
Abstract
IL-17-producing T helper (Th17) cells have been implicated in the pathogenesis of many inflammatory and autoimmune diseases. Targeting the effector cytokines IL-17 and GM-CSF secreted by autoimmune Th17 cells has been shown to be effective for the treatment of the diseases. Understanding a molecular basis of Th17 differentiation and effector functions is therefore critical for the regulation of the pathogenicity of tissue Th17 cells in chronic inflammation. Here, we discuss the roles of proinflammatory cytokines and environmental stimuli in the control of Th17 differentiation and chronic tissue inflammation by pathogenic Th17 cells in humans and in mouse models of autoimmune diseases. We also highlight recent advances in the regulation of pathogenic Th17 cells by gut microbiota and immunometabolism in autoimmune arthritis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CD4+ T helper (Th) cells play pivotal roles in the tissue destruction of many inflammatory and autoimmune diseases such as multiple sclerosis (MS), rheumatoid arthritis (RA), psoriasis, and inflammatory bowel diseases [1]. Among Th subsets, interleukin-17 (IL-17)-producing Th (Th17) cells are classified as an inflammatory Th subset, resulting in chronic tissue inflammation and subsequent organ failure [2, 3]. Indeed, some biologic agents targeting the effector cytokines of Th17 cells have been approved for the treatment of certain immune-mediated diseases and we will see the expansion of diseases, which could be treatable by them. Therefore, understanding the differentiation and pathogenic functions of Th17 cells is crucial for the development of a novel immunotherapy for Th17-associated inflammatory diseases. Here, we focus on how Th17 cells acquire the terminal effector function, in particular, the pathogenicity in disease models and how environmental cues modulate Th17 cells that orchestrate chronic tissue inflammation in SKG mice, an animal model of RA. Finally, we discuss the role of gut microbiota and immunometabolism in modulating Th17 cells of patients with RA.
Induction of Th17 cells
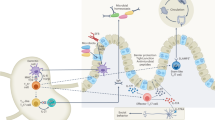
The differentiation and pathogenic functions of Th17 cells are regulated by numerous internal and external signals (Fig. 1). The differentiation of Th17 cells from naïve Th cells is initiated by stimulation with professional antigen-presenting cells (APCs) and particular cytokines including IL-6, IL-21, and TGFβ [4,5,6,7]. Following the upregulation of the lineage-defining transcription factors RORγt and RORα mediated by IL-6-JAK-STAT3 axis [8,9,10,11,12], Th17 cells produce the signature cytokines IL-17A, IL-17F, and IL-22, which are essential for mucosal host defense against extracellular bacteria and fungi by inducing anti-microbial peptides from epithelial cells and also recruit neutrophils by inducing chemokines under inflammation [13]. Hence, the deficiency of IL-6, IL-21, TGFβ, or RORγt impairs the differentiation of Th17 cells and subsequent Th17-mediated immunity [6, 8, 14]. However, the effect of TGFβ signaling in vitro is complicated with respects to Th17 effector function and could inhibit the function of autoimmune Th17 cells [15].

The regulation of Th17 differentiation and effector function
Among negative regulators of Th17 induction such as Th1- and Th2-inducing cytokines, IL-2 is a key repressor of Th17 differentiation and IL-2-mediated STAT5 activation specifically inhibits production of IL-17 [16]. By contrast, phosphatase and tensin homolog (PTEN) and Aiolos (encoded by Ikzf3), which repress IL-2 expression in T cells, have been identified to promote the development of Th17 cells [17, 18].
Natural Th17 cells are normally present in the gut in a microbiota-dependent manner, maintain tissue homeostasis, and fight against pathogenic microbes [19]. This defense mechanism is mainly mediated by IL-17 and IL-22, which increase anti-microbial peptides from gut epithelial cells [20]. Intriguingly, Th17 cells in Peyer’s patches can be converted into T follicular helper cells with high levels of Bcl6 and IL-21 expression, which support the differentiation of IgA-secreting B cells with antigen-specific properties [21]. These gut-specific Th17 cells are termed as “non-pathogenic Th17 cells” and are not associated with autoimmune reactions to self-antigens. However, when self-reactive Th cells are accidentally primed under Th17 conditions, additional environmental cues are able to modulate the effector profiles of autoimmune Th17 cells that cause the pathogenic outcome of targeted organs.
Pro-inflammatory cytokines to modulate the pathogenic function of Th17 cells
IL-1 and IL-23 are pro-inflammatory cytokines and are well-characterized as an enhancer and stabilizer of effector Th17 cells in autoimmune models, which predominantly express their corresponding receptors IL-1R1 and IL-23R, respectively [22,23,24,25,26,27,28]. Consistent with the importance of IL-1 and IL-23 in in vivo models, in vitro-polarized Th17 cells by stimulation with IL-6 and TGFβ are not able to induce Th17 cell-driven experimental autoimmune encephalomyelitis (EAE), an animal model of MS, whereas Th17 cells induced by IL-1β, IL-6, and IL-23 acquire the pathogenicity and elicit EAE [15]. One of the possible explanations about the difference between these in vitro conditions is that the treatment with IL-6 and TGFβ induces anti-inflammatory cytokine IL-10 in Th17 cells whereas IL-23 is critical for induction of the endogenous cytokine TGFβ3 by developing Th17 cells in addition to restraining IL-10 and in turn induces the pathogenicity of Th17 cells with high levels of T-bet, IL-23R, and GM-CSF [29, 30]. Furthermore, the single-cell RNA-sequencing analysis of ex vivo Th17 cells causing chronic inflammation in the central nervous system (CNS) of EAE mice identified Gpr65, Toso, and Plzp as novel genes promoting Th17 pathogenicity and CD5 antigen-like (CD5L) as a repressor of Th17 cell-mediated disease [31, 32]. IL-1 and IL-23 signaling also modulate the effector profile of Th17 cells through regulation of JunB and SOCS family members and induce highly pathogenic IL-17+ IFNγ+ and IL-17+ GM-CSF+ double-positive T cells, which have been shown to be originated from Th17 cells using a fate mapping strain [33,34,35,36]. It is of note that IL-23 is not required for the differentiation and maintenance of “non-pathogenic” Th17 cells in the gut and the functional plasticity toward T follicular helper cells [21].
Since naïve T cells do not express IL-1R and IL-23R [23, 37], their expression occurs during the initiation of Th17 differentiation in the presence of IL-6 whose signaling upregulates IL-1R1 and IL-23R expression via RORγt binding to the Il1r1 locus and STAT3 binding to the Il23r locus, respectively [15, 24, 38]. STAT3 activation also increases miR-183-96-182 cluster, which dampens Foxo1 expression, a negative regulator of IL-1R1 and IL-23R expression [38]. Protein C receptor also represses IL-1R and IL-23R expression on Th17 cells [39]. On the other hand, RBPJ (downstream of Notch signaling) promotes IL-23R expression as a positive regulator and therefore RBPJ KO Th17 cells fail to show the pathogenicity [40].
Through integrating these positive/negative regulators and a positive feed-forward loop by IL-1β and IL-23 stimulation, Th17 differentiation are finally stabilized along with upregulation of IL-1R and IL-23R [15, 41]. Thus, IL-23R expression is the hallmark of effector Th17 cells and its signaling promotes Blimp-1 (encoded by Prdm1) expression in vivo and the pathogenicity of Th17 cells by increasing IL-17 and GM-CSF production in RORγt-, STAT3-, and p300-dependent manners [42].
Environmental stimuli to modulate the pathogenic function of Th17 cells
Environmental factors are implicated in the increased prevalence of autoimmune and allergic diseases, some of which are induced by Th17 cells. External signals such as environmental toxins, metabolic stress, and osmotic pressure substantially affect the immune system including the pathogenicity of Th17 cells.
The aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor and senses many environmental toxins and endogenous ligands such as tryptophan metabolites. AhR is specifically induced under Th17 culture conditions and AhR ligation enhances production of IL-17 and IL-22 by effector Th17 cells [43, 44].
Metabolic cues regulate the function and differentiation of innate and adaptive immune cells. Metabolic demands dramatically increase during T cell activation and proliferation. The transcription factor hypoxia-inducible factor 1 (HIF-1), a key metabolic sensor, controls, in particular, a glycolytic pathway during Th17 differentiation and the effector function of Th17 cells through directly activating RORγt and IL-17 [45, 46]. The kinase complex mTORC1 is also known to be a central regulator of transcriptional pathways mediated by metabolic stress and contributes to the differentiation and effector function of Th subsets. The pathogenic phenotype of Th17 cells expressing T-bet and IFN-γ is partly regulated by mTORC1 signaling and therefore deletion of mTORC1 after Th17 differentiation reduces EAE severity [47].
A high salt diet widely spreads over the world and is associated with modern diseases. Serum glucocorticoid kinase 1 (SGK1), a serine/threonine kinase, can be induced in a high salt concentration under Th17 culture conditions and promotes IL-23R expression and stabilization of Th17 cells by deactivating Foxo1, the antagonist of Th17 cells. Thus, SGK1 activation in Th17 cells promotes autoimmune Th17 responses by upregulating GM-CSF [48, 49]. Taken together, environmental factors have a robust impact on accelerating the pathogenic function of Th17 cells.
GM-CSF, a key pathogenic cytokine in autoimmune tissue inflammation
GM-CSF is recently highlighted as the pathogenic cytokine of Th17 cells. The role of GM-CSF in EAE model was first reported in 2001, in which blockade of GM-CSF showed resistance to the EAE induction, but the critical source of GM-CSF in immune cells was not investigated in details [50]. There was the first report that among Th subsets infiltrating into the CNS after EAE induction, some of Th cells showed IL-17A+ GM-CSF+ double-positive producer [51]. The critical function of IL-23 signaling directing encephalitogenic Th17 cells has been reported to drive GM-CSF production, which causes local tissue inflammation [41, 52]. Because T cells do not express GM-CSF receptor [41], GM-CSF affects non-T cells. GM-CSF first acts on CNS-infiltrating myeloid cells such as dendritic cells (DCs), monocytes, and macrophages which in turn secrete pro-inflammatory cytokines such as IL-6 and IL-23, both of which upregulate IL-23R expression, amplifying IL-23-mediated pathogenic circuit to directly cause neurological pathogenicity and establishing local tissue inflammation by recruiting inflammatory macrophages in the CNS [51, 53]. GM-CSF also activates CCR2+ monocytes, monocyte-derived DCs and microglia in the brain to produce IL-1β [54, 55]. Since microglia have a potential to produce IL-23, they could participate the IL-23-IL-17 immune axis in Th17 cell-mediated tissue inflammation [56].
The transcription factor Bhlhe40, whose expression is initiated by CD28 signaling and enhanced by IL-1R1 signaling in T cells, has been identified as the direct driver of GM-CSF expression and Bhlhe40 KO mice were shown to be resistant to EAE due to impaired production of GM-CSF from pathogenic T cells [57, 58].
We recently identified special AT-rich binding protein 1 (Satb1), a genome organizer, as a crucial regulator of the pathogenic function of encephalitogenic tissue Th17 cells, while Satb1 was dispensable for the differentiation of Th17 cells [59]. To elucidate a specific role of Satb1 in Th17 cells, we generated Il17aCreR26ReYFPSatb1fl/fl conditional knock out (Th17Satb1CKO) mice, in which Cre-mediated deletion of Satb1 occurs in Th17 cells upon their differentiation into IL-17-expressing eYFP+ CD4+ T cells. We found that Th17Satb1CKO mice after EAE induction had impaired Th17 cells with low levels of GM-CSF expression and as a result, Th17Satb1CKO mice were resistant to EAE. Mechanistically, Satb1 specifically bound to the active promoter region of the Bhlhe40 locus and upregulated GM-CSF production in encephalitogenic Th17 cells. This machinery was pathogenic Th17-dependent in inflamed tissue because Satb1-sufficient gut Th17 cells did not express GM-CSF. We also observed that in vitro re-stimulation of draining LN eYFP+ Th17 cells from EAE mice with IL-23, but not IL-1β or IL-6 increased Satb1 expression, whereas TGF-β restrained its effect, suggesting that IL-23 signaling in chronic inflammation upregulates Satb1 expression in Th17 cells. In addition, Satb1 specifically promoted the effector function of Th17 cells in the CNS by inhibiting PD-1 expression. Considering that IL-1 signaling directly upregulates Bhlhe40, IL-1 and IL-23 signaling synergistically enhance GM-CSF production and the encephalitogenicity of Th17 cells by increasing Bhlhe40 and Satb1 expression, respectively.
Human Th17 cells and their role in neuroinflammation
Similar to mouse Th17 cells, the differentiation of human Th17 cells in vitro requires IL-1, IL-6, IL-23, and TGFβ. Several studies initially demonstrated that IL-1β, IL-6, and IL-23 but not TGFβ were sufficient to induce the differentiation of human Th17 cells [60, 61]. However, careful assessments later reconciled the role of TGFβ in human Th17 differentiation, showing that TGFβ, IL-23, and IL-1β (or IL-6) under serum-free conditions were essential in driving Th17 differentiation because culture medium contained serum-derived TGFβ or AhR ligands [62, 63]. Consistent with mouse Th17 cells, IL-23 plays the major role in human Th17 differentiation as human naïve CD4+ T cells can immediately respond to IL-23 and the IL-23R expression is further upregulated by IL-23 signals in the presence of additional IL-1β [62, 64]. Furthermore, dominant-negative mutations in STAT3, a key transcription factor downstream of IL-6 and IL-23 signaling, were responsible for disease manifestations of hyper-immunoglobulin E syndrome and impaired IL-17 production and the differentiation of Th17 cells, supporting roles of STAT3 in IL-6 and IL-23 signaling pathways and Th17 differentiation in humans [65, 66].
Before the discovery of Th17 subset, IL-17 was identified as the highest-ranking gene expressed in the CNS of MS patients [67]. It is of note that accumulation of Th17 cells were the first wave of T cells infiltrating the CNS [68], followed by the infiltration of DCs, macrophages, and other cells, which further promote and sustain tissue inflammation. In addition, a number of studies have shown that a single nucleotide polymorphism (SNP) in IL-23R is linked to a number of human autoimmune diseases [1], indicating that IL-23 signaling is essential for evoking a pathogenic feature in Th17 cells. IL-23 is also associated with the risk of MS [69, 70]. Since GM-CSF have a pivotal role in Th17 cells for encephalitogenicity in mice [41, 52], the findings that there were elevated levels of GM-CSF in the cerebrospinal fluid and serum of active MS patients with relapsing-remitting type and increased GM-CSF production of T cell from the peripheral blood and brain lesion of MS suggest a crucial role of GM-CSF in disease manifestations and tissue inflammation [71, 72].
The pathogenicity of Th17 cells in autoimmune arthritis models
RA is a systemic autoimmune disease that affects about 1% of general population worldwide, characterized by chronic joint inflammation and bone destruction [73]. Although the exact pathogenesis remains to be determined, it has been recognized that Th subsets play an important role in RA pathogenesis based on human leukocyte antigen (HLA)-DRB1 identified as the strongest disease risk gene, abundant T cells and macrophages infiltrating into synovial membrane in RA patients, and presence of circulating autoantibody such as rheumatoid factor (RF) and anti-citrullinated peptide antibodies (ACPA) [73,74,75,76]. When the Th1/Th2 paradigm dominated in the pathogenesis of autoimmune diseases before the discovery of Th17 cells, RA as well as MS was previously thought to be Th1-mediated diseases. However, the levels of Th-1-mediated cytokines such as IFN-γ in RA synovium were relatively low compared with those of TNF-α, IL-1, or IL-6 derived from inflammatory macrophage- and fibroblast-like synoviocytes (FLS) [77, 78]. The importance of these macrophage- and FLS-derived proinflammatory cytokines in RA is evident based on the efficacy of anti-cytokine therapy, such as anti-TNF or anti-IL-6 therapy, which brought a paradigm shift in RA treatment.
The discovery of Th17 cells have shed new insights into how inflammatory Th subsets contribute to the initiation of RA and form a proinflammatory cytokine network, leading to chronic inflammation, in particular, in animal models of autoimmune arthritis. There are several murine models to understand the pathogenesis of RA. Firstly, collagen-induced arthritis (CIA), which is induced by immunizing mice with type II collagen and whose pathogenicity is totally dependent on the generation of anti-collagen autoantibodies, is one of the well-established arthritis models. K/BxN mice, another model of arthritis, develop spontaneous arthritis mediated by the arthritogenic anti-glucose-6-phosphate isomerase (GPI) autoantibody [79, 80]. It has been demonstrated that IL-17 plays a pathogenic role to a greater or lesser extent in these murine models. However, with regard to T cell-dependent, but not autoantibody-dependent autoimmune arthritis, SKG mice are a suitable model to focus how T cells mediate autoimmune arthritis [81,82,83].
The SKG strain of mice on a BALB/c background bears a point mutation in ζ-associated protein-70 (ZAP-70), a key TCR-proximal signaling molecule, and spontaneously develops Th cell-mediated autoimmune arthritis in a microbially conventional environment, which immunopathologically resembles human RA [84, 85]. The mutation of the ZAP-70 gene alters the threshold for positive and negative selection of T cells in the thymus, leading to the production of self-reactive (arthritogenic) T cells and development of autoimmune arthritis [84,85,86]. SKG mice also frequently develop extra-articular lesions such as interstitial pneumonitis, rheumatoid nodules and vasculitides and show the production of autoantibodies such as RF and ACPAs, as seen in human RA [84]. Interestingly, SKG mice do not develop spontaneous arthritis under a specific-pathogen-free condition, yet can be induced by stimulation of innate immunity via Toll-like receptors, the Dectin pathway, or complement activation pathways, for example, by injection of fungal products such as zymosan and mannan, or, by provoking homeostatic proliferation of self-reactive T cells under lyphopenic conditions [87, 88]. Synovial inflammation in SKG mice is characterized by abundant infiltration of Th cells, which can adoptively transfer the disease into lymphopenic mice such as Rag2−/− mice, showing the disease dependency on Th cells. Recently, we succeeded to newly identify an arthritogenic self-antigen, 60S ribosomal protein L23a (RPL23A), by isolating arthritogenic effector Th cells from SKG arthritic joints and screening their TCR repertoires with an arthritogenic potential in retrogenic mice system. In addition, anti-RPL23A autoantibody was specifically detected in sera from patients with RA, which would support a similar molecular basis of the pathogenicity between SKG arthritis and human RA [89].
We reported that arthritis in SKG mice was highly dependent on Th17 cells since the cell transfer of IL-17-deficient SKG Th cells into T cell-deficient mice completely failed to induce arthritis. Supporting this finding, IL-6-deficient SKG mice were highly resistance to arthritis induction due to impaired T cell differentiation into Th17 cells. Although Th1 cells along with Th17 cells are also detected in SKG inflamed synovium, IFN-γ deficient SKG mice rather exacerbate arthritis, because Th17 differentiation is inhibited by Th1- or Th2-associated cytokines such as IFN-γ or IL-4 and IFN-γ deficient conditions expand arthritogenic Th17 cells [82].
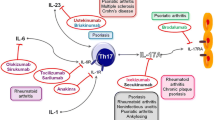
We have proposed a possible mechanism of how self-reactive (arthritogenic) Th cells become effector Th17 cells, migrate to synovium and initiate joint inflammation in SKG mice. Self-reactive T cells become activated in the periphery via recognition of class II MHC/self-peptide complexes expressed by APCs, and stimulate APCs through CD40/CD40L interaction to upregulate CD80/CD86, which further activate these T cells to proliferate. Activated APCs secrete a large amount of IL-6 (also IL-1, IL-23, and TNFα), together with surrounding tissue-derived TGF-β, that induces the differentiation of effector Th17 cells. Expanded arthritogenic Th17 cells predominantly express CCR6 and migrate to joints in response to CCL20, the ligand of CCR6, which is secreted by activated FLSs. Indeed, treatment with anti-CCR6 blocking mAb significantly inhibits the infiltration of Th17 cells into joints and reduces the severity of arthritis in SKG mice. In vitro, CCL20 expression in synoviocytes is promoted by IL-17, IL-1β, or TNFα, whereas IFN-γ or IL-4 inhibits its expression. Thus, once arthritogenic Th17 cells are activated and recruited into joints to initiate inflammation, synoviocytes further recruit Th17 cells in a feed-forward mechanism by which CCL20 production is augmented by proinflammatory cytokines such as IL-17, IL-1β, or TNFα derived from both activated synoviocytes and Th17 cells (Fig. 2). Expression of CCR6 in Th17 cells and CCL20 in synoviocytes are also observed in RA patients with a significant correlation between the amounts of IL-17 and CCL20 in RA joints [90]. Furthermore, recent genome-wide association study (GWAS) studies identified CCR6 as a disease susceptibility gene of RA, which together implies the pathogenicity of Th17 in RA and a shared mechanism of Th17 recruitment in inflamed joints [91].

A possible mechanism of self-reactive Th17 differentiation in SKG mice
Although Th17 cells are responsible for initiating autoimmune arthritis, it remained unclear how Th17 cells participate in “chronic” tissue inflammation. Recently, we demonstrated that GM-CSF is a crucial mediator in forming chronic joint inflammation in SKG mice and how Th17 cells orchestrate this “GM-CSF-cytokine network.” GM-CSF can be produced by various cell types including endothelial cells, fibroblasts, and activated T cells upon receiving immune stimuli, and is a key proinflammatory cytokine for the activation of macrophages and dendritic cells [92]. GM-CSF is abundantly seen in RA synovium, which is a reasonable observation to explain highly activated macrophages in RA joints, indicating its importance in the pathogenesis of RA [93, 94]. As expected, GM-CSF was crucial for arthritis induction in SKG mice since Csf2−/− SKG mice completely failed to develop arthritis, regardless of the presence of activated Th17 cells [95]. Adoptive T cell transfer experiment showed that T cell-derived GM-CSF, although it augmented arthritis, was dispensable for inducing arthritis, while non-T cell-derived GM-CSF was indispensable. Using bone marrow chimeras, the crucial source of GM-CSF was identified in radio-resistant stromal cells, including FLSs, and synovial-resident innate lymphoid cells (ILCs). Inhibition or loss of GM-CSF production in either radio-resistant stromal cells or ILCs significantly reduced the severity of arthritis. In vitro, FLSs upregulate GM-CSF secretion in response to recombinant IL-17 stimulation. In addition, adoptive transfer of wild type (WT), but not Il17a−/− SKG Th cells into Rag2−/− mice significantly induced Csf2 (also Ccl20 and Il6), in synoviocytes, which together imply that arthritogenic Th17 cells in joints stimulate FLSs via IL-17 and promote GM-CSF production. ILCs reside in healthy joints of SKG (or even other healthy mouse strains), but when arthritis occurs, GM-CSF-producing ILCs specifically expand in the joints. Predominant population of GM-CSF-producing synovial ILCs expresses Gata-3 and/or IL-13, which are the master transcription factor and signature cytokine of ILC2s. Indeed, in vitro, when synovial ILCs are treated with a combination of IL-2 and IL-33, GM-CSF production is significantly enhanced as well as IL-13 and IL-5 production. Furthermore, these synovial ILCs highly express Toll-like receptor 9 (TLR9) and synergistically upregulate GM-CSF production in response to CpG DNA, a ligand of TLR9, in combination with IL-33, but not CpG DNA alone. Expression of functional TLR9 in synovial ILCs indicates that they may sense mitochondrial DNA, possibly released as one of endogenous damage-associated molecular patterns (DAMPs). Taken together, unlike mechanisms of GM-CSF production in FLSs, synovial ILCs sense IL-2, IL-33, and self DNA, which can be produced by arthritogenic Th17 cells and released from necrotic cells in inflamed joints, leading to GM-CSF production (Fig. 3) [95].

Th17 cells orchestrate chronic joint inflammation in SKG mice
In the SKG arthritis model, induction of arthritis is fully dependent on Th17 cells. The key early event to initiate joint inflammation seems to be stimulating tissue stromal cells via IL-17 produced by arthritogenic Th17 cells migrating into joints. Expanded Th17 cells then orchestrate a GM-CSF-centric cytokine network, which results in the activation of synovial macrophages, leading to chronic inflammation and joint destruction.
Human Th17 cells in rheumatoid arthritis
As we have discussed above, there are several observations that support the pathogenesis of Th cells in RA. The efficacy of CTLA4-Ig treatment in RA indicates a central role of Th cells. In addition, recent GWAS study further identified RA risk loci that are linked to T cell function (e.g., PTPN22, CD28, CD40), and are even more specific to Th17-associated molecules such as CCR6 [91, 96,97,98,99,100]. However, the role of Th17 cells in RA is not as clear as animal models yet. Many reports agree that levels of IL-17 are increased in synovial fluid (SF) and synovial tissue (ST) in RA [101,102,103,104]. Kirkham et al. reported that the levels of IL-17 expression in ST are predictive of joint damage in RA patients, which could be the result of enhanced osteoclasts activation mediated by IL-17-rich environment [105]. However, it is controversial whether Th17 cells increase in the peripheral blood, SF, or ST in RA [106,107,108,109,110]. Furthermore, it was reported in ST that the vast majority of IL-17-secreting cells were not Th17 cells, which is one of the major differences from animal models of arthritis. The exact sources of IL-17 in ST remain unclear [101, 111]. Hueber et al. reported that the major source of IL-17 was synovial mast cells, which promote IL-17 production in stimuli by TNFα, IgG complexes or C5a, whereas Kan et al. later reported that the number of IL-17-producing mast cells and the frequency of IL-17-producing mast cells among all the IL-17+ cells in ST were comparable between RA and osteoarthritis (OA) patients, raising a question to the contribution of synovial mast cells as a source of IL-17 [111, 112]. Furthermore, the frequencies of Th1 cells among CD4+ T cells in SF and ST of RA patients are much larger than those of Th17 cells [81, 110]. However, the plasticity of Th17 cells toward Th1-like cells may be able to give a possible explanation for this observation. Nistala et al. showed the presence of IFN-γ-producing Th17 cells (hereafter Th17/Th1) in SF from patients with juvenile idiopathic arthritis (JIA), which express both Th1 and Th17 transcription factors such as T bet and RORC2 [113]. They also demonstrated in vitro that Th17 cells can be converted to Th17/Th1-cell phenotype in IL-12high TGFβlow conditions that resembles the SF environment of JIA. Furthermore, TCRβ (TRBV) repertoire analysis revealed that synovial Th17/Th1 cells share TCRβ repertoire oligoclonality with Th17 cells, indicating that Th17/Th1 cells may be deviated from Th17 cells. Moreover, CD161, one of the surface marker of Th17 cells in human as well as CCR6, is expressed not only in Th17/Th1 cells, but also in a number of Th1 cells in SF. These findings together suggest that a certain subpopulation of IFN-γ-producing CD4+ T cells in arthritic joints may originate from Th17 cells, which shift to Th1-like phenotype via an intermediate state of Th17/Th1 cells, when they encounter a IL-12high TGFβlow environmental cue in the local inflammatory site. In fact, there are accumulating evidence that IFN-γ+ ex-Th17 cells are enriched in SF of JIA and RA in comparison to peripheral blood [113,114,115]. In addition, recent studies revealed that approximately 80% of GM-CSF-producing CD4+ T cells in joints of JIA and RA co-express IFN-γ, but rarely express IL-17 [116, 117]. The majority of these IFN-γ+ GM-CSF+ CD4+ T cells express CD161, indicating that the subpopulation of IFN-γ+ ex-Th17 cells also actively produce GM-CSF [117].
As Th17 cells have been highlighted in the pathogenesis of RA, IL-17 inhibitors as a new potential biologic agent were trialed for RA treatment. In phase II trials, treatment with anti-IL-17 antibodies, secukinumab or ixekizumab, have demonstrated preliminary efficacy in RA with biologic-naïve or who failed to respond to TNF inhibitors or methotrexate [118,119,120,121]. However, in phase III trials, IL-17 inhibition showed no incremental benefit over the biologic agents currently approved to non-responders for TNF inhibitors [122, 123]. Although there is no clinical trial that has focused on evaluating the efficacy of IL-17 inhibitors in the onset or early stage of RA, these results indicate that IL-17 inhibitors alone are not sufficient enough to suppress ongoing chronic inflammation in established RA. Notably, it has been reported that higher levels of IL-17 in SF exist at the early stage of RA compared with the established stage and those of IL-17 in sera rather decreases after the onset of RA [124, 125]. Taking these findings together, Th17 cells may have different roles at different phases of RA. Th17 cells initiate joint inflammation via IL-17 by stimulating FLSs, promoting osteoclast differentiation and recruiting abundant neutrophils and more Th17 cells. However, soon after the onset of inflammation, a number of Th17 cells may become IFN-γ+ ex-Th17 cells in response to surrounding cytokine environments in the arthritic joints and simultaneously begin to actively secrete GM-CSF together with other GM-CSF-producing cells such as activated FLSs and synovial ILCs that synergistically provoke synovial macrophages to secrete a large amount of tissue destructive molecules and proinflammatory cytokines such as TNF-α or IL-6, leading to chronic inflammation. Thus, the pathogenesis of Th17 cells in RA may shift from “IL-17-producer,” as an initiator of the disease, into “GM-CSF-producer,” as an organizer of chronic inflammation. GM-CSF has been highlighted as a key mediator in RA based on recent clinical trials using GM-CSF inhibitors for RA patients [126,127,128,129]. From our point of view, IL-17 inhibitors may show a better clinical outcome for early onset RA patients, although it is not easy to identify and test such patients in a clinical trial.
Psoriasis, psoriatic arthritis (PsA), and ankylosing arthritis (AS) have been implicated in Th17-mediated autoimmune diseases in humans. Unlike RA, IL-17 inhibition shows marked clinical efficacies in psoriasis or PsA patients (also IL-12/23p40 inhibition) and AS patients, indicating the proof of concept in the pathogenesis of IL-17 in these diseases [130,131,132,133,134]. Further studies will be required to understand how IL-17-type/disease-specific tissue inflammation changes before and after the treatment with IL-17 inhibitors.
Regulation of Th17 cells by gut microbiota and immunometabolism in arthritis
There are accumulating evidence that the dysbiosis of gut microbiota is associated with various autoimmune diseases. Studies have shown that altered intestinal microbiota is observed not only in inflammatory bowel diseases, but also in organ-specific or systemic autoimmune diseases that affect internal organs, including type 1 diabetes, MS, and RA [135,136,137,138,139,140]. Autoimmune arthritis in murine models is also dependent on gut microbiota, for example, both K/BxN and SKG mice spontaneously develop arthritis in conventional conditions, but arthritis cannot be induced under germ-free (GF) conditions [87, 141]. Several studies have shown gut dysbiosis in RA patients. By using 16s rRNA gene sequencing, two reports revealed that Prevotella copri was significantly increased in RA patients, while Bacteroides was reduced. Maeda et al. demonstrated the disease-modifying role of P. copri in arthritis by generating gnotobiote SKG mice with P. copri, showing that P. copri induces CD4+ T cells in the large intestine to differentiate into Th17 cells, leading to production of arthritogenic Th17 cells and trigger autoimmune arthritis [138]. In contrast, another Prevotella species showed a beneficial role in humans. Marietta et al. demonstrated that Prevotella histicola, which is one of the commensal bacteria in the human gut, reduced the severity of arthritis in CIA model of HLA-DQ8-humanized mice [142]. Mice treated with P. histicola showed increased regulatory T cells (Treg) in the gut, suppressed antigen-specific Th17 response, and reduced the levels of prionflammatory cytokines, including IL-17 or TNF-α. These findings are intriguing in that therapeutic intervention of gut dysbiosis-targeting specific species may potentially be a new treatment for RA by regulating Th17 or Treg cells.
Immunometabolism plays a key role in the regulation of autoimmune T cells and T cell-mediated autoimmunity, including RA. Intriguingly, RA T cells fail to upregulate a glycolytic activity due to the insufficient induction of the key glycolytic enzyme PFKFB3, and shunt glucose toward the pentose-phosphate pathway. Impaired glycolysis induces a pyruvatelowATPlow intracellular environment and increase the production of NADPH, resulting in reduction of reactive oxygen species (ROS) and upregulation of fatty acid synthesis [143, 144]. Using a chimeric mouse model generated by implanting human synovium and transferring human T cells into NOD/Scid/IL2Rγnull mice, Shen et al. reported that these altered metabolic conditions lead to overexpression of TKS5, a podosome scaffolding protein, which enables them to form tissue-invasive membrane structures, resulting in enhancement of T cell invasion into synovium [144]. Expression levels of TKS5 in activated CD4+ CD45RA+ T cells in RA patients were correlated with their disease activities. They demonstrated in the chimeric mice that regulating these altered metabolic conditions by supplementation of pyruvate or inhibiting fatty acid synthesis successfully rewired the tissue-invasive behavior of RA T cells. In addition, another report from the same group showed that reduction of ROS promotes T cell maldifferentiation into Th17 and Th1 cells [145]. ROS restoration by menadione treatment in the chimeric mice, significantly reduced IFN-γ or IL-17-producing T cells in the synovium. These observations suggest that regulating T cell metabolic condition may successfully control proinflammatory differentiation and behavior of RA T cells.
Concluding remark
The discovery of Th17 cells brought us a new insight into a molecular basis of autoimmune disorders beyond the Th1/Th2 paradigm and clinical applications for various immune-mediated diseases. However, it remains elusive how Th17 cells mediate tissue injuries and orchestrate chronic tissue inflammation at different target organs. A comprehensive single-cell atlas that will characterize the pathogenicity of Th17 cells at different inflamed organs will open a new avenue for a novel immunotherapy, which enables us to specifically manipulate the function of pathogenic Th17 cells in autoimmune disease, but preserve the immune homeostasis, for instance, in the gut, mediated by physiological Th17 cells.
Change history
29 April 2019
Unfortunately, an error occurred in the following passus of the article. The word “receptor” was missing in the sentence “Because T cells do not express GM-CSF receptor [41], GM-CSF affects non-T cells.”
References
Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14(9):585–600. https://doi.org/10.1038/nri3707
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6(11):1123–1132. https://doi.org/10.1038/ni1254
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6(11):1133–1141. https://doi.org/10.1038/ni1261
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441(7090):235–238. https://doi.org/10.1038/nature04753
Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441(7090):231–234. https://doi.org/10.1038/nature04754
Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24(2):179–189. https://doi.org/10.1016/j.immuni.2006.01.001
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8(9):967–974. https://doi.org/10.1038/ni1488
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126(6):1121–1133. https://doi.org/10.1016/j.cell.2006.07.035
Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C (2008) T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28(1):29–39. https://doi.org/10.1016/j.immuni.2007.11.016
Zhong Z, Wen Z, Darnell JE Jr (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264(5155):95–98
Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S (1998) Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol 161(9):4652–4660
Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C (2007) STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282(13):9358–9363. https://doi.org/10.1074/jbc.C600321200
Littman DR, Rudensky AY (2010) Th17 and regulatory T cells in mediating and restraining inflammation. Cell 140(6):845–858. https://doi.org/10.1016/j.cell.2010.02.021
Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A (1998) IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol 28(7):2178–2187. https://doi.org/10.1002/(SICI)1521-4141(199807)28:07<2178::AID-IMMU2178>3.0.CO;2-D
Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O'Shea JJ (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467(7318):967–971. https://doi.org/10.1038/nature09447
Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O'Shea JJ (2007) Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26(3):371–381. https://doi.org/10.1016/j.immuni.2007.02.009
Kim HS, Jang SW, Lee W, Kim K, Sohn H, Hwang SS, Lee GR (2017) PTEN drives Th17 cell differentiation by preventing IL-2 production. J Exp Med 214(11):3381–3398. https://doi.org/10.1084/jem.20170523
Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, Georgopoulos K, Kuchroo VK (2012) Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol 13(8):770–777. https://doi.org/10.1038/ni.2363
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139(3):485–498. https://doi.org/10.1016/j.cell.2009.09.033
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203(10):2271–2279. https://doi.org/10.1084/jem.20061308
Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, Stockinger B (2013) Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol 14(4):372–379. https://doi.org/10.1038/ni.2552
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421(6924):744–748. https://doi.org/10.1038/nature01355
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201(2):233–240. https://doi.org/10.1084/jem.20041257
Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C (2009) Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30(4):576–587. https://doi.org/10.1016/j.immuni.2009.02.007
Schiffenbauer J, Streit WJ, Butfiloski E, LaBow M, Edwards C 3rd, Moldawer LL (2000) The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin Immunol 95(2):117–123. https://doi.org/10.1006/clim.2000.4851
Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203(7):1685–1691. https://doi.org/10.1084/jem.20060285
Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y (2006) Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol 18(2):399–407. https://doi.org/10.1093/intimm/dxh379
McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O'Shea JJ, Cua DJ (2009) The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol 10(3):314–324. https://doi.org/10.1038/ni.1698
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ (2007) TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 8(12):1390–1397. https://doi.org/10.1038/ni1539
Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK (2012) Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13(10):991–999. https://doi.org/10.1038/ni.2416
Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK, Park H, Regev A (2015) Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163(6):1400–1412. https://doi.org/10.1016/j.cell.2015.11.009
Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, Kuchroo VK (2015) CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163(6):1413–1427. https://doi.org/10.1016/j.cell.2015.10.068
Carr TM, Wheaton JD, Houtz GM, Ciofani M (2017) JunB promotes Th17 cell identity and restrains alternative CD4(+) T-cell programs during inflammation. Nat Commun 8(1):301. https://doi.org/10.1038/s41467-017-00380-3
Hasan Z, Koizumi SI, Sasaki D, Yamada H, Arakaki N, Fujihara Y, Okitsu S, Shirahata H, Ishikawa H (2017) JunB is essential for IL-23-dependent pathogenicity of Th17 cells. Nat Commun 8:15628. https://doi.org/10.1038/ncomms15628
Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, Pear WS, Hatton RD, Weaver CT (2015) IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol 16(3):286–295. https://doi.org/10.1038/ni.3099
Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B (2011) Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 12(3):255–263. https://doi.org/10.1038/ni.1993
Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13(5):715–725
Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, Sabouri-Ghomi M, Xu S, Zheng P, Xiao C, Dong C (2016) The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression. Immunity 44(6):1284–1298. https://doi.org/10.1016/j.immuni.2016.05.015
Kishi Y, Kondo T, Xiao S, Yosef N, Gaublomme J, Wu C, Wang C, Chihara N, Regev A, Joller N, Kuchroo VK (2016) Protein C receptor (PROCR) is a negative regulator of Th17 pathogenicity. J Exp Med 213(11):2489–2501. https://doi.org/10.1084/jem.20151118
Meyer Zu Horste G, Wu C, Wang C, Cong L, Pawlak M, Lee Y, Elyaman W, Xiao S, Regev A, Kuchroo VK (2016) RBPJ controls development of pathogenic Th17 cells by regulating IL-23 receptor expression. Cell Rep 16(2):392–404. https://doi.org/10.1016/j.celrep.2016.05.088
El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A (2011) The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 12(6):568–575. https://doi.org/10.1038/ni.2031
Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, Blumenschein WM, Sukumar S, Haines CJ, Sadekova S, McClanahan TK, McGeachy MJ, O'Shea JJ, Cua DJ (2016) Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells. Immunity 44(1):131–142. https://doi.org/10.1016/j.immuni.2015.11.009
Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL (2008) Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453(7191):65–71. https://doi.org/10.1038/nature06880
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453(7191):106–109. https://doi.org/10.1038/nature06881
Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, Luo W, Zeller K, Shimoda L, Topalian SL, Semenza GL, Dang CV, Pardoll DM, Pan F (2011) Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146(5):772–784. https://doi.org/10.1016/j.cell.2011.07.033
Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H (2011) HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208(7):1367–1376. https://doi.org/10.1084/jem.20110278
Karmaus PWF, Chen X, Lim SA, Herrada AA, Nguyen TM, Xu B, Dhungana Y, Rankin S, Chen W, Rosencrance C, Yang K, Fan Y, Cheng Y, Easton J, Neale G, Vogel P, Chi H (2018) Metabolic heterogeneity underlies reciprocal fates of TH17 cell stemness and plasticity. Nature. https://doi.org/10.1038/s41586-018-0806-7
Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK (2013) Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496(7446):513–517. https://doi.org/10.1038/nature11984
Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA (2013) Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496(7446):518–522. https://doi.org/10.1038/nature11868
McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, Reid HH, Bernard CCA (2001) Granulocyte macrophage colony-stimulating factor. J Exp Med 194(7):873–882. https://doi.org/10.1084/jem.194.7.873
Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M (2008) GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med 205(10):2281–2294. https://doi.org/10.1084/jem.20071119
Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B (2011) RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12(6):560–567. https://doi.org/10.1038/ni.2027
Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD (2007) Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol 178(8):5245–5252. https://doi.org/10.4049/jimmunol.178.8.5245
Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, Clausen BE, Jung S, Greter M, Becher B (2015) The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity 43(3):502–514. https://doi.org/10.1016/j.immuni.2015.08.010
Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN (2006) GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol 178(1):39–48. https://doi.org/10.4049/jimmunol.178.1.39
Li J, Gran B, Zhang G-X, Ventura ES, Siglienti I, Rostami A, Kamoun M (2003) Differential expression and regulation of IL-23 and IL-12 subunits and receptors in adult mouse microglia. J Neurol Sci 215(1–2):95–103. https://doi.org/10.1016/s0022-510x(03)00203-x
Martinez-Llordella M, Esensten JH, Bailey-Bucktrout SL, Lipsky RH, Marini A, Chen J, Mughal M, Mattson MP, Taub DD, Bluestone JA (2013) CD28-inducible transcription factor DEC1 is required for efficient autoreactive CD4+ T cell response. J Exp Med 210(8):1603–1619. https://doi.org/10.1084/jem.20122387
Lin CC, Bradstreet TR, Schwarzkopf EA, Jarjour NN, Chou C, Archambault AS, Sim J, Zinselmeyer BH, Carrero JA, Wu GF, Taneja R, Artyomov MN, Russell JH, Edelson BT (2016) IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J Exp Med 213(2):251–271. https://doi.org/10.1084/jem.20150568
Yasuda K, Kitagawa Y, Kawakami R, Isaka Y, Watanabe H, Kondoh G, Kohwi-Shigematsu T, Sakaguchi S, Hirota K (2019) Satb1 regulates the effector program of encephalitogenic tissue Th17 cells in chronic inflammation. Nat Commun 10(1):549. https://doi.org/10.1038/s41467-019-08404-w
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007) Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 8(9):942–949. https://doi.org/10.1038/ni1496
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal MR (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8(9):950–957. https://doi.org/10.1038/ni1497
Manel N, Unutmaz D, Littman DR (2008) The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol 9(6):641–649. https://doi.org/10.1038/ni.1610
Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V (2008) A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol 9(6):650–657. https://doi.org/10.1038/ni.1613
Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ (2007) Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum 56(9):2936–2946. https://doi.org/10.1002/art.22866
Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H (2007) Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448(7157):1058–1062. https://doi.org/10.1038/nature06096
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O'Shea J, Holland SM, Paul WE, Douek DC (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452(7188):773–776. https://doi.org/10.1038/nature06764
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8(5):500–508. https://doi.org/10.1038/nm0502-500
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK (2007) IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448(7152):484–487. https://doi.org/10.1038/nature05970
Vaknin-Dembinsky A, Balashov K, Weiner HL (2006) IL-23 is increased in dendritic cells in multiple sclerosis and Down-regulation of IL-23 by antisense Oligos increases dendritic cell IL-10 production. J Immunol 176(12):7768–7774. https://doi.org/10.4049/jimmunol.176.12.7768
Wen SR, Liu GJ, Feng RN, Gong FC, Zhong H, Duan SR, Bi S (2012) Increased levels of IL-23 and osteopontin in serum and cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 244(1–2):94–96. https://doi.org/10.1016/j.jneuroim.2011.12.004
Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O (1998) Profile of cerebrospinal fluid and serum cytokines in patients with relapsing remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol:373–382
Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, Mari ER, Safavi F, Leist TP, Zhang GX, Rostami A (2015) Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-beta therapy. J Immunol 194(11):5085–5093. https://doi.org/10.4049/jimmunol.1403243
Smolen JS, Aletaha D, McInnes IB (2016) Rheumatoid arthritis. Lancet 388(10055):2023–2038. https://doi.org/10.1016/S0140-6736(16)30173-8
Gregersen PK, Silver J, Winchester RJ (1987) The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 30(11):1205–1213
Gonzalez-Gay MA, Garcia-Porrua C, Hajeer AH (2002) Influence of human leukocyte antigen-DRB1 on the susceptibility and severity of rheumatoid arthritis. Semin Arthritis Rheum 31(6):355–360
Lundy SK, Sarkar S, Tesmer LA, Fox DA (2007) Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther 9(1):202. https://doi.org/10.1186/ar2107
Firestein GS, Zvaifler NJ (1987) Peripheral blood and synovial fluid monocyte activation in inflammatory arthritis. II Low levels of synovial fluid and synovial tissue interferon suggest that gamma-interferon is not the primary macrophage activating factor. Arthritis Rheum 30(8):864–871
Firestein GS, Zvaifler NJ (1990) How important are T cells in chronic rheumatoid synovitis? Arthritis Rheum 33(6):768–773
Courtenay JS, Dallman MJ, Dayan AD, Martin A, Mosedale B (1980) Immunisation against heterologous type II collagen induces arthritis in mice. Nature 283(5748):666–668
Matsumoto I, Staub A, Benoist C, Mathis D (1999) Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 286(5445):1732–1735
Ito Y, Usui T, Kobayashi S, Iguchi-Hashimoto M, Ito H, Yoshitomi H, Nakamura T, Shimizu M, Kawabata D, Yukawa N, Hashimoto M, Sakaguchi N, Sakaguchi S, Yoshifuji H, Nojima T, Ohmura K, Fujii T, Mimori T (2009) Gamma/delta T cells are the predominant source of interleukin-17 in affected joints in collagen-induced arthritis, but not in rheumatoid arthritis. Arthritis Rheum 60(8):2294–2303. https://doi.org/10.1002/art.24687
Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S (2007) T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med 204(1):41–47. https://doi.org/10.1084/jem.20062259
Iwanami K, Matsumoto I, Tanaka-Watanabe Y, Inoue A, Mihara M, Ohsugi Y, Mamura M, Goto D, Ito S, Tsutsumi A, Kishimoto T, Sumida T (2008) Crucial role of the interleukin-6/interleukin-17 cytokine axis in the induction of arthritis by glucose-6-phosphate isomerase. Arthritis Rheum 58(3):754–763. https://doi.org/10.1002/art.23222
Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, Sakaguchi S (2003) Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 426(6965):454–460. https://doi.org/10.1038/nature02119
Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y, Kanai C, Moriizumi E, Nomura T, Nakamura T, Sakaguchi S (2004) Distinct contribution of IL-6, TNF-alpha, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest 114(4):582–588. https://doi.org/10.1172/JCI21795
Tanaka S, Maeda S, Hashimoto M, Fujimori C, Ito Y, Teradaira S, Hirota K, Yoshitomi H, Katakai T, Shimizu A, Nomura T, Sakaguchi N, Sakaguchi S (2010) Graded attenuation of TCR signaling elicits distinct autoimmune diseases by altering thymic T cell selection and regulatory T cell function. J Immunol 185(4):2295–2305. https://doi.org/10.4049/jimmunol.1000848
Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T, Sakihama T, Hirota K, Tanaka S, Nomura T, Miki I, Gordon S, Akira S, Nakamura T, Sakaguchi S (2005) A role for fungal {beta}-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med 201(6):949–960. https://doi.org/10.1084/jem.20041758
Hashimoto M, Hirota K, Yoshitomi H, Maeda S, Teradaira S, Akizuki S, Prieto-Martin P, Nomura T, Sakaguchi N, Kohl J, Heyman B, Takahashi M, Fujita T, Mimori T, Sakaguchi S (2010) Complement drives Th17 cell differentiation and triggers autoimmune arthritis. J Exp Med 207(6):1135–1143. https://doi.org/10.1084/jem.20092301
Ito Y, Hashimoto M, Hirota K, Ohkura N, Morikawa H, Nishikawa H, Tanaka A, Furu M, Ito H, Fujii T, Nomura T, Yamazaki S, Morita A, Vignali DA, Kappler JW, Matsuda S, Mimori T, Sakaguchi N, Sakaguchi S (2014) Detection of T cell responses to a ubiquitous cellular protein in autoimmune disease. Science 346(6207):363–368. https://doi.org/10.1126/science.1259077
Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S (2007) Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 204(12):2803–2812. https://doi.org/10.1084/jem.20071397
Kochi Y, Okada Y, Suzuki A, Ikari K, Terao C, Takahashi A, Yamazaki K, Hosono N, Myouzen K, Tsunoda T, Kamatani N, Furuichi T, Ikegawa S, Ohmura K, Mimori T, Matsuda F, Iwamoto T, Momohara S, Yamanaka H, Yamada R, Kubo M, Nakamura Y, Yamamoto K (2010) A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet 42(6):515–519. https://doi.org/10.1038/ng.583
Gasson JC (1991) Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood 77(6):1131–1145
Alvaro-Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K, Firestein GS (1991) Cytokines in chronic inflammatory arthritis. VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production and gene expression in rheumatoid arthritis and its regulation by IL-1 and tumor necrosis factor-alpha. J Immunol 146(10):3365–3371
Wright HL, Bucknall RC, Moots RJ, Edwards SW (2012) Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatology (Oxford) 51(3):451–459. https://doi.org/10.1093/rheumatology/ker338
Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, Watanabe H, Kondoh G, Tanaka A, Yasuda K, Kopf M, Potocnik AJ, Stockinger B, Sakaguchi N, Sakaguchi S (2018) Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity 48(6):1220–1232 e1225. https://doi.org/10.1016/j.immuni.2018.04.009
Terao C, Raychaudhuri S, Gregersen PK (2016) Recent advances in defining the genetic basis of rheumatoid arthritis. Annu Rev Genomics Hum Genet 17:273–301. https://doi.org/10.1146/annurev-genom-090314-045919
Kim K, Bang SY, Lee HS, Bae SC (2017) Update on the genetic architecture of rheumatoid arthritis. Nat Rev Rheumatol 13(1):13–24. https://doi.org/10.1038/nrrheum.2016.176
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, Graham RR, Manoharan A, Ortmann W, Bhangale T, Denny JC, Carroll RJ, Eyler AE, Greenberg JD, Kremer JM, Pappas DA, Jiang L, Yin J, Ye L, Su DF, Yang J, Xie G, Keystone E, Westra HJ, Esko T, Metspalu A, Zhou X, Gupta N, Mirel D, Stahl EA, Diogo D, Cui J, Liao K, Guo MH, Myouzen K, Kawaguchi T, Coenen MJ, van Riel PL, van de Laar MA, Guchelaar HJ, Huizinga TW, Dieude P, Mariette X, Bridges SL Jr, Zhernakova A, Toes RE, Tak PP, Miceli-Richard C, Bang SY, Lee HS, Martin J, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Rantapaa-Dahlqvist S, Arlestig L, Choi HK, Kamatani Y, Galan P, Lathrop M, consortium R, consortium G, Eyre S, Bowes J, Barton A, de Vries N, Moreland LW, Criswell LA, Karlson EW, Taniguchi A, Yamada R, Kubo M, Liu JS, Bae SC, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Raychaudhuri S, Stranger BE, De Jager PL, Franke L, Visscher PM, Brown MA, Yamanaka H, Mimori T, Takahashi A, Xu H, Behrens TW, Siminovitch KA, Momohara S, Matsuda F, Yamamoto K, Plenge RM (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506(7488):376–381. https://doi.org/10.1038/nature12873
Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, Li Y, Kurreeman FA, Zhernakova A, Hinks A, Guiducci C, Chen R, Alfredsson L, Amos CI, Ardlie KG, Consortium B, Barton A, Bowes J, Brouwer E, Burtt NP, Catanese JJ, Coblyn J, Coenen MJ, Costenbader KH, Criswell LA, Crusius JB, Cui J, de Bakker PI, De Jager PL, Ding B, Emery P, Flynn E, Harrison P, Hocking LJ, Huizinga TW, Kastner DL, Ke X, Lee AT, Liu X, Martin P, Morgan AW, Padyukov L, Posthumus MD, Radstake TR, Reid DM, Seielstad M, Seldin MF, Shadick NA, Steer S, Tak PP, Thomson W, van der Helm-van Mil AH, van der Horst-Bruinsma IE, van der Schoot CE, van Riel PL, Weinblatt ME, Wilson AG, Wolbink GJ, Wordsworth BP, Consortium Y, Wijmenga C, Karlson EW, Toes RE, de Vries N, Begovich AB, Worthington J, Siminovitch KA, Gregersen PK, Klareskog L, Plenge RM (2010) Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 42(6):508–514. https://doi.org/10.1038/ng.582
Eyre S, Bowes J, Diogo D, Lee A, Barton A, Martin P, Zhernakova A, Stahl E, Viatte S, McAllister K, Amos CI, Padyukov L, Toes RE, Huizinga TW, Wijmenga C, Trynka G, Franke L, Westra HJ, Alfredsson L, Hu X, Sandor C, de Bakker PI, Davila S, Khor CC, Heng KK, Andrews R, Edkins S, Hunt SE, Langford C, Symmons D, Biologics in Rheumatoid Arthritis G, Genomics Study S, Wellcome Trust Case Control C, Concannon P, Onengut-Gumuscu S, Rich SS, Deloukas P, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Arlsetig L, Martin J, Rantapaa-Dahlqvist S, Plenge RM, Raychaudhuri S, Klareskog L, Gregersen PK, Worthington J (2012) High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet 44(12):1336–1340. https://doi.org/10.1038/ng.2462
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P (1999) Human interleukin-17: a T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum 42(5):963–970. https://doi.org/10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9):1345–1352. https://doi.org/10.1172/JCI5703
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W (2000) High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol 164(5):2832–2838
Shahrara S, Huang Q, Mandelin AM 2nd, Pope RM (2008) TH-17 cells in rheumatoid arthritis. Arthritis Res Ther 10(4):R93. https://doi.org/10.1186/ar2477
Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ (2006) Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum 54(4):1122–1131. https://doi.org/10.1002/art.21749
Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, Skapenko A (2010) Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum 62(10):2876–2885. https://doi.org/10.1002/art.27622
Penatti A, Facciotti F, De Matteis R, Larghi P, Paroni M, Murgo A, De Lucia O, Pagani M, Pierannunzii L, Truzzi M, Ioan-Facsinay A, Abrignani S, Geginat J, Meroni PL (2017) Differences in serum and synovial CD4+ T cells and cytokine profiles to stratify patients with inflammatory osteoarthritis and rheumatoid arthritis. Arthritis Res Ther 19(1):103. https://doi.org/10.1186/s13075-017-1305-1
van Hamburg JP, Asmawidjaja PS, Davelaar N, Mus AM, Colin EM, Hazes JM, Dolhain RJ, Lubberts E (2011) Th17 cells, but not Th1 cells, from patients with early rheumatoid arthritis are potent inducers of matrix metalloproteinases and proinflammatory cytokines upon synovial fibroblast interaction, including autocrine interleukin-17A production. Arthritis Rheum 63(1):73–83. https://doi.org/10.1002/art.30093
Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P (2008) Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum 58(8):2307–2317. https://doi.org/10.1002/art.23655
Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y (2008) Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis 67(9):1299–1304. https://doi.org/10.1136/ard.2007.080341
Hueber AJ, Asquith DL, Miller AM, Reilly J, Kerr S, Leipe J, Melendez AJ, McInnes IB (2010) Mast cells express IL-17A in rheumatoid arthritis synovium. J Immunol 184(7):3336–3340. https://doi.org/10.4049/jimmunol.0903566
Kan J, Mishima S, Kashiwakura J, Sasaki-Sakamoto T, Seki M, Saito S, Ra C, Tokuhashi Y, Okayama Y (2016) Interleukin-17A expression in human synovial mast cells in rheumatoid arthritis and osteoarthritis. Allergol Int 65(Suppl):S11–S16. https://doi.org/10.1016/j.alit.2016.04.007
Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj-Elliott M, Wedderburn LR (2010) Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci U S A 107(33):14751–14756. https://doi.org/10.1073/pnas.1003852107
Basdeo SA, Cluxton D, Sulaimani J, Moran B, Canavan M, Orr C, Veale DJ, Fearon U, Fletcher JM (2017) Ex-Th17 (nonclassical Th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells. J Immunol 198(6):2249–2259. https://doi.org/10.4049/jimmunol.1600737
Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, Frosali F, Querci V, Simonini G, Barra G, Piccinni MP, Liotta F, De Palma R, Maggi E, Romagnani S, Annunziato F (2011) Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum 63(8):2504–2515. https://doi.org/10.1002/art.30332
Yamada H, Haraguchi A, Sakuraba K, Okazaki K, Fukushi JI, Mizu-Uchi H, Akasaki Y, Esaki Y, Kamura S, Fujimura K, Kondo M, Miyahara H, Nakashima Y, Yoshikai Y (2017) Th1 is the predominant helper T cell subset that produces GM-CSF in the joint of rheumatoid arthritis. RMD Open 3(1):e000487. https://doi.org/10.1136/rmdopen-2017-000487
Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR, Nistala K (2014) T cell expression of granulocyte-macrophage colony-stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheum 66(7):1955–1960. https://doi.org/10.1002/art.38647
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, Aelion JA, Lee SH, Codding CE, Kellner H, Ikawa T, Hugot S, Mpofu S (2013) Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis 72(6):863–869. https://doi.org/10.1136/annrheumdis-2012-201601
Genovese MC, Greenwald M, Cho CS, Berman A, Jin L, Cameron GS, Benichou O, Xie L, Braun D, Berclaz PY, Banerjee S (2014) A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheum 66(7):1693–1704. https://doi.org/10.1002/art.38617
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Aelion JA, Lee SH, Codding CE, Kellner H, Ikawa T, Hugot S, Ligozio G, Mpofu S (2014) One-year efficacy and safety results of secukinumab in patients with rheumatoid arthritis: phase II, dose-finding, double-blind, randomized, placebo-controlled study. J Rheumatol 41(3):414–421. https://doi.org/10.3899/jrheum.130637
Genovese MC, Braun DK, Erickson JS, Berclaz PY, Banerjee S, Heffernan MP, Carlier H (2016) Safety and efficacy of open-label subcutaneous Ixekizumab treatment for 48 weeks in a phase II study in biologic-naive and TNF-IR patients with rheumatoid arthritis. J Rheumatol 43(2):289–297. https://doi.org/10.3899/jrheum.140831
Blanco FJ, Moricke R, Dokoupilova E, Codding C, Neal J, Andersson M, Rohrer S, Richards H (2017) Secukinumab in active rheumatoid arthritis: a phase III randomized, double-blind, active comparator- and placebo-controlled study. Arthritis Rheum 69(6):1144–1153. https://doi.org/10.1002/art.40070
Dokoupilova E, Aelion J, Takeuchi T, Malavolta N, Sfikakis PP, Wang Y, Rohrer S, Richards HB (2018) Secukinumab after anti-tumour necrosis factor-alpha therapy: a phase III study in active rheumatoid arthritis. Scand J Rheumatol 47(4):276–281. https://doi.org/10.1080/03009742.2017.1390605
Raza K, Falciani F, Curnow SJ, Ross EJ, Lee CY, Akbar AN, Lord JM, Gordon C, Buckley CD, Salmon M (2005) Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res Ther 7(4):R784–R795. https://doi.org/10.1186/ar1733
Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, Rantapaa Dahlqvist S (2010) Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum 62(2):383–391. https://doi.org/10.1002/art.27186
Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, Berenfus VY, Vladeva S, Rech J, Rubbert-Roth A, Korkosz M, Rekalov D, Zupanets IA, Ejbjerg BJ, Geiseler J, Fresenius J, Korolkiewicz RP, Schottelius AJ, Burkhardt H (2015) MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis 74(6):1058–1064. https://doi.org/10.1136/annrheumdis-2013-204816
Burmester GR, Weinblatt ME, IB MI, Porter D, Barbarash O, Vatutin M, Szombati I, Esfandiari E, Sleeman MA, Kane CD, Cavet G, Wang B, Godwood A, Magrini F, Group ES (2013) Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis 72(9):1445–1452. https://doi.org/10.1136/annrheumdis-2012-202450
Burmester GR, McInnes IB, Kremer J, Miranda P, Korkosz M, Vencovsky J, Rubbert-Roth A, Mysler E, Sleeman MA, Godwood A, Sinibaldi D, Guo X, White WI, Wang B, Wu CY, Ryan PC, Close D, Weinblatt ME, investigators EEs (2017) A randomised phase IIb study of mavrilimumab, a novel GM-CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann Rheum Dis 76(6):1020–1030. https://doi.org/10.1136/annrheumdis-2016-210624
Weinblatt ME, McInnes IB, Kremer JM, Miranda P, Vencovsky J, Guo X, White WI, Ryan PC, Godwood A, Albulescu M, Close D, Burmester GR (2018) A randomized phase IIb study of Mavrilimumab and Golimumab in rheumatoid arthritis. Arthritis Rheum 70(1):49–59. https://doi.org/10.1002/art.40323
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C, Group ES, Group FS (2014) Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med 371(4):326–338. https://doi.org/10.1056/NEJMoa1314258
Gordon KB, Leonardi CL, Lebwohl M, Blauvelt A, Cameron GS, Braun D, Erickson J, Heffernan M (2014) A 52-week, open-label study of the efficacy and safety of ixekizumab, an anti-interleukin-17A monoclonal antibody, in patients with chronic plaque psoriasis. J Am Acad Dermatol 71(6):1176–1182. https://doi.org/10.1016/j.jaad.2014.07.048
IB MI, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, van der Heijde D, Landewe R, Conaghan PG, Gottlieb AB, Richards H, Pricop L, Ligozio G, Patekar M, Mpofu S, Group FS (2015) Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 386(9999):1137–1146. https://doi.org/10.1016/S0140-6736(15)61134-5
Mease P, van der Heijde D, Landewe R, Mpofu S, Rahman P, Tahir H, Singhal A, Boettcher E, Navarra S, Meiser K, Readie A, Pricop L, Abrams K (2018) Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis 77(6):890–897. https://doi.org/10.1136/annrheumdis-2017-212687
Pavelka K, Kivitz A, Dokoupilova E, Blanco R, Maradiaga M, Tahir H, Pricop L, Andersson M, Readie A, Porter B (2017) Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: a randomized, double-blind phase 3 study, MEASURE 3. Arthritis Res Ther 19(1):285. https://doi.org/10.1186/s13075-017-1490-y
Needell JC, Zipris D (2016) The role of the intestinal microbiome in type 1 diabetes pathogenesis. Curr Diab Rep 16(10):89. https://doi.org/10.1007/s11892-016-0781-z
Miyake S, Kim S, Suda W, Oshima K, Nakamura M, Matsuoka T, Chihara N, Tomita A, Sato W, Kim SW, Morita H, Hattori M, Yamamura T (2015) Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One 10(9):e0137429. https://doi.org/10.1371/journal.pone.0137429
Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, Huttenhower C, Littman DR (2013) Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2:e01202. https://doi.org/10.7554/eLife.01202
Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, Hirota K, Matsushita M, Furuta Y, Narazaki M, Sakaguchi N, Kayama H, Nakamura S, Iida T, Saeki Y, Kumanogoh A, Sakaguchi S, Takeda K (2016) Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheum 68(11):2646–2661. https://doi.org/10.1002/art.39783
Vaahtovuo J, Munukka E, Korkeamaki M, Luukkainen R, Toivanen P (2008) Fecal microbiota in early rheumatoid arthritis. J Rheumatol 35(8):1500–1505
Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, Lan Z, Chen B, Li Y, Zhong H, Xie H, Jie Z, Chen W, Tang S, Xu X, Wang X, Cai X, Liu S, Xia Y, Li J, Qiao X, Al-Aama JY, Chen H, Wang L, Wu QJ, Zhang F, Zheng W, Li Y, Zhang M, Luo G, Xue W, Xiao L, Li J, Chen W, Xu X, Yin Y, Yang H, Wang J, Kristiansen K, Liu L, Li T, Huang Q, Li Y, Wang J (2015) The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 21(8):895–905. https://doi.org/10.1038/nm.3914
Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D (2010) Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32(6):815–827. https://doi.org/10.1016/j.immuni.2010.06.001
Marietta EV, Murray JA, Luckey DH, Jeraldo PR, Lamba A, Patel R, Luthra HS, Mangalam A, Taneja V (2016) Suppression of inflammatory arthritis by human gut-derived Prevotella histicola in humanized mice. Arthritis Rheum 68(12):2878–2888. https://doi.org/10.1002/art.39785
Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM (2013) Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 210(10):2119–2134. https://doi.org/10.1084/jem.20130252
Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, Weyand CM (2017) Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol 18(9):1025–1034. https://doi.org/10.1038/ni.3808
Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, Weyand CM (2016) Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med 8(331):331ra338. https://doi.org/10.1126/scitranslmed.aad7151
Funding
This work was supported by the JSPS Grants-in-Aid for Scientific Research (K.H.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
This article is a contribution to the special issue on The Pathogenicity of Acquired Immunity in Human Diseases - Guest Editor: Kiyoshi Hirahara
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: The word “receptor” was missing in the sentence “Because T cells do not express GM-CSF receptor [41], GM-CSF affects non-T cells.” . Full information regarding the corrections made can be found in the erratum/correction for this article.
Rights and permissions
About this article

Cite this article
Yasuda, K., Takeuchi, Y. & Hirota, K. The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol 41, 283–297 (2019). https://doi.org/10.1007/s00281-019-00733-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-019-00733-8