38]. Significantly, AGE-induced stimulation of the cell surface receptor for advanced glycation end products (RAGE) is an important activator of the transcription factors nuclear factor kappa B (NF-κB), signal transducer and activator of transcription (STAT), and activator protein 1 (AP-1) [39] causing inflammation and pericyte loss in DR. Taken together, elevated blood glucose provokes mitochondrial dysfunction, oxidative stress, and AGE formation, all of which can have deleterious effects on retinal cellular function [38, 39].

Pathogenesis of diabetic eye disease. Schematic representation of pathogenic mechanisms leading to sight-threatening endpoints of DR: PDR and DME. Metabolic alterations are sensed by multiple retinal cells resulting in glial dysfunction, which induces inflammation, secretion of pro-inflammatory cytokines and growth factors, and downstream signaling of diverse cascades, all leading to neuronal apoptosis and retinal vascular dysfunction. Hallmarks of disease include blood-retinal barrier breakdown (inducing vascular permeability and macular edema formation), vasoregression, and consecutive hypoxia, causing microvascular abnormalities that trigger DR progression through the different severity stages. AGE, advanced glycation end products; PKC, protein kinase C; RAS, renin-angiotensin system
Neurovascular changes in the diabetic retina
Metabolic changes, oxidative stress, and AGE accumulation affect multiple retinal cell types that are implicated in the development of DR. This condition is associated with early changes in both the neural and vascular compartments of the retina. Apoptosis of ganglion cells has been observed in both diabetic rodent retina and in human patients [40, 41] and has been associated with thinning of retinal nerve fiber layer [42,43,44]. Death or dysfunction of the neural retina promotes reactive gliosis, indicated by increased expression of glial fibrillary acidic protein (GFAP) in retinal astrocytes and Müller cells in both the early diabetic rodent and human retina [45, 46]. These neuroglial changes are accompanied and exacerbated by a sequence of profound alterations occurring in the retinal vasculature: thickening of the basement membrane [47], microaneurysm formation [48], pericyte loss [49], vasoregression [20], and capillary dropout [50], which may eventually cause isolated hypoxia and retinal ischemia.
The first vascular changes in the retina that are observed on histopathology are thickening of the basement membrane—an endothelial injury that leads to the disruption of the TJs- and pericyte cell death [33]. The consequences of pericyte loss are vascular tone dysregulation, and the growth and proliferation of endothelial cells owing to the deficit of transforming growth factor-β (TGF- β) produced by pericytes [13]. These changes underpin the development of microaneurysms and dot intraretinal hemorrhages, two of the earliest clinical manifestations (and disease hallmarks) which can be observed by ophthalmoscopy in early NPDR. The thickening of the basement membrane and the disruption of the TJ are determining factors in the leakage of the retinal capillaries. The thickened basement membrane is dysfunctional and permits the passage of the intravascular content (proteins, lipids, inflammatory mediators, and other plasma constituents) to the interstitial space [33]. On the other hand, vascular endothelial growth factor (VEGF-A) and pro-inflammatory cytokines (IL-1β, TNF-α, IL-6, IL-8, MCP-1, etc.) secreted by glial cells (Müller cells and microglia), RPE, and macrophages also have critical roles in early microvascular impairment and tissue dysfunction [46]. Sustained secretion of inflammatory mediators may lead to an early and persistent chronic inflammatory state in the retina that results in upregulation of adhesion factors such as intercellular adhesion molecule 1 (ICAM-1) on the endothelial cell surface, leukocyte activation, leukocyte adhesion to the vascular endothelium (a process known as leukostasis) [13, 14], and alteration of the BRB, which then leads to increased vascular permeability [22]. The clinical consequence of this process is the occurrence of hard exudates, which represent the leakage of plasma constituents and mainly consist of lipids and proteins. Severe BRB breakdown produces DME, in which fluid accumulates within the macular retina, potentially compromising vision [49, 50] (Fig. 5).

DME pathophysiology. Within the diabetic retina, multiple metabolic and cellular changes are induced as a result of hyperglycemia, oxidative stress, AGE accumulation, and the presence of oxidized lipids. Together, these abnormalities promote neurovascular changes which lead to the activation of microglia, dropout of pericytes, and disruption of endothelial tight junctions, creating breakdown of the blood-retinal barrier and accumulation of fluid within the tissue. Inflammatory breakdown of the blood-retinal barrier and pericyte loss is driven by the coordinated actions of a number of cytokines and growth factors, including VEGF-A, Ang 2, IL-6, and IL-1β produced by activated glial cells and the dysfunctional endothelium itself. As a consequence of inflammatory stress, endothelial cells upregulated expression of adhesion molecules such as ICAM-1, promoting extravasation of inflammatory cells such as monocytes, which further exacerbate the inflammatory status of the tissue driving increasingly severe diabetic macular edema
Leukostasis can also contribute to retinal capillary occlusion and thrombogenesis. The resulting retinal ischemia is the second sight-threatening diabetic complication. Histolopathological analyses have shown that areas of angiographic nonperfusion in vivo frequently co-localize to regions full of acellular capillaries, basement membrane tubes devoid of viable endothelial cells or pericytes [13]. These acellular capillaries are a hallmark of advanced, severe DR. Progressive capillary nonperfusion and subsequent retinal ischemia cause disease progression to PDR, in which new (albeit abnormal) blood vessels develop in the retina or in the optic disk [33].
Role of VEGF in diabetic eye disease
The VEGF family consists of multiple related proteins, VEGF-A, B, C, D, E (virally encoded), and placental growth factor (PlGF) [51], each of which plays a role in the regulation of vasculogenesis and lymphangiogenesis [52]. VEGF-A—the prototypical member of the VEGF family—has been classically described for its key roles in angiogenesis, exerting cellular effects by binding to high affinity tyrosine kinase VEGF-receptors (VEGFR-1 and 2) [52]. VEGF-A was also originally independently identified as vascular permeability factor (VPF) due to its ability to increase vascular permeability during inflammation and pathological angiogenesis [53]. There are multiple VEGF-A isoforms, which differ in amino acid sequence, tissue location, and diffusability. The most important isoform with respect to the pathological effects of VEGF within the diabetic retina is VEGFA-165, a cell secreted and heparin-binding isoform [52]. Therapies inhibiting VEGF activity are used to treat both DR and DME, resulting in improved visual outcomes [54, 55].
VEGF-A can be produced by multiple retinal cell types (Müller glia, endothelial cells, RPE, and astrocytes) and the expression of VEGF-A increases in these cells in response to hyperglycemia [56], hypoxia (via the hypoxia inducible factor HIF-1α) [57], AGE stimulation [58], and inflammatory cytokines such as IL-1β [59].
Indeed, although hypoxia is the most well-characterized factor that promotes VEGF gene expression, chronic hyperglycemia, AGEs, and pro-inflammatory cytokines such as IL-1β and IL-6 could upregulate VEGF mRNA expression [59]. These findings may explain why VEGF is overexpressed in DME despite the absence of overt hypoxia [54].
Müller glia-dependent production of VEGF-A has been demonstrated to be instrumental for the pathophysiological consequences of diabetes in the retinae of animal models, with VEGF-A-deficient diabetic mice displaying attenuated vascular leakage, leukostasis, and pericyte dropout [60]. Interestingly, combining photoreceptor overexpression of human VEGF-A165 with induction of diabetes in the Akimba mouse (Ins2AkitaVEGF+/-) model produces capillary nonperfusion, neovascularization, and edema [61], characteristic signs of DED. VEGF-A exerts pro-proliferative and pro-permeability effects on vascular endothelial cells by binding to VEGF-R2. Upon VEGF-R2 activation, the tyrosine kinase activity of the receptor increases, resulting in activation of multiple downstream signaling pathways including phosphatidylinositol 3-kinase (PI3K)-Akt, protein kinase C (PKC), endothelial nitric oxide synthase (eNOS), and mitogen-activated protein kinases/extracellular signal-regulated kinases (MAPK/ERK) [62]. In order to induce increased vascular permeability, VEGF-A-activated PKC phosphorylates the occludin [62] and VE-cadherin [63] components of the tight and adherens junctions respectively, resulting in their translocation from the cell-cell barrier to intracellular compartments [64, 65]. Consequently, the BRB becomes compromised, an essential pathomechanism during DME. In addition, VEGF-A has a direct mitogenic effect on vascular endothelial cells [66] and promotes alteration of the extracellular matrix required for neovascular processes by stimulation of matrix metalloprotease expression [67]. Both VEGF-A-regulated mechanisms are likely important for the development of PDR. Finally, VEGF-A also has further pathophysiological roles in the induction of leukostasis via upregulation of ICAM-1 expression [68]. Taken together, VEGF-A responses in the retina result in increased vascular permeability, retinal leukostasis, and neovascularization, which is characteristic of increasingly severe DR.
Angiopoietins
Recent clinical studies have highlighted the promising therapeutic efficacy of simultaneous inhibition of VEGF-A and angiopoietin 2 (Ang-2) in DME [69]. The Ang–tyrosine kinase with immunoglobulin-like domains (Tie) signaling pathway regulates vessel homeostasis and controls vascular permeability, inflammation, and angiogenic responses [70]. Angiopoietins are a family of growth factors that bind to the endothelial receptor tyrosine kinase Tie-2 and regulate vascular development and function [71]. Interestingly, binding of either Ang-1 or Ang-2 to Tie-2 exerts opposing cellular effects. Ang-1 acts as the cognate agonist of Tie-2, promoting receptor autophosphorylation and activation of the downstream PI3K/Akt and ERK signaling pathways [72]. This signaling promotes the stabilization of new and established vessels through the recruitment of pericytes and inhibition of vascular permeability induced by inflammatory cytokines [72]. With particular significance for inflammatory processes in DR, following ligand-induced activation Tie-2 also interacts with the NF-ĸB inhibitory protein TNF Alpha Induced Protein 3 (TNFAIP3). By suppressing the transcriptional activity of NFĸB via TNFAIP3, Ang-1-induced Tie-2 activation exerts anti-inflammatory and anti-apoptotic effects. Together, the signaling pathways promoted by Ang-1-induced Tie-2 activation stabilize new and established vessels through the recruitment of pericytes and inhibit vascular permeability induced by inflammatory cytokines [73]. Conversely, under adverse conditions such as hyperglycemia, hypoxia, or oxidative stress, Ang-2 is upregulated. Ang-2 also binds Tie-2, but it can have partial agonistic or antagonistic activity depending on the cellular context [73]. During inflammation, Ang-2 competitively binds to Tie-2 inhibiting Ang-1 signaling [74], leading to vascular destabilization, pericyte dropout, breakdown of the BRB, and aggravation of inflammation [75]. Indeed, as shown in the study conducted by Rangasamy and colleagues, Ang-2 is involved in the breakdown of the BRB through alteration of VE-cadherin at cell-cell contact in endothelial cells, suggesting that the Ang-2/Tie-2 system could serve as an alternative therapeutic target in DME [76]. Moreover, in Ang-2-deficient mice, vascular permeability induced by multiple inflammatory cytokines or angiogenic growth factors is attenuated, highlighting the role of Ang-2 in permitting vascular permeability under conditions of tissue stress. Blocking Ang-2 may also stabilize the vasculature by preventing pericyte loss and inhibiting Ang-2/integrin receptor-mediated endothelial tip cell sprouting [77]. A strategy of combination therapy via inhibition of both VEGF and Ang-2 could more effectively treat DME and prevent the recurrence of this condition than anti-VEGF monotherapy [77].
Cytokines and chemokines
Inflammatory molecules such as IL-6 [78], IL-1β, IL-8 [79], TNF-α [80], ICAM-1 [81, 82], vascular cell adhesion molecule 1 (VCAM-1) [82], integrin b-2 (CD-18) [83], and MCP-1 [84] have been reported to play pivotal roles in the pathogenesis of DR [85].
Interleukin-6
IL-6 is a key cytokine featuring redundancy and pleiotropic activity [86]. It plays a pivotal role in host defense against environmental stress such as infection and tissue injury. Dysregulated, persistent IL-6 production has been implicated in the development of various chronic inflammatory diseases and even cancers [87, 88]. IL-6 has broad biological activities in different target cells, including acute-phase protein synthesis and regulation of various components of the innate and adaptive immune responses [88]. IL-6 is promptly produced by monocytes, macrophages, and microglia after stimulation of Toll-like receptors (TLRs) which recognize distinct pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) such as those derived from oxidative stress [30, 89, 90]. This acute IL-6 expression is critical in host defense by stimulating various cell populations and promoting differentiation of specific cell subsets. For instance, IL-6 together TGF-β preferentially promotes differentiation of IL-17-producing T helper cells (Th17) and inhibits TGF-β-induced regulatory T cell (Treg) differentiation [91]. The resultant Th17/Treg imbalance leads to breakage of immunological tolerance and is of pathological importance for the development of various chronic inflammatory diseases [91]. Th17 cells further produce IL-17, IL-6, and TNF-α, and these cytokines perpetuate inflammation by stimulating fibroblasts, endothelial cells, and macrophages to produce chemokines, with the subsequent recruitment of more neutrophils and macrophages to the retina, which results in tissue damage and perpetuation of chronic inflammation [92, 93].
Signal transduction is mediated by the IL-6 receptor (IL-6R) system containing two functional proteins known as IL-6R and glycoprotein 130 (gp130) [94]. Binding of IL-6 to IL-6R results in the formation of the IL-6/IL-6R complex and subsequent homodimerization of gp130, a membrane-bound protein which is the signal transducing receptor for IL-6 and its related family members. IL-6 signaling initiated by transmembrane IL-6R (only expressed in a few cell types, including hepatocytes, neutrophils, monocytes, macrophages, and some lymphocytes) is known as IL-6 classic or cis-signaling. However, the soluble form of IL-6R (sIL-6R) can also bind IL-6 and form a complex with gp130 subunits in cells that lack the transmembrane IL-6R, a process known as IL-6 trans-signaling [95]. Both modes of IL-6R signaling lead to gp130 activation of Jak1, Jak2, and Tyk2, which in turn induces phosphorylation of STAT1 and STAT3 as well as activation of the MAPK cascades (Fig. 6). It has been hypothesized that IL-6 responses via the membrane-bound IL-6R are rather protective and regenerative whereas IL-6 trans-signaling appears to be involved in the pro-inflammatory activities of this cytokine [95].

IL-6 signaling. IL-6 activates cellular signaling using either cis- (reliant on expression of cell surface receptor) or trans- (via soluble IL-6 receptor) mechanisms. Both mechanisms require expression of the co-receptor gp130 and lead to activation of the Jak-STAT3 and Ras-MEK-ERK-AP1(Fos + Jun) signaling pathways. STAT3 activity promotes expression of VEGF-A, inflammatory mediators, and disruption of the endothelial barrier, whilst Fos+Jun promote expression of matrix metalloproteases. Together, these effects can cause multiple sequelae of diabetic retinopathy
The IL-6 signaling pathway plays a prominent role in the pathogenesis of DR and other inflammatory eye diseases [96, 97] due to its involvement in endothelial cell dysfunction and vascular inflammation [98]. Several lines of evidence suggest that IL-6 is a crucial mediator of both VEGF-mediated and inflammatory vascular leakage, through either direct or indirect effects on the vascular endothelium [99]. Interactions between IL-6 and endothelial cells regulate recruitment of leukocytes and expression of inflammatory proteins. Indeed, IL-6 can promote increased vascular permeability via direct barrier-disrupting effects on endothelial and epithelial cells or by inducing production of other cytokines and growth factors including the pro-permeability and pro-angiogenic factor VEGF [98, 99]. In the preclinical setting, Valle et al. recently reported that IL-6 trans-signaling causes barrier disruption in human retinal microvascular endothelial cells (HRMECs) via upregulation of ICAM-1 an endothelial cell specific leukocyte adhesion molecule which plays an important role in leukostasis and immune response [98]. Their findings suggest that IL-6 trans-signaling causes reactive oxygen species (ROS)-induced damage, mitochondrial dysfunction, inflammation, apoptosis, and the loss of barrier function in HRMECs, and blockade of IL-6 trans-signaling prevents inflammation and endothelial barrier disruption [98]. In another in vitro study, Yun et al. demonstrate that IL-6-induced STAT3 activation increases endothelial permeability through the downregulation of ZO-1 and occludin, which are main components of intercellular TJ forming the iBRB [100]. On the other hand, IL-6 has also been found to promote ocular angiogenesis in different cell lines via induction of VEGF production [101]. Moreover, in the murine laser-induced choroidal neovascularization (CNV) model, antibody-based blockade of the IL-6R or genetic ablation of IL-6 led to a significant suppression of CNV and vascular leakage [102]. CNV generation was accompanied by activation of the transcription factor STAT3 in choroidal endothelial cells and macrophages, and IL-6R blockade resulted in suppression of CNV formation [102] and inhibition of subretinal fibrosis [103].
From the clinical perspective, vitreous and aqueous humor concentrations of IL-6 are frequently elevated in patients with retinal diseases such as DED [104] neovascular age-related macular degeneration (nAMD) [102, 103], uveitis [105], and retinal vein occlusion (RVO) [106], where this cytokine has been suggested to play a fundamental pathogenic role. In uveitis patients, the IL-6R inhibitor tocilizumab has shown notable efficacy in various small clinical studies, particularly in cases of uveitis-related macular edema refractory to other therapies [97, 105]. In addition, from the DED standpoint, a post-hoc analysis of the READ3 study—in which DME patients were randomized to receive 0.5 or 2.0 mg ranibizumab and aqueous humor samples were collected in a prospective manner—has revealed that those patients who presented elevated aqueous humor levels of IL-6 at baseline ended up with worse visual outcomes when compared to those with initial low aqueous IL-6 levels [107]. These data suggest that aqueous IL-6 concentration may have prognostic therapeutic implications, potentially representing a candidate biomarker for a less favorable response to anti-VEGF therapy [108, 109].
Interleukin-1β
IL-1β is a master pro-inflammatory cytokine normally produced in response to infection, tissue injury, or immunologic challenge [110]. It plays a central role in mediating autoinflammatory syndromes [111], cardiovascular diseases [112, 113], and progression of cancer [114]. Even though its secretion comes from a wide variety of cell types, the vast majority of studies have focused on its production by cells of the innate immune system such as monocytes and macrophages [115]. It is formed as an inactive precursor termed pro-IL-1β in response to danger signals such as PAMPs or DAMPs [30]. Pro-IL-1β is then cleaved by the protease caspase 1, whose activation occurs via assembling of a multi-protein complex known as the inflammasome [111]. The best characterized inflammasome is the cytosolic NACHT, LRR, and PYD domains-containing protein 3 (NALP3), also known as cryopirine [115].
From the ocular perspective, the pathogenic role of IL-1β in DED is well established [116]. Apart from inducing vascular leakage, IL-1β contributes to vasoregression and neurodegeneration, which makes it a prominent target for the treatment of DME and for disease prevention in the early stages of DR [116]. In previous reports, IL-1β was shown to induce a number of alterations in the BRB including leukocyte recruitment, increased permeability, and changes in endothelial cell morphology and function [21, 116,117,118,119]. Activity of caspase-1 (the protease which converts the inactive proform of IL-1β into the active inflammatory cytokine) is increased in retinas of diabetic mice, galactose-fed mice, diabetic humans, and in retinal Müller cells incubated in elevated glucose concentration [119]. Inhibition of caspase-1 using minocycline inhibited the diabetes-induced increase in IL-1β in retina and prevented degeneration of retinal capillaries in those animals [119]. As a further confirmation of the role of IL-1β in DED, in vivo studies showed that diabetic mice lacking the IL-1 receptor (IL-1R) were protected from degeneration of retinal capillaries [119].
Another well-known action of IL-1β is to activate NF-κB. In response to the binding of IL-1β to IL-1R, a complex sequence of intracellular signaling reactions results in the activation of NF-κB via the JNK and p38 MAPK pathways (Fig. 7), which ultimately promote the expression of IL-6 and other pro-inflammatory cytokines (IL-8, MCP-1, TNF-α, etc.) [116]. Yoshida et al. proved that IL-1β is a potent inducer of IL-6 expression in cultured human Müller cells, a cell type considered to be a major source of IL-1β and other inflammatory factors in the retina [89]. This finding has led to the use of serum IL-6 as a surrogate biomarker of systemic IL-1β inhibition by approved IL-1 inhibitors such as anakinra (IL-1R antagonist) and canakinumab (anti-IL-1β monoclonal antibody) in several clinical trials for indications such as cardiovascular disease, as outlined in the CANTOS trial [120,121,122].

IL-1β signaling. IL-1β activates cellular signaling by binding to the cell surface IL-1R1 and promoting activation of Myd88 and MAPKs leading to increased transcriptional activity of NF-ĸB, AP-1 (Fos + Jun), and CREB. NF-ĸB promotes upregulation of adhesion molecules including ICAM-1 and inflammatory gene expression, whilst Fos + Jun promote expression of matrix disrupting enzymes such as MMP9. Finally, CREB promotes inflammatory cell survival and inflammatory gene expression
Apart from its potent pro-inflammatory capacity, IL-1β can also induce angiogenesis [123]. Notably, both IL-1β and VEGF upregulate each other in endothelial cells and are both essential to induce a proangiogenic response [124, 125]. Further, preclinical studies have shown that IL-1 inhibition by the recombinant IL-1R antagonist anakinra not only significantly reduced the development of new subretinal vessels in a rodent model of laser-induced CNV [126, 127] but also ameliorated endothelial dysfunction in streptozocin-induced diabetic rats [128].
In humans, augmented levels of IL-1β have been reported in ocular fluids of patients with DR/DME in numerous studies [129,130,131]. From the clinical perspective, the safety and efficacy of canakinumab were tested in a small open-label study conducted in DR patients [132], in which the authors noted that systemic IL-1β inhibition with canakinumab in patients with PDR showed only a modest regression of new vessels but a consistent reduction in vascular leakage and resolution of macular edema in those patients concomitantly presenting PDR with DME [132].
Interleukin 8
IL-8 is a pro-inflammatory chemokine and a member of the CXC chemokine family [133]. It is recognized as an important activator and chemoattractant for neutrophils, monocytes, and lymphocytes [133], and also an important mediator of neovascularization [134]. Increased levels of IL-8 have been found in ocular fluids of patients with advanced DR such as PDR [135, 136] and DME [137]. Interestingly, IL-8 positively correlated with severity of macular edema caused by DR but not with branch RVO [138], suggesting that IL-8 plays an influential role in the development of DME, specifically. High vitreous levels of IL-8 in DR indicate a role for this chemokine in the recruitment of neutrophils and monocytes into the vitreous humor, contributing to the intraocular neovascularization process characteristic of PDR [136]. Preclinically, Liu et al. demonstrated that IL-1β caused a significant increase of the production of IL-8 which may further enhance the pathogenic effect of IL-1β in DR [139]. This in vitro study shows that IL-1β potently stimulates IL-8 expression in Müller cells mainly via the p38 MAPK and ERK1/2 pathways, which illustrates the significant overlap and cross-talk between different cytokines and chemokines that occurs during the inflammatory response in the diabetic retina.
Tumor necrosis factor alpha
TNF-α is a pro-inflammatory cytokine that has been implicated in immune-mediated, cardiovascular and neoplastic diseases [140]. This monocyte/macrophage-derived factor is produced by microglia, Müller cells, macrophages, neutrophils, and T cells in response to various stimuli [140]. TNF-α exerts a variety of biological effects including upregulation of adhesion molecules, proliferation, differentiation, and cell death, and is thought to be strongly involved in the pathogenesis of DR and intraocular inflammation, among others [141]. Numerous in vitro and in vivo studies have shown that TNF-α increases leukocyte adhesion to retinal endothelium and augments BRB permeability [141,142,143,144]. Aveleira and colleagues showed that TNF-α acts through PKCζ/NF-κB to reduce the expression and alter the distribution of the TJ proteins claudin-5 and ZO-1 in bovine retinal endothelial cells (BRECs), which results in increased cell permeability [141]. In this study, glucocorticoid treatment completely prevented the TNF-α–induced rise in retinal endothelial cell permeability through both transactivation of the glucocorticoid receptor and transrepression of the NF-κB signaling pathway. Moreover, the inhibition of TNF-α with etanercept inhibits NF-κB activation, leukostasis, and BRB breakdown in diabetic rat retinas [142].
Two clinical studies have investigated the potential efficacy of TNF-α blockade (local and systemic) in the context of VTDR. Firstly, Tsilimbaris et al. conducted an uncontrolled prospective pilot study to investigate the effect of intravitreal administration of etanercept in DME refractory to laser therapy [145]. Seven patients received 2 consecutive intravitreal injections of 2.5 mg of etanercept with a 2-week interval. This small study did not reveal any improvement in the DME clinical course after 3 months of follow-up, and even a slight worsening of hard exudates and fluorescein leakage was observed in certain cases. Conversely, Sfikakis et al. coordinated a single-center, double-blind, randomized, placebo-controlled, crossover study, where 11 patients with sight-threatening DME refractory to laser received infliximab (5 mg/kg) intravenous infusions at weeks 0, 2, 6, and 14. In this study, a moderate visual improvement by a mean of 5 letters was found at week 16 in infliximab-treated eyes when compared to placebo. Infliximab was well tolerated with no serious adverse events reported [146].
Monocyte chemoattractant protein-1
MCP-1, also called CCL2, is a member of the CC chemokine family and plays a vital role in retinal inflammation in the context of DM [147]. Numerous cell types including monocytes, macrophages, fibroblasts, and astrocytes, as well as endothelial, epithelial, smooth muscle, mesangial, and microglial cells, are involved in MCP-1 production and secretion, either directly or following stimulation by growth factors, cytokines, or oxidative stress [147]. MCP-1 stimulates recruitment and activation of monocytes and macrophages. In addition, this chemokine has a potent contribution to fibrosis and angiogenesis induction stimulating the production of VEGF [148]. Of note, MCP-1 expression is also regulated by NF-κB [147].
Based on the available clinical and experimental data, secretion of the MCP-1 as a potent chemotactic factor by different types of retinal cells and through attachment to its receptor (CCR2) plays a significant role in recruitment and accumulation of monocytes/macrophages, alteration in the retinal vascular permeability, ROS formation, cell injury, inflammation and angiogenesis, as well as contribution to DR pathogenesis [84, 149]. Several clinical studies have demonstrated higher MCP-1 levels in the ocular fluids compared to the serum in DR patients, supporting a local retinal production based on the MCP-1 expression pattern [84, 150]. Furthermore, a significant association was found between vitreous levels of MCP-1 and the severity of DR [84].
Plasma kallikrein
The plasma kallikrein kinin system (KKS) is activated during vascular injury, where it mediates important functions in the inflammatory response, blood flow, and coagulation [151, 152]. Recent findings from human vitreous proteomics and preclinical studies in diabetic animal models have implicated the KKS in contributing to DR. Vitreous fluid from patients with advanced stages of DR contains increased levels of KKS components, including plasma kallikrein (PKal) [152]. Activation of the intraocular KKS induces retinal vascular permeability, vasodilation, and retinal thickening leading to macular edema. Both bradykinin B1 (B1R) and B2 (B2R) receptors are expressed in the human retina, and preclinical experiments have shown that administration of PKal inhibitors and B1R antagonists to diabetic rats ameliorates retinal vascular hyperpermeability and inflammation [153]. All together, these findings suggest that components of KKS are potential therapeutic targets for DME and several clinical trials targeting this pathway are underway [154, 155].
Integrins
Other targets involved in the diabetic inflammatory cascade that have raised interest for drug development are the integrins. The integrin family are cell adhesion receptors that mediate cell-cell and cell-extracellular matrix interactions. They are involved in a wide variety of biological activities (e.g., cell differentiation, adhesion, shape, migration, motility, invasion, proliferation, and survival) and have also been associated with various pathological processes, such as vascular leakage, inflammation, neovascularization, inflammation, and fibrosis [156]. Integrins are formed through noncovalent association of two transmembrane glycoproteins, the α- and the β subunit. These adhesion molecules mediate leukocyte interaction with the vascular endothelium and have been long implicated in the pathogenesis of DED, making them attractive targets for drug development [156,157,158]. We have seen that leukocytes play a major role in the degeneration of retinal capillaries in DR, as DM causes a significant increase in leukocyte adhesion to the retinal microvasculature (leukostasis). Selective antagonism of that adhesion by expression of neutrophil inhibitory factor has been shown to revert the diabetes-induced degeneration of retinal capillaries [156]. For instance, leukocyte integrin αmβ2 (also known as CD11b/CD18 or MAC1), a protein mediating adhesion between leukocytes and endothelial cells, has been shown to facilitate such damage to the retinal endothelium by activating leukocytes [159]. Thus, prevention of leukocyte adhesion to retinal endothelial cells is a potential therapeutic target to treat DR.
Current therapies for DME and DR: standard of care and unmet need
The management of DR and DME must include recommendations regarding the benefits of a healthy diet and lifestyle that includes exercise, weight vigilance, and control of associated risk factors such as dyslipidemia and arterial hypertension. Indeed, results from multiple studies have demonstrated the value of controlling blood glucose, serum lipid levels, and blood pressure in patients with DM [1, 3]. This may decrease the risk of developing DED in some patients [160, 161]. Moreover, the Diabetes Control and Complications Trial (DCCT) showed that the development and progression of DR in patients with type 1 DM can be delayed when the HbA1c is optimized [162].
As aforementioned, DME is a leading cause of visual impairment that is associated with retinal barrier dysfunction and the consequent accumulation of fluid within the central retina [11]. When it involves the center of the macula (center-involving DME), therapeutic intervention is recommended because of the significant risk of visual loss [163]. The classical treatment for DME is focal laser photocoagulation. However, this intervention may have a number of associated ocular complications (i.e., possible transient initial decrease in central vision, paracentral scotomas if laser burns have been placed close to the fovea, permanent central scotoma from inadvertent foveal burns, etc.) [164]. Besides, current data from various well-designed studies (READ-2, DRCR.net Protocol I, among others) demonstrate that intravitreal VEGF inhibitors provide a more effective treatment for DME than monotherapy with laser surgery [165, 166]. Multiple clinical trials have demonstrated the benefit of anti-VEGF therapy for center-involving DME and, at the present time, anti-VEGF agents are the initial treatment choice and standard of care, with both ranibizumab (Lucentis®; Genentech, South San Francisco, CA, USA) and aflibercept (Eylea®; Regeneron Pharmaceuticals, Tarrytown, NY, USA) being approved by the US Food and Drug Administration (FDA) and the European Medicines Agency for this indication. Bevacizumab (Avastin®, Genentech, South San Francisco, California, USA) is also used off-label by retina specialists for the treatment of DME and other retinal conditions. Most patients require near-monthly administration of intravitreal therapy with anti-VEGF agents during the first 12 months of treatment, with fewer injections needed in subsequent years to maintain disease remission [166, 167].
It is indisputable that the development and widespread use of ocular anti-VEGF therapy has been revolutionary in improving visual outcomes of DME patients. However, almost half of those patients fail to sufficiently respond to anti-VEGF therapy [167, 168], and the need for frequent injections creates a considerable treatment burden and high costs for both patients and health-care systems across the world [169]. Anti-VEGF agents also fail to resolve macular edema in more than 20% of patients [169] suggesting that there are alternative mediators contributing to its pathophysiology other than VEGF. In these patients, pro-inflammatory cytokines may be the main causative factors, and therefore treatment with anti-inflammatory agents could be a more effective therapeutic strategy [170]. Moreover, the clinical efficacy of intravitreal corticosteroids (triamcinolone acetonide, dexamethasone, fluocinolone) in patients with DME also highlights the prominent role of inflammation in this condition [171]. Nonetheless, in spite of their considerable effectiveness in reducing retinal inflammation and improving vision in the short term, corticosteroids are associated with a high rate of ocular side effects related to chronic use including augmented intraocular pressure (IOP) and increased risk of developing glaucoma and cataract formation, accelerating the need for cataract surgery in up to one third of patients with DR and DME [171, 172]. Triamcinolone is a synthetic corticosteroid suspension that has been used as local therapy to treat ophthalmic pathologies for many decades, although it is not licensed for treating DME. In fact, Triamcinolone acetonide has been shown to temporarily improve vision in eyes with DME [173] but failed to clinically compare favorably to laser therapy [174]. In addition, It is known to have a high rate of complications (rise in IOP and accelerated cataract formation), which is why it is usually reserved for DME refractory to other therapies [175].
In recent years, a number of long-acting, slow-release corticosteroid implants have been developed to reduce the side effects and prolong the duration of action, with 3 of them being now FDA-approved for use in DME: the fluocinolone acetonide intravitreal implants Retisert® (Bausch & Lomb, Rochester, NY, USA; 0.59 mg, approximate duration up to 30 months) and Iluvien® (Alimera Sciences, Alpharetta, GA, USA; 0.19 mg, approximate duration up to 24 months), as well as the dexamethasone intravitreal implant Ozurdex® (Allergan, Irvine, CA, USA; 0.7 mg, approximate duration 4–6 months) [171]. In the multicenter 3-year long FAME trial, intravitreal inserts of fluocinolone acetonide resulted in significant visual improvement in patients with DME [176]. However, the majority of patients treated with fluocinolone acetonide developed cataract, and the incidence of incisional glaucoma surgery was 4.8% (low dose) and 8.1% (high dose) [176]. Recently, the FDA approved this drug for DME in those patients who have been treated with a course of corticosteroids and did not show significant IOP rise. Another steroid, dexamethasone, has been investigated in the MEAD study, which examined the efficacy and safety of the slow-release intravitreal dexamethasone implant [177] There was more than 3-line visual improvement in about 22% patients in the dexamethasone group compared to the sham group (12% patients). On the down side, Cataract formation was seen in up to 68% of the patients on dexamethasone, and IOP rises could be managed with topical medications [177]. Because of the known increased rate of elevated IOP and cataract formation, the use of intravitreal steroids in clinical practice is presently reserved as a second-line treatment in center-involving DME patients [175, 178].
With regard to the management of DR, laser panretinal photocoagulation (PRP) is considered the mainstay of treatment for PDR and may also be contemplated for certain high-risk patients with severe NPDR [178, 179]. Nevertheless, despite its notable efficacy in preventing visual loss in vision-threatening advanced DR, the destructive nature of laser is associated with significant complications including peripheral visual field defects, difficulties with light-dark adaptation, night blindness, and loss of contrast sensitivity. Furthermore, PRP itself can be incompletely effective in some eyes and can also induce or exacerbate macular edema [179]. Of note, evidence from the phase 3 trials leading to FDA-approval of ranibizumab RIDE/RISE [180] and aflibercept (VISTA/VIVID) [181] for the treatment of center-involving DME not only revealed remarkable efficacy at improving macular anatomy and function but also demonstrated that VEGF blockade can impact far more than just retinal edema. Indeed, it was found that anti-VEGF therapy can slow progression of DR [182] and even improve DR severity by 2 steps in more than 36.5% of treated patients after 24 months versus only 5.4% after sham injections [182], which led to FDA ranibizumab’s approval for all forms of DR (with or without DME) in 2017 and aflibercept’s in 2019. In light of these studies, the American Academy of Ophthalmology Preferred Practice Pattern committee now states that there is sufficient evidence for the treatment of DR with anti-VEGF treatment [178].
On the other hand, corticosteroids have also been demonstrated to impact DR. As a matter of fact, in the paired FAME phase 3 clinical trials, fluocinolone acetonide implant was observed to cause a significant delay in progression of PDR [183]. At the 3-year FAME end point, with study participants having received a mean of 1.3–1.4 implant treatments, around 29% of sham-treated patients compared with approximately 17% of fluocinolone acetonide-treated patients progressed to PDR [183]. Additionally, a pilot study also showed reduction in peripheral retinal ischemia in patients with DME treated with intravitreal dexamethasone implant [184].
In summary, regardless of their potential benefit in ameliorating various disease features, corticosteroids are at the moment not approved for the treatment of DR and are recommended only as second-line therapy for patients with DME due to their less favorable safety profile in comparison to anti-VEGF or laser therapy [178]. Nevertheless, in applying these findings to clinical practice factors such as frequency of follow-up, treatment cost and patient preference must be considered in addition to the safety and efficacy outcomes for decision-making concerning treatment choice.
Translational research in diabetic eye disease
Evidence that low-grade inflammation plays a critical role in the pathogenesis of DR and DME has unraveled the potential of novel targets for the development of better medicines for our patients [85]. Anti-inflammatory compounds such as intravitreal, suprachoroidal, or topical glucocorticoids, inflammatory molecule inhibitors and new therapies targeting alternative pathways (cytokines, angiopoietins, integrins, plasma kallikrein, bradykinin, etc.) may be considered alone or in combination with VEGF inhibitors to improve efficacy and/or reduce the rate of administration [185].
As previously discussed, anti-VEGF intravitreal therapies caused a major paradigm shift for the treatment of DED. Nevertheless, there is still a high unmet need in this space as the required intense treatment frequency of approved anti-VEGF agents as well as the elevated rates of incomplete response pose a major health-care challenge for patients, physicians, and payers [186]. What is more, notwithstanding their clinical benefit, anti-VEGF drugs do not address the inflammatory component of the disease, which may explain the lack of clinically meaningful response observed in some patients and stresses the need for targeting alternative pathways implicated in DR/DME pathogenesis [85]. It has also been hypothesized that the disease may be driven by VEGF early in the disease, but in chronic stages, further micro-environmental changes may necessitate targeting of multiple inflammatory mediators other than just VEGF [85].
Faricimab (formerly RG7716) is a novel anti-Ang-2/anti-VEGF bispecific monoclonal antibody precisely designed for intraocular use [187]. It is engineered using Roche’s proprietary CrossMAb technology (Basel, Switzerland) and simultaneously binds VEGF-A and Ang-2 with high affinity and specificity [188]. Faricimab was investigated in the phase 2 BOULEVARD clinical trial in patients with DME, which met its primary endpoint demonstrating clinically meaningful response and superior visual acuity gains compared with ranibizumab [187]. Additionally, treatment with faricimab resulted in central subfield thickness (CST) reduction, DR severity score improvements, and extended durability of effect (Fig. 8). Currently, two phase 3 clinical trials, YOSEMITE (NCT03622580) and RHINE (NCT03622593), are ongoing to further test the efficacy, durability, and safety of faricimab for DME.

Ultra-wide field fluorescein angiography (UWF-FA) and SD-OCT images of a DME patient who participated in the BOULEVARD study and received treatment with Faricimab. a, b UWF-FA and SD-OCT corresponding to baseline visit. a UWF-F shows moderate NPDR with numerous microaneurysms in 4 quadrants and areas of capillary dropout with retinal nonperfusion; b SD-OCT shows cystoid macular edema with intraretinal cysts and presence of subretinal fluid. c, d Week 24 images, after 6 monthly doses of Faricimab IVT. c UWF-FA shows notable DR disease severity improvement after Faricimab therapy. d SD-OCT: complete fluid resolution, leaving a dry macula
Numerous other anti-inflammatory targets (integrin or cytokine blockers) are currently being investigated in early clinical trials for the treatment of DED, either in monotherapy or in combination with anti-VEGF agents. These novel anti-inflammatory compounds may be more selective and safer than steroids, which in theory could make them more suitable for the long-term treatment required for chronic diseases such as DR and DME, albeit more evidence is needed in regard to their safety and efficacy. These aspects will expectantly be clarified in later clinical trials, some of which are already underway or planned.
Conclusions
Diabetic eye disease is a global public health problem and a leading cause of blindness. Therapies such as anti-VEGF have been a major breakthrough in the management of vision-threatening stages of DR and are considered first line therapy for DME [190,191]. Whilst these are widely recognized effective drugs, there is a substantial proportion of patients who do not present clinically meaningful improvements in vision with such therapies. Consequently, there remains tremendous opportunity for improving outcomes for our patients. Henceforth, what are the avenues for future research? We know that anti-VEGF monotherapy is not a cure and new therapeutics targeting other pathways are needed. Apart from angiogenesis, several other mechanisms orchestrate the pathogenesis of DR and DME such as inflammation and neurodegeneration. More research into systemic and vitreous biomarkers (microRNAs, proteomics, metabolomics) may help discover additional novel pathways which may be suitable for drug development. With this critical information, a more personalized approach to treatment could be developed to achieve better efficacy and more optimal cost-effectiveness. Nowadays, clinicians can choose intravitreal injections of a limited number of anti-VEGF or corticosteroid agents albeit treatment selection is not based on pathophysiological reasoning. Intraocular fluids (e.g., aqueous or vitreous humor) obtained during the first intravitreal injection procedure could be useful for determining whether the predominant factor is inflammation or VEGF [16, 189]. This in turn could allow physicians to select a more rational and potentially more cost-effective treatment, with the aim of one day conducting precision medicine in ophthalmology—a strategy that has proved successful in the oncology field. Improved understanding of the underlying pathophysiological mechanisms of DED to find innovative therapeutic strategies, as well as early clinical intervention, will be pivotal to prevent DR-associated blindness worldwide.
Abbreviations
- AGEs:
-
advanced glycation end products
- BRB:
-
blood-retinal barrier
- DME:
-
diabetic macular edema
- NPDR:
-
nonproliferative diabetic retinopathy
- PDR:
-
proliferative diabetic retinopathy
- VEGF:
-
vascular endothelial growth factor
References
Antonetti DA, Klein R, Gardner TW (2012) Diabetic retinopathy. N Engl J Med 366:1227–1239
Nair P, Aiello LP, Gardner TW, Jampol LM, Ferris FL III (2016) Report from the NEI/FDA Diabetic Retinopathy Clinical Trial Design and Endpoints Workshop. Invest Ophthalmol Vis Sci 57:5127–5142
Cheung N, Mitchell P, Wong TY (2010) Diabetic retinopathy. Lancet 376:124–136
Bourne RR, Stevens GA, White RA, Smith JL, Flaxman SR, Price H et al (2013) Causes of vision loss worldwide, 1990-2010: a systematic analysis. Lancet Glob Health 1:e339–e349
Lee R, Wong TY, Sabanayagam C (2015) Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis (Lond) 2:17
Resnikoff S, Pascolini D, Etya’ale D, Kocur I, Pararajasegaram R, Pokharel GP, Mariotti SP (2004) Global data on visual impairment in the year 2002. Bull World Health Organ 82:844–851
Zhang X, Saaddine JB, Chou CF, Cotch MF, Cheng YJ, Geiss LS, Gregg EW, Albright AL, Klein BE, Klein R (2010) Prevalence of diabetic retinopathy in the United States, 2005-2008. JAMA 304:649–656
Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, Chen SJ, Dekker JM, Fletcher A, Grauslund J, Haffner S, Hamman RF, Ikram MK, Kayama T, Klein BE, Klein R, Krishnaiah S, Mayurasakorn K, O'Hare JP, Orchard TJ, Porta M, Rema M, Roy MS, Sharma T, Shaw J, Taylor H, Tielsch JM, Varma R, Wang JJ, Wang N, West S, Xu L, Yasuda M, Zhang X, Mitchell P, Wong TY, Meta-Analysis for Eye Disease (META-EYE) Study Group (2012) Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 35:556–564
ETDRS (1991) Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Early treatment diabetic retinopathy study research group. Ophthalmology 98:766–785
Wilkinson CP, Ferris FL 3rd, Klein RE, Lee PP, Agardh CD, Davis M, Dills D, Kampik A, Pararajasegaram R, Verdaguer JT, Global Diabetic Retinopathy Project Group (2003) Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 110:1677–1682
Das A, McGuire PG, Rangasamy S (2015) Diabetic macular edema: pathophysiology and novel therapeutic targets. Ophthalmology 122:1375–1394
Fong DS, Aiello LP, Ferris FL 3rd, Klein R (2004) Diabetic retinopathy. Diabetes Care 27:2540–2553
Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18:1450–1452
Adamis AP, Berman AJ (2008) Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol 30:65–84
Rübsam A, Parikh S, Fort PE (2018) Role of inflammation in diabetic retinopathy. Int J Mol Sci 19(4)
Simó-Servat O, Hernández C, Simó R (2012) Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediat Inflamm 2012:872978
Dong N, Xu B, Wang B, Chu L (2013) Study of 27 aqueous humor cytokines in patients with type 2 diabetes with or without retinopathy. Mol Vis 19:1734–1746
Mochizuki M, Sugita S, Kamoi K (2013) Immunological homeostasis of the eye. Prog Retin Eye Res 33:10–27
Cunha-Vaz JG (1997) The blood ocular barriers: past, present, and future. Doc Ophthalmol 93:149e157
Rizzolo LJ, Peng S, Luo Y, Xiao W (2011) Integration of tight junctions and claudins with the barrier functions of the retinal pigment epithelium. Prog Retin Eye Res 30:296–323
Klaassen I, Van Noorden CJ, Schlingemann RO (2013) Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 34:19–48
Cunha-Vaz JG (1983) Studies on the pathophysiology of diabetic retinopathy. The blood retinal barrier in diabetes. Diabetes 32:20e27
Akhtar-Schäfer I, Wang L, Krohne TU, Xu H, Langmann T (2018) Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol Med 10(10)
Arroba AI, Valverde ÁM (2017) Modulation of microglia in the retina: new insights into diabetic retinopathy. Acta Diabetol 54:527–533
Dick AD, Carter D, Robertson M, Broderick C, Hughes E, Forrester JV, Liversidge J (2003) Control of myeloid activity during retinal inflammation. J Leukoc Biol 74:161–166
Karlstetter M, Kopatz J, Aslanidis A, Shahraz A, Caramoy A, Linnartz-Gerlach B, Lin Y, Luckoff A, Fauser S, Duker K et al (2017) Polysialic acid blocks mononuclear phagocyte reactivity, inhibits complement activation, and protects from vascular damage in the retina. EMBO Mol Med 9:154–166
Zhang J, Gerhardinger C, Lorenzi M (2002) Early complement activation and decreased levels of glycosylph sphatidylinositol-anchored complement inhibitors in human and experimental diabetic retinopathy. Diabetes 51:3499–3504
Mohamed IN, Ishrat T, Fagan SC, El-Remessy AB (2015) Role of inflammasome activation in the pathophysiology of vascular diseases of the neurovascular unit. Antioxid Redox Signal 22:1188–1206
Xu H, Chen M (2017) Diabetic retinopathy and dysregulated innate immunity. Vis Res 139:39–46
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454:428–435
Yu Y, Lyons TJ (2005) A lethal tetrad in diabetes: hyperglycemia, dyslipidemia, oxidative stress, and endothelial dysfunction. Am J Med Sci 330:227–232
White NH, Cleary PA, Dahms W, Goldstein D, Malone J, Tamborlane WV, Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Research Group (2001) Beneficial effects of intensive therapy of diabetes during adolescence: outcomes after the conclusion of the Diabetes Control and Complications Trial (DCCT). J Pediatr 139:804–812
Wong TY, Cheung CM, Larsen M, Sharma S, Simó R (2016) Diabetic retinopathy. Nat Rev Dis Primers 2:16012
Kowluru RA (2005) Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal 7:1581–1587
Kowluru RA, Mishra M (2015) Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Biophys Acta 1852:2474–2483
Hammes HP, Martin S, Federlin K, Geisen K, Brownlee M (1991) Aminoguanidine treatment inhibits the development of experimental diabetic retinopathy. Proc Natl Acad Sci U S A 88:11555–11558
Hammes HP, Alt A, Niwa T, Clausen JT, Bretzel RG, Brownlee M, Schleicher ED (1999) Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia 42:728–736
Stitt A, Gardiner TA, Alderson NL, Canning P, Frizzell N, Duffy N, Boyle C, Januszewski AS, Chachich M, Baynes JW, Thorpe SR (2002) The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 51:2826–2832
Zong H, Ward M, Stitt AW (2011) AGEs, RAGE, and diabetic retinopathy. Curr Diab Rep 11:244–252
Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW (1998) Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest 102:783–791
Kern TS, Barber AJ (2008) Retinal ganglion cells in diabetes. J Physiol 586:4401–4408
HW v D, Kok PH, Garvin M, Sonka M, Devries JH, Michels RP, van Velthoven ME, Schlingemann RO, Verbraak FD, Abràmoff MD (2009) Selective loss of inner retinal layer thickness in type 1 diabetic patients with minimal diabetic retinopathy. Invest Ophthalmol Vis Sci 50:3404–3409
Lopes de Faria JM, Russ H, Costa VP (2002) Retinal nerve fibre layer loss in patients with type 1 diabetes mellitus without retinopathy. Br J Ophthalmol 86:725–728
van Dijk HW, Verbraak FD, Kok PH, Garvin MK, Sonka M, Lee K, Devries JH, Michels RP, van Velthoven ME, Schlingemann RO, Abràmoff MD, Lieth E, Barber A, Xu B, Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy et al (2010) Decreased retinal ganglion cell layer thickness in patients with type 1 diabetes. Invest Ophthalmol Vis Sci 51:3660–3665
Lieth E, Barber AJ, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM (1998) Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes 47:815–820
Simó R, Stitt AW, Gardner TW (2018) Neurodegeneration in diabetic retinopathy: does it really matter? Diabetologia 61:1902–1912
Roy S, Bae E, Amin S, Kim D (2015) Extracellular matrix, gap junctions, and retinal vascular homeostasis in diabetic retinopathy. Exp Eye Res 133:58–68
Hasegawa N, Nozaki M, Takase N, Yoshida M, Ogura Y (2016) New insights into microaneurysms in the deep capillary plexus detected by optical coherence tomography angiography in diabetic macular edema. Invest Ophthalmol Vis Sci 57:OCT348–OCT355
Pfister F, Feng Y, vom Hagen F, Hoffmann S, Molema G, Hillebrands JL, Shani M, Deutsch U, Hammes HP (2008) Pericyte migration: a novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes 57:2495–2502
Ebneter A, Wolf S, Zinkernagel MS (2016) Prognostic significance of foveal capillary drop-out and previous panretinal photocoagulation for diabetic macular oedema treated with ranibizumab. Br J Ophthalmol 100:365–370
Holmes DI, Zachary I (2005) The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol 6:209
Apte RS, Chen DS, Ferrara N (2019) VEGF in signaling and disease: beyond discovery and development. Cell 176:1248–126425
Connolly DT (1991) Vascular permeability factor: a unique regulator of blood vessel function. J Cell Biochem 47:219–223
Simó R, Sundstrom JM, Antonetti DA (2014) Ocular anti-VEGF therapy for diabetic retinopathy: the role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care 37:893–899
Virgili G, Parravano M, Evans JR, Gordon I, Lucenteforte E (2018) Anti-vascular endothelial growth factor for diabetic macular oedema: a network meta-analysis. Cochrane Database Syst Rev 10:CD007419
Sone H, Kawakami Y, Okuda Y et al (1996) Vascular endothelial growth factor is induced by long-term high glucose concentration and up-regulated by acute glucose deprivation in cultured bovine retinal pigmented epithelial cells. Biochem Biophys Res Commun 221:193–198
Kurihara T, Westenskow PD, Friedlander M (2014) Hypoxia-inducible factor (HIF)/vascular endothelial growth factor (VEGF) signaling in the retina. Adv Exp Med Biol 801:275–281
Treins C, Giorgetti-Peraldi S, Murdaca J, Van Obberghen E (2001) Regulation of vascular endothelial growth factor expression by advanced glycation end products. J Biol Chem 276:43836–43841
Nagineni CN, Kommineni VK, William A, Detrick B, Hooks JJ (2012) Regulation of VEGF expression in human retinal cells by cytokines: implications for the role of inflammation in age-related macular degeneration. J Cell Physiol 227:116–126
Wang J, Xu X, Elliott MH, Zhu M, Le YZ (2010) Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 59:2297–2305
Rakoczy EP, Ali Rahman IS, Binz N, Li CR, Vagaja NN, de Pinho M, Lai CM (2010) Characterization of a mouse model of hyperglycemia and retinal neovascularization. Am J Pathol 177:2659–2670
Murakami T, Frey T, Lin C, Antonetti DA (2012) Protein kinase cβ phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes 61:1573–1583
Esser S, Lampugnani MG, Corada M, Dejana E, Risau W (1998) Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 111:1853–1865
AJ B, Antonetti DA, Gardner TW (2000) Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. Invest Ophthalmol Vis Sci 41:3561–3568
Gavard J, Gutkind JS (2006) VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol 8:1223–1234
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246:1306–1309
Rodrigues M, Xin X, Jee K, Babapoor-Farrokhran S, Kashiwabuchi F, Ma T, Bhutto I, Hassan SJ, Daoud Y, Baranano D, Solomon S, Lutty G, Semenza GL, Montaner S, Sodhi A (2013) VEGF secreted by hypoxic Müller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy. Diabetes 62:3863–3873
Lu M, Perez VL, Ma N, Miyamoto K, Peng HB, Liao JK, Adamis AP (1999) VEGF increases retinal vascular ICAM-1 expression in vivo. Invest Ophthalmol Vis Sci 40:1808–1812
Sahni J, Patel SS, Dugel PU, Khanani AM, Jhaveri CD, Wykoff CC, Hershberger VS, Pauly-Evers M, Sadikhov S, Szczesny P, Schwab D, Nogoceke E, Osborne A, Weikert R, Fauser S (2019) Simultaneous inhibition of angiopoietin-2 and vascular endothelial growth factor-a with faricimab in diabetic macular edema: BOULEVARD Phase 2 Randomized Trial. Ophthalmologyhttps://doi.org/10.1016/j.ophtha.2019.03.023
Scholz A, Plate KH, Reiss Y (2015) Angiopoietin-2: a multifaceted cytokine that functions in both angiogenesis and inflammation. Ann N Y Acad Sci 1347:45–51
Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y (1995) Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature 376:70–74
Yuan HT, Khankin EV, Karumanchi SA, Parikh SM (2009) Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol 29:2011–2022
Thurston G, Daly C (2012) The complex role of angiopoietin-2 in the angiopoietin-tie signaling pathway. Cold Spring Harb Perspect Med 2:a006550
Hackett SF, Ozaki H, Strauss RW et al (2000) Angiopoietin 2 expression in the retina: upregulation during physiologic and pathologic neovascularization. J Cell Physiol 184:275–284
Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U (2004) Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes 53:1104–1110
Rangasamy S, Srinivasan R, Maestas J, McGuire PG, Das A (2011) A potential role for angiopoietin 2 in the regulation of the blood–retinal barrier in diabetic retinopathy. Invest Ophthalmol Vis Sci 52:3784–3791
Saharinen P, Eklund L, Alitalo K (2017) Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov 16:635–661
Yao Y, Li R, Du J, Long L, Li X, Luo N (2019) Interleukin-6 and diabetic retinopathy: a systematic review and meta-analysis. Curr Eye Res 44:564–574
Dong L, Bai J, Jiang X, Yang MM, Zheng Y, Zhang H, Lin D (2017) The gene polymorphisms of IL-8(-251T/A) and IP-10(-1596C/T) are associated with susceptibility and progression of type 2 diabetic retinopathy in northern Chinese population. Eye (Lond) 31:601–607
Yao Y, Li R, Du J, Li X, Zhao L, Long L, Li D, Lu S (2018) Tumor necrosis factor-α and diabetic retinopathy: review and meta-analysis. Clin Chim Acta 485:210–217
Zhu D, Zhu H, Wang C, Yang D (2014) Intraocular soluble intracellular adhesion molecule-1 correlates with subretinal fluid height of diabetic macular edema. Indian J Ophthalmol 62:295–298
Blum A, Pastukh N, Socea D, Jabaly H (2018) Levels of adhesion molecules in peripheral blood correlate with stages of diabetic retinopathy and may serve as biomarkers for microvascular complications. Cytokine 106:76–79
Barouch FC, Miyamoto K, Allport JR, Fujita K, Bursell SE, Aiello LP, Luscinskas FW, Adamis AP (2000) Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Vis Sci 41:1153–1158
Taghavi Y, Hassanshahi G, Kounis NG, Koniari I, Khorramdelazad H (2019) Monocyte chemoattractant protein-1 (MCP-1/CCL2) in diabetic retinopathy: latest evidence and clinical considerations. J Cell Commun Signal https://doi.org/10.1007/s12079-018-00500-8
Tang J, Kern TS (2011) Inflammation in diabetic retinopathy. Prog Retin Eye Res 30:343–358
Kishimoto T (1989) The biology of interleukin-6. Blood 74:1–10
Hunter CA, Jones SA (2015) IL-6 as a keystone cytokine in health and disease. Nat Immunol 16:448–457
Calabrese LH, Rose-John S (2014) IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol 10:720–727
Yoshida S, Sotozono C, Ikeda T, Kinoshita S (2001) Interleukin-6 (IL-6) production by cytokine stimulated human Müller cells. Curr Eye Res 22:341–347
Altmann C, Schmidt MHH (2018) The role of microglia in diabetic retinopathy: inflammation, microvasculature defects and neurodegeneration. Int J Mol Sci 19(1)https://doi.org/10.3390/ijms19010110
Kimura A, Kishimoto T (2010) IL-6: regulator of Treg/Th17 balance. Eur J Immunol 40:1830–1835
Tanaka T, Narazaki M, Kishimoto T (2014) IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6:a016295
Nish SA, Schenten D, Wunderlich FT, Pope SD, Gao Y, Hoshi N, Yu S, Yan X, Lee HK, Pasman L, Brodsky I, Yordy B, Zhao H, Brüning J, Medzhitov R (2014) T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife 3:e01949
Garbers C, Aparicio-Siegmund S, Rose-John S (2015) The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol 34:75–82
Rose-John S (2012) IL-6 trans-signaling via the soluble IL-6 receptor: importance for the proinflammatory activities of IL-6. Int J Biol Sci 8:1237–1247
Zahir-Jouzdani F, Atyabi F, Mojtabavi N (2017) Interleukin-6 participation in pathology of ocular diseases. Pathophysiology 24:123–131
Mesquida M, Leszczynska A, Llorenç V, Adán A (2014) Interleukin-6 blockade in ocular inflammatory diseases. Clin Exp Immunol 176:301–309
Valle ML, Dworshak J, Sharma A, Ibrahim AS, Al-Shabrawey M, Sharma S (2019) Inhibition of interleukin-6 trans-signaling prevents inflammation and endothelial barrier disruption in retinal endothelial cells. Exp Eye Res 178:27–36
Ye EA, Steinle JJ (2017) miR-146a suppresses STAT3/VEGF pathways and reduces apoptosis through IL-6 signaling in primary human retinal microvascular endothelial cells in high glucose conditions. Vis Res 139:15–22
Yun JH, Park SW, Kim KJ, Bae JS, Lee EH, Paek SH, Kim SU, Ye S, Kim JH, Cho CH Endothelial STAT3 activation increases vascular leakage through downregulating tight junction proteins: implications for diabetic retinopathy. J Cell Physiol 232:1123–1134
Cohen T, Nahari D, Cerem LW, Neufeld G, Levi BZ (1996) Interleukin 6 induces the expression of vascular endothelial growth factor. J Biol Chem 271(2):736–741
Izumi-Nagai K, Nagai N, Ozawa Y, Mihara M, Ohsugi Y, Kurihara T, Koto T, Satofuka S, Inoue M, Tsubota K, Okano H, Oike Y, Ishida S (2007) Interleukin-6 receptor-mediated activation of signal transducer and activator of transcription-3 (STAT3) promotes choroidal neovascularization. Am J Pathol 170:2149–2158
Sato K, Takeda A, Hasegawa E, Jo YJ, Arima M, Oshima Y, Ryoji Y, Nakazawa T, Yuzawa M, Nakashizuka H, Shimada H, Kimura K, Ishibashi T, Sonoda KH (2018) Interleukin-6 plays a crucial role in the development of subretinal fibrosis in a mouse model. Immunol Med 41:23–29
Funatsu H, Noma H, Mimura T, Eguchi S (2012) Vitreous inflammatory factors and macular oedema. Br J Ophthalmol 96:302–304
Mesquida M, Molins B, Llorenç V, de la Maza MS, Adán A (2017) Targeting interleukin-6 in autoimmune uveitis. Autoimmun Rev 16:1079–1089
Noma H, Funatsu H, Mimura T, Harino S, Hori S (2009) Vitreous levels of interleukin-6 and vascular endothelial growth factor in macular edema with central retinal vein occlusion. Ophthalmology 116:87–93
Sepah YJ, Nguyen QD, Do DV, Day B, Wakshull E, Stoilov I (2019) Trends for poorer vision outcomes in nAMD and DME patients with higher aqueous humor levels of IL-6. Investigative Ophthalmology & Visual Science vol.59 1077
Affridi R, Halim MS, Sadiq MA, Hassan M, Agarwal A, Do DV, Nguyen QD, Sepah J. (2017) Can the levels of inflammatory cytokines in the anterior chamber of eyes with diabetic macular edema predict response to therapy? ARVO Annual meeting abstract 2017
Chalam KV, Grover S, Sambhav K, Balaiya S, Murthy RK (2014) Aqueous interleukin-6 levels are superior to vascular endothelial growth factor in predicting therapeutic response to bevacizumab in age-related macular degeneration. J Ophthalmol 2014:502174
Dinarello CA, van der Meer JW (2013) Treating inflammation by blocking interleukin-1 in humans. Semin Immunol 25:469–484
Church LD, Cook GP, McDermott MF (2008) Primer: inflammasomes and interleukin 1beta in inflammatory disorders. Nat Clin Pract Rheumatol 4:34–42
Libby P (2017) Interleukin-1 Beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol 70:2278–2289
Dinarello CA, Donath MY, Mandrup-Poulsen T (2010) Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 17:314–321
Bent R, Moll L, Grabbe S, Bros M (2018) Interleukin-1 Beta—a friend or foe in malignancies? Int J Mol Sci 19(8)
Lopez-Castejon G, Brough D (2011) Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev 22:189–195
Kowluru RA, Odenbach S (2004) Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol 88:1343–1347
Kern TS (2007) Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res 2007:95103
Claudio L, Martiney JA, Brosnan CF (1994) Ultrastructural studies of the blood-retina barrier after exposure to interleukin-1 beta or tumor necrosis factor-alpha. Lab Investig 70:850–861
Vincent JA, Mohr S (2007) Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 56:224–230
Ridker PM, Everett BM, Thuren T, JG MF, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, JJP K, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, PRF R, RPT T, Libby P, Glynn RJ, CANTOS Trial Group (2017) Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131
ILARIS® (canakinumab) U.S. Prescribing Information, Novartis
KINERET® (anakinra) U.S. Prescribing Information: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/103950s5136lbl.pdf Accessed 27 May 2019
Amano K, Okigaki M, Adachi Y, Fujiyama S, Mori Y, Kosaki A, Iwasaka T, Matsubara H (2004) Mechanism for IL-1 beta-mediated neovascularization unmasked by IL-1 beta knock-out mice. J Mol Cell Cardiol 36(4):469–480
Maruyama K, Mori Y, Murasawa S, Masaki H, Takahashi N, Tsutusmi Y, Moriguchi Y, Shibazaki Y, Tanaka Y, Shibuya M, Inada M, Matsubara H, Iwasaka T (1999) Interleukin-1 beta upregulates cardiac expression of vascular endothelial growth factor and its receptor KDR/flk-1 via activation of protein tyrosine kinases. J Mol Cell Cardiol 31:607–617
Carmi Y, Dotan S, Rider P et al (2013) The role of IL-1β in the early tumor cell-induced angiogenic response. J Immunol 190:3500–3509
Olson JL, Courtney RJ, Rouhani B et al (2009) Intravitreal anakinra inhibits choroidal neovascular membrane growth in a rat model. Ocul Immunol Inflamm 17:195–200
Lavalette S, Raoul W, Houssier M et al (2011) Interleukin-1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol 178:2416–2423
Vallejo S, Palacios E, Romacho T, Villalobos L, Peiro C, Sanchez-Ferrer CF (2014) The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovasc Diabetol 13:158
Zhou J, Wang S, Xia X (2012) Role of intravitreal inflammatory cytokines and angiogenic factors in proliferative diabetic retinopathy. Curr Eye Res 37:416–420
Mao C, Yan H (2014) Roles of elevated intravitreal IL-1β and IL-10 levels in proliferative diabetic retinopathy. Indian J Ophthalmol 62:699–701
Lim SW, Bandala-Sanchez E, Kolic M, Rogers SL, McAuley AK, Lim LL, Wickremasinghe SS (2018) The influence of intravitreal ranibizumab on inflammation-associated cytokine concentrations in eyes with diabetic macular edema. Invest Ophthalmol Vis Sci 59:5382–5390
Stahel M, Becker M, Graf N, Michels S (2016) Systemic interleukin 1 beta inhibition in proliferative diabetic retinopathy: a prospective open-label study using canakinumab. Retina 36:385–391
Xie K (2001) Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev 12:375–391
Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM (1992) Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258:1798–1801
Canataroglu H, Varinli I, Ozcan AA, Canataroglu A, Doran F, Varinli S (2005) Interleukin (IL)-6, interleukin (IL)-8 levels and cellular composition of the vitreous humor in proliferative diabetic retinopathy, proliferative vitreoretinopathy, and traumatic proliferative vitreoretinopathy. Ocul Immunol Inflamm 13:375–381
Hernández C, Segura RM, Fonollosa A, Carrasco E, Francisco G, Simó R (2005) Interleukin-8, monocyte chemoattractant protein-1 and IL-10 in the vitreous fluid of patients with proliferative diabetic retinopathy. Diabet Med 22:719–722
Jonas JB, Jonas RA, Neumaier M, Findeisen P (2012) Cytokine concentration in aqueous humor of eyes with diabetic macular edema. Retina 32:2150–2157
Lee WJ, Kang MH, Seong M, Cho HY (2012) Comparison of aqueous concentrations of angiogenic and inflammatory cytokines in diabetic macular oedema and macular oedema due to branch retinal vein occlusion. Br J Ophthalmol 96:1426–1430
Liu X, Ye F, Xiong H, Hu D, Limb GA, Xie T, Peng L, Yang W, Sun Y, Zhou M, Song E, Zhang DY (2014) IL-1beta upregulates IL-8 production in human Muller cells through activation of the p38 MAPK and ERK1/2 signaling pathways. Inflammation 37:1486e1495
Hehlgans T, Pfeffer K (2005) The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 115:1–20
Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA (2010) TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 59:2872e2882
Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Döhmen S, Adamis AP (2002) Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J 16:438–440
Sagara M, Satoh J, Zhu XP, Takahashi K, Fukuzawa M, Muto G, Muto Y, Toyota T (1994) Inhibition with N-acetylcysteine of enhanced production of tumor necrosis factor in streptozotocin-induced diabetic rats. Clin Immunol Immunopathol 71:333–337
Behl Y, Krothapalli P, Desta T, DiPiazza A, Roy S, Graves DT (2008) Diabetes-enhanced tumor necrosis factor-alpha production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. Am J Pathol 172:1411e1418
Tsilimbaris MK, Panagiotoglou TD, Charisis SK, Anastasakis A, Krikonis TS, Christodoulakis E (2007) The use of intravitreal etanercept in diabetic macular oedema. Semin Ophthalmol 22:75–79
Sfikakis PP, Grigoropoulos V, Emfietzoglou I, Theodossiadis G, Tentolouris N, Delicha E, Katsiari C, Alexiadou K, Hatziagelaki E, Theodossiadis PG (2010) Infliximab for diabetic macular edema refractory to laser photocoagulation: a randomized, double-blind, placebo-controlled, crossover, 32-week study. Diabetes Care 33(7):1523–1528
Deshmane SL, Kremlev S, Amini S, Sawaya BE (2009) Monocyte chemoattractant Protein-1 (MCP-1): an overview. Interferon Cytokine Res 29:313–326
Hong KH, Ryu J, Han KH (2005) Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 105:1405–1407
Rangasamy S, McGuire PG, Franco Nitta C, Monickaraj F, Oruganti SR, Das A (2014) Chemokine mediated monocyte trafficking into the retina: role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS One 9:e108508
Tashimo A, Mitamura Y, Nagai S, Nakamura Y, Ohtsuka K, Mizue Y, Nishihira J (2004) Aqueous levels of macrophage migration inhibitory factor and monocyte chemotactic protein-1 in patients with diabetic retinopathy. Diabet Med 21:1292–1297
Feener EP, Zhou Q, Fickweiler W (2013) Role of plasma kallikrein in diabetes and metabolism. Thromb Haemost 110(3):434–441
Liu J, Feener EP (2013) Plasma kallikrein-kinin system and diabetic retinopathy. Biol Chem 394:319–328
Pouliot M, Talbot S, Sénécal J, Dotigny F, Vaucher E, Couture R (2012) Ocular application of the kinin B1 receptor antagonist LF22-0542 inhibits retinal inflammation and oxidative stress in streptozotocin-diabetic rats. PLoS One 7(3):e33864
Pruneau D, Bélichard P, Sahel JA, Combal JP (2010) Targeting the kallikrein-kinin system as a new therapeutic approach to diabetic retinopathy. Curr Opin Investig Drugs 11:507–514
Kita T, Clermont AC, Murugesan N, Zhou Q, Fujisawa K, Ishibashi T, Aiello LP, Feener EP (2015) Plasma Kallikrein-Kinin system as a VEGF-independent mediator of diabetic macular edema. Diabetes 64(10):3588–3599
Joy SS, Siddiqui K (2018) Molecular and pathophysiological mechanisms of diabetic retinopathy in relation to adhesion molecules. Curr Diabetes Rev
Quiroz-Mercado H (2018) Randomized, Prospective, double-masked, controlled phase 2b trial to evaluate the safety and efficacy of ALG-1001 (Luminate) in diabetic macular edema. Paper presented at: ARVO Annual meeting 2018
Hu TT, Vanhove M, Porcu M, Van Hove I, Van Bergen T, Jonckx B, Barbeaux P, Vermassen E, Feyen JHM (2019) The potent small molecule integrin antagonist THR-687 is a promising next-generation therapy for retinal vascular disorders. Exp Eye Res 180:43–52
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158:147-52
Tuomilehto J, Lindström J, Eriksson JG, Valle TT, Hämäläinen H, Ilanne-Parikka P, Keinänen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, Salminen V, Uusitupa M, Finnish Diabetes Prevention Study Group (2001) Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med 344:1343–1350
Ren C, Liu W, Li J, Cao Y, Xu J, Lu P (2019) Physical activity and risk of diabetic retinopathy: a systematic review and meta-analysis. Acta Diabetol
Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Schmidt-Erfurth U, Garcia-Arumi J, Bandello F, Berg K, Chakravarthy U, Gerendas BS, Jonas J, Larsen M, Tadayoni R, Loewenstein A (2017) Guidelines for the Management of Diabetic Macular Edema by the European Society of Retina Specialists (EURETINA). Ophthalmologica 237(4):185–222
Early Treatment Diabetic Retinopathy Study Research Group (1987) Treatment techniques and clinical guidelines for photocoagulation of diabetic macular edema. Early Treatment Diabetic Retinopathy Study Report Number 2. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology 94:761–774
Elman MJ, Qin H, Aiello LP, Diabetic retinopathy clinical research Network et al (2012) Intravitreal ranibizumab for diabetic macular edema with prompt versus deferred laser treatment: three-year randomized trial results. Ophthalmology 119:2312–2318
Tan GS, Cheung N, Simó R, Cheung GC, Wong TY (2017) Diabetic macular oedema. Lancet Diabetes Endocrinol 5:143–155
Cai S, Bressler NM (2017) Aflibercept, bevacizumab or ranibizumab for diabetic macular oedema: recent clinically relevant findings from DRCR.net Protocol T. Curr Opin Ophthalmol 28:636–643
Pieramici DJ, Wang PW, Ding B, Gune S (2016) Visual and anatomic outcomes in patients with diabetic macular edema with limited initial anatomic response to ranibizumab in RIDE and RISE. Ophthalmology 123:1345–1350
Krick TW, Bressler NM (2018) Recent clinically relevant highlights from the Diabetic Retinopathy Clinical Research Network. Curr Opin Ophthalmol 29:199–205
Figueras-Roca M, Sala-Puigdollers A, Zarranz-Ventura J, Alba-Linero C, Alforja S, Esquinas C, Molins B, Adán A (2019) Anatomic response to intravitreal dexamethasone implant and baseline aqueous humor cytokine levels in diabetic macular edema. Invest Ophthalmol Vis Sci 60:1336–1343
Whitcup SM, Cidlowski JA, Csaky KG, Ambati J (2018) Pharmacology of corticosteroids for diabetic macular edema. Invest Ophthalmol Vis Sci 59:1–12
Cunningham MA, Edelman JL, Kaushal S (2008) Intravitreal steroids for macular edema: the past, the present, and the future. Surv Ophthalmol 53:139–149
Gillies MC, Sutter FK, Simpson JM, Larsson J, Ali H, Zhu M (2006) Intravitreal triamcinolone for refractory diabetic macular edema: two-year results of a double-masked, placebo-controlled, randomized clinical trial. Ophthalmology 113:1533–1538
Diabetic Retinopathy Clinical Research Network (2008) A randomized trial comparing intravitreal triamcinolone acetonide and focal/grid photocoagulation for diabetic macular edema. Ophthalmology 115:1447–1449 (1449.e1–10)
Wong TY, Sun J, Kawasaki R, Ruamviboonsuk P, Gupta N, Lansingh VC, Maia M, Mathenge W, Moreker S, Muqit MMK, Resnikoff S, Verdaguer J, Zhao P, Ferris F, Aiello LP, Taylor HR (2018) Guidelines on diabetic eye care: the International Council of Ophthalmology Recommendations for Screening, Follow-up, Referral, and Treatment Based on Resource Settings. Ophthalmology 125:1608–1622
Campochiaro PA, Brown DM, Pearson A, Ciulla T, Boyer D, Holz FG, Tolentino M, Gupta A, Duarte L, Madreperla S, Gonder J, Kapik B, Billman K, Kane FE, FAME Study Group (2011) Long-term benefit of sustained-delivery fluocinolone acetonide vitreous inserts for diabetic macular edema. Ophthalmology 118:626–635 e2
Boyer DS, Yoon YH, Belfort R Jr, Bandello F, Maturi RK, Augustin AJ et al (2014) Three-year, randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with diabetic macular edema. Ophthalmology 121:1904–1914
American Academy of Ophthalmology Retina/Vitreous Panel, Hoskins Center for Quality Eye Care (2017) Diabetic retinopathy preferred practice pattern – Updated 2017 https://www.aao.org/preferred-practice-pattern/diabetic-retinopathy-ppp-updated-2017. Accessed 23-25 Apr 2019
Bressler NM, Beck RW, Ferris FL 3rd (2011) Panretinal photocoagulation for proliferative diabetic retinopathy. N Engl J Med 365:1520e1526
Nguyen QD, Brown DM, Marcus DM, RISE and RIDE Research Group et al (2012) Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology 119:789–801
Brown DM, Schmidt-Erfurth U, Do DV, Holz FG, Boyer DS, Midena E, Heier JS, Terasaki H, Kaiser PK, Marcus DM, Nguyen QD, Jaffe GJ, Slakter JS, Simader C, Soo Y, Schmelter T, Yancopoulos GD, Stahl N, Vitti R, Berliner AJ, Zeitz O, Metzig C, Korobelnik JF (2015) Intravitreal aflibercept for diabetic macular edema: 100-week results from the VISTA and VIVID studies. Ophthalmology 122:2044–2052
Ip MS, Domalpally A, Hopkins JJ, Wong P, Ehrlich JS (2012) Long-term effects of ranibizumab on diabetic retinopathy severity and progression. Arch Ophthalmol 130:1145–1152
Pearson PA, Comstock TL, Ip M, Callanan D, Morse LS, Ashton P, Levy B, Mann ES, Eliott D (2011) Fluocinolone acetonide intravitreal implant for diabetic macular edema: a 3-year multicenter, randomized, controlled clinical trial. Ophthalmology 118:1580–1587
Querques L, Parravano M, Sacconi R, Rabiolo A, Bandello F, Querques G (2017) Ischemic index changes in diabetic retinopathy after intravitreal dexamethasone implant using ultra-widefield fluorescein angiography: a pilot study. Acta Diabetol 54:769–773
Das A, McGuire PG, Monickaraj F (2016) Novel pharmacotherapies in diabetic retinopathy: current status and what's in the horizon? Indian J Ophthalmol 64:4–13
Whitehead M, Wickremasinghe S, Osborne A, Van Wijngaarden P, Martin KR (2018) Diabetic retinopathy: a complex pathophysiology requiring novel therapeutic strategies. Expert Opin Biol Ther 18:1257–1270
Sahni J, Patel SS, Dugel PU, Khanani AM, Jhaveri CD, Wykoff CC, Hershberger VS, Pauly-Evers M, Sadikhov S, Szczesny P, Schwab D, Nogoceke E8, Osborne A, Weikert R, Fauser S (2019) Simultaneous inhibition of angiopoietin-2 and vascular endothelial growth factor-A with faricimab in diabetic macular edema: BOULEVARD Phase 2 Randomized Trial. Ophthalmology
Regula JT, Lundh von Leithner P, Foxton R, Barathi VA, Chui Ming GC, Tun SBB, Wey YS, Iwata D, Dostalek M, Moelleken J, Stubenrauch KG, Nogoceke E, Widmer G, Strassburger P, Koss MJ, Klein C, Shima DT, Hartmann G (2019) Targeting key angiogenic pathways with a bispecific CrossMAb optimized for neovascular eye diseases. EMBO Mol Med
Felfeli T, Juncal VR, Hillier RJ, Mak MY, Wong DT, Berger AR, Kohly RP, Kertes PJ, Eng K, Boyd SR, Altomare F, Giavedoni LR, Muni RH (2019) Aqueous humor cytokines and long-term response to anti-vascular endothelial growth factor therapy in diabetic macular edema. Am J Ophthalmol
Diabetic Retinopathy Clinical Research Network, Elman MJ, Aiello LP, Beck RW et al (2010) Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 117:1064–1077
Mitchell P, Bandello F, Schmidt-Erfurth U, Lang GE, Massin P, Schlingemann RO, Sutter F, Simader C, Burian G, Gerstner O, Weichselberger A, RESTORE study group (2011) The RESTORE study: ranibizumab monotherapy or combined with laser versus laser monotherapy for diabetic macular edema. Ophthalmology 118(4):615–625
Acknowledgments
The authors would like to thank Jayashree Sahni, Meike Pauly-Evers, and Robert Weikert for the BOULEVARD study data and manuscript review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MM, FD, and SF are employed by F. Hoffmann-La Roche Ltd., Basel, Switzerland.
Additional information
This article is a contribution to the special issue on Inflammation and Type 2 Diabetes - Guest Editor: Marc Y. Donath
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mesquida, M., Drawnel, F. & Fauser, S. The role of inflammation in diabetic eye disease. Semin Immunopathol 41, 427–445 (2019). https://doi.org/10.1007/s00281-019-00750-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-019-00750-7