
Abstract
Adverse cutaneous drug reactions are recognized as being major health problems worldwide causing considerable costs for health care systems. Most adverse cutaneous drug reactions follow a benign course; however, up to 2 % of all adverse cutaneous drug eruptions are severe and life-threatening. These include acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS), Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN). Physicians should be aware of specific red flags to rapidly identify these severe cutaneous drug eruptions and initiate appropriate treatment. Besides significant progress in clinical classification and treatment, recent studies have greatly enhanced our understanding in the pathophysiology of adverse cutaneous drug reactions. Genetic susceptibilities to certain drugs have been identified in SJS/TEN patients, viral reactivation in DRESS has been elucidated, and the discovery of tissue resident memory T cells helps to better understand the recurrent site-specific inflammation in patients with fixed drug eruption.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As a result of improved treatment outcomes, longer patient survival, extended treatment courses, and polymedication of an ageing population, exposure to drugs has increased in frequency and duration. As a consequence, the likelihood of drug sensitization is rising with subsequent increases of adverse drug reactions (ADR). Of all organs affected by ADR, the skin is most frequently involved [1]. Cutaneous adverse reactions to drugs are observed in 0.1–1 % of patients during pre-marketing clinical trials, and post-marketing analyses suggest that their incidence can be as high as 1–8 % for certain types of drugs (NSAIDS, antibiotics, antiepileptics) [1]. The incidence of these reactions amongst hospitalized patients ranges from 1 to 3 %. The majority of adverse cutaneous drug eruptions are benign in nature, mostly occurring as maculopapular eruptions or urticaria [2]. Nonetheless, studies suggest that roughly a third of drug eruptions require hospital management and are classified as severe, although fortunately only 2 % of cutaneous drug eruptions are really life-threatening [1]. These include acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS), Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN). Although the pathomechanism of the benign and severe forms of cutaneous drug eruptions remains incompletely understood, great progress in this field of medicine has been made in the past few years. Improvements range from the clinical classification that is essential for a better understanding of cutaneous ADR to the identification of genetic susceptibilities to certain drugs and consequently the implementation of the first preventive genetic screening measures for selected patient groups and drug classes [1]. Allergologic workup to identify the culprit agent includes skin tests (prick, intradermal, and epicutaneous testing [3]), in vitro assays (basophil activation tests [4], lymphocyte activation tests [5], measurements of drug-induced cytokine production (e.g., Enzyme-Linked ImmunoSpot (ELISpot)) [6–8]), and/or in some cases, serum measurement of drug-specific IgEs [9]. The aim of this review is to give a current overview of the field of cutaneous drug eruptions with a special focus on the pathogenesis and immunopathology.
Benign drug-induced maculopapular rash
Drug-induced exanthematous reactions of the skin are the most common hypersensitivity reactions. They have been reported to occur in approximately 2 % of hospitalized patients [10–12]. Cutaneous exanthematous drug reactions most frequently present themselves clinically as a maculopapular rash (MPR), but they can also present in eczematoid-, psoriasiform-, or lichenoid-like pattern. The MPR is characterized as erythematous maculae and/or papules, which are symmetrically distributed on the trunk and extremities (Fig. 1). In contrast to severe adverse drug reactions like SJS and TEN, MPR is not associated with skin detachment. However, some cutaneous lesions might develop into bullous lesions. Furthermore, in MPR, the mucosae are not involved. The exanthematous lesions show a quite characteristic symmetric distribution, most often appearing on the ventral and dorsal trunk before expanding to the proximal extremities (Fig. 1). In some exanthemas with a more papular phenotype, a distribution starting on the extremities has been observed. Another pattern is characterized by a distribution of the rash to the large body folds (e.g., intertriginous, perigenital, and perianal area) sparing the central parts of the trunk. This particular pattern is observed in the so-called symmetrical drug-related intertriginous and flexural exanthema (SDRIFE or so-called Baboon syndrome [13], which will be discussed in detail further below). The chronology of the appearance of lesions in drug-induced MPR is quite characteristic and of importance in the clinical diagnosis [2]. When exposed to the drug for the first time, the skin eruption will be delayed until after a sensitization phase of at least 5 to 7 days. Typically, full-blown skin lesions form around the eight to the tenth day after the first contact with the sensitizing agent. In the following week, drug-reactive cells expand. In previously exposed and sensitized patients, renewed exposure to the same drug results in the appearance of the first skin lesions within 6 to 12 h. If typical wheals and flares appear within a few hours after drug intake, such urticarial lesions might be a first sign of a more severe anaphylactic reaction and caution should be taken before the next dose. The drug classes of pharmacological agents responsible for the majority of cutaneous adverse reactions include antibiotics, anti-infectious, and tuberculostatic drugs as well as anticonvulsant and antihypertensive agents. In contrast, there are some drugs that are very rarely associated with an adverse cutaneous reaction, such as antihistamines, digoxin, local anesthetics, steroid hormones, acetylsalicylic acid, acetaminophen, and coumarins [10].

Clinical pattern of a maculopapular rash after the intake of amoxicillin in an EBV-positive patient
Pathophysiology
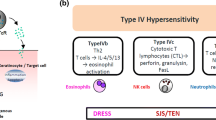
Drug-induced exanthemas are often considered as immunologically mediated hypersensitivity reactions, although such a mechanism can only be proven in a minority of cases. The underlying pathophysiological mechanisms are manifold. In many cases, an immunological type IV hypersensitivity reaction according to Coombs and Gell is the underlying pathomechanism [14]. Recently, a further subclassification of type IV hypersensitivity reactions into types IVa to IVd has been proposed (Table 1) [15]. Type IVa corresponds to a Th1-type immune reaction with macrophages as major effector cells secreting interferon-γ and stimulating a pro-inflammatory response via TNF-α and IL-12 (e.g., allergic contact dermatitis) (Fig. 2). Type IVb corresponds to a Th2-type immune response involving in particular cytokines IL-4, IL-13, and IL-5, which promote B cell expansion and subsequent plasma cell activation with production of IgE and IgG4. This type IVb pathomechanism can explain the eosinophil-rich inflammation that may be seen in many drug-induced exanthemas and is especially relevant in the pathogenesis of DRESS (Fig. 2). In type IVc reactions, the T cells themselves are the effector cells. Direct cytotoxicity is mediated by granzyme B, granulysin, and, in cells expressing Fas (CD95), by FAS ligand (CD95L) (Fig. 2). This pathomechanism is observed in maculopapular exanthemas, but more often in severe cutaneous drug reactions like SJS/TEN. Type IVd reactions are mediated by CXCL8 (interleukin 8) and granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing T cells, which recruit neutrophilic granulocytes and prevent their apoptosis. This pathomechanism appears to play a major role in AGEP (Fig. 2). Cofactors in the elicitation of exanthemas include concomitant viral infections, particularly infections involving viruses of the herpes family such as EBV, CMV, and HHV-6 [16], as well as HIV. Patients with certain autoimmune disorders such as systemic lupus erythematosus also have a higher incidence of ADR [17, 18].

Pathophysiological mechanisms underlying adverse cutaneous drug reactions. Maculopapular rash (MPR), drug reaction with eosinophilia and systemic symptoms (DRESS), acute generalized exanthematous pustulosis (AGEP), Stevens-Johnson syndrome (SJS)/ toxic epidermal necrolysis (TEN). APC antigen-presenting cell, TRM resident memory T cell, TEM effector memory T cell, GM-CSF granulocyte-macrophage colony-stimulating factor, Eo eosinophilic granulocyte, NK cell natural killer cell, Neu neutrophilic granulocyte
Management
Identification and rapid discontinuation of the culprit drug are the most important therapeutic measures. Depending on the nature and intensity of signs and symptoms, topical corticosteroids and systemic antihistamines for symptom relief, especially itch control, can be helpful. In severe cases, treatment with systemic corticosteroids over a short period of time is indicated.
Localized forms of drug-induced exanthemas
Symmetrical drug-related intertriginous and flexural exanthema
SDRIFE, or Baboon Syndrome, is a drug-related exanthema symmetrically located on the flexural areas (e.g., axillae, bottoms) [13]. SDRIFE means symmetrical drug-related intertriginous and flexural exanthema. In opposition to other drug-induced exanthematous reactions, men are more often affected than women. Perigenital and perianal involvement is associated with the involvement of the large body folds, such as the axilla, the elbows, and the knees. Papules, pustules, or vesicles are rarely found, although erythematous patches and plaques are typical. Systemic symptoms such as high fever, malaise, and visceral organ involvement are rare. The exanthema can subsequently result in rather heavy desquamation. Aminopenicillins are the most frequent causative agent, but other drugs have also been associated [13]. The chronology of SDRIFE development is still subject to controversy, with long reaction times of up to 7 days between initial exposure and onset of lesions having been reported, which could indicate a new sensitization. A T cell-mediated allergic reaction seems to be the most frequent pathomechanism. Rapid withdrawal of the causative pharmaceutical agent and administration of topical or systemic corticosteroids are recommended.
Fixed drug eruption
Solitary or few well-circumscribed, round and/or oval erythematous macules and plaques with dusky centers on the skin and/or mucous membrane are the most common lesions found in fixed drug eruption (FDE) (Fig. 3). Rarely, the lesions may evolve to become bullous. One pathognomonic characteristic of FDE is the site-specific reoccurrence of lesions with each new exposition to the causative agent. Usually, lesions appear within 30 min to 8 h after exposition [19]. The appearance of FDE lesions is often preceded and/or accompanied by a sensation of itching or burning. FDE typically resolves after discontinuation of the causative drug, leaving a circumscribed hyperpigmented area at the site of resolved lesions. Although systemic manifestations are usually absent in cases with solitary FDE lesions, multiple lesions are often associated with systemic symptoms including malaise, high fever, nausea, and arthralgia [20–23]. Most FDE lesions occur after orally administered rather than parenterally administered drugs: the most common agents are pseudoephedrine, trimethoprim, tetracycline, barbiturates, sulfonamide, mefenamic acid, acetylsalicylic acid, phenolphthalein, ibuprofen, and oxyphenbutazone. Recent data suggests that the characteristic recurrent site-specific inflammation can be at least partially explained by the role played by tissue sessile immune cells, so-called resident memory T cells (Fig. 2). Tissue resident memory T cells (TRM) provide long-lasting specific immunity to infection (e.g., herpes virus) and remain resident in the skin for a long period of time after antigen/drug exposure. They are also associated with recurring site-specific inflammatory diseases (e.g., cutaneous T cell lymphoma, psoriasis, FDE) [24–27]. The skin-infiltrating T cell phenotypes in FDE lesion are strikingly similar to TRM, as various murine in vivo studies have shown [28–30]. One hypothesis is that TRM (both self- and drug-antigen-reactive) home to the inflammatory lesions in FDE as an immunological response and remain resident after inflammation subsides. Recurrent exposition would then reflect the subsequent reactivation of these TRM in response to local or systemic inflammatory signals. Similarly, the involvement of new skin areas previously unaffected could reflect the silent distribution of sensitized effector memory T cells from the initially involved site or draining lymph nodes to previously unaffected areas. This hypothesis is further supported by the fact that patch testing only yields diagnostic evaluable results when performed at the skin site involved during previous FDE, but not in uninvolved skin. Therapeutic recommendations include identification and cessation of the causative drug. Topical application of corticosteroids over a short period of time is usually sufficient to clear the cutaneous inflammation.

Fixed drug eruption after the intake of thiazide
Stevens-Johnson syndrome and toxic epidermal necrolysis—a disease spectrum of severe cutaneous drug reactions
Toxic epidermal necrolysis (TEN) and Stevens-Johnson Syndrome (SJS) are rare but severe medical emergencies. The average reported mortality rate for SJS is between 1 and 5 %, and it increases up to 25–35 % in patients with TEN. SJS was first described by two US physicians, Stevens and Johnson. In 1922, they observed an acute mucocutaneous syndrome in two young boys, which was characterized by purulent conjunctivitis, severe stomatitis with extensive mucosal necrosis, and purpuric macules. This condition became known as SJS and was recognized as a severe mucocutaneous disease with a prolonged course and potentially lethal outcome. In most cases, it is drug-induced and it should be distinguished from erythema multiforme (EM) majus. A previously undescribed eruption resembling scalding of the skin was named toxic epidermal necrolysis in 1956 by the Scottish dermatologist Alan Lyell [31]. The association of TEN with exposure to certain medications only became clear as more patients with TEN were reported in the years following Lyell’s original publication. Increasing evidence strongly suggests that SJS and TEN are two ends of a spectrum of severe epidermolytic adverse cutaneous drug reactions differing only by the extent of skin detachment and should not be classified as two separate clinical entities [1]. SJS and TEN are rare with an incidence of roughly 1.9 cases per million inhabitants per year [32, 33]. There are several factors that seem to impact on the incidence of SJS and TEN: regional differences in drug prescription, the genetic background of patients (HLA, metabolizing enzymes), the coexistence of cancer, concomitant radiotherapy, and certain infectious diseases (e.g., HIV) [34, 35].
Non-specific symptoms such as fever, stinging eyes, and discomfort upon swallowing precede the cutaneous onset of TEN and SJS by hours to days. Cutaneous lesions in TEN and SJS usually first appear in not only the presternal region and the face but also the palms and soles. The involvement of the buccal, genital, and/or ocular mucosa, characterized by erythema and erosions, occurs in more than 90 % of patients, and in some cases the respiratory and gastrointestinal tract is also affected [36, 37]. Ocular involvement is frequent [38, 39]. Early skin lesions often present as erythematous and livid macules, which may or may not show slight infiltration and have a tendency to coalesce rapidly and evolve into tense bullae (Fig. 4). As the disease progresses, lesions form large, confluent areas of epidermal detachment. The extent of skin involvement is a major prognostic factor. However, only necrotic, already detached skin (e.g., blisters, erosions) or detachable skin (Nikolsky positive) should be included in the estimation of the extent of skin involvement. Bastuji-Garin et al. proposed classifying patients into three groups according to the degree of skin detachment: 1–10 % defined as SJS, 11–30 % defined as SJS/TEN overlap, and greater than 30 % defined as TEN [40]. Sequelae are common in SJS and TEN and include cutaneous hyper- and hypopigmentation (62.5 % of cases), nail dystrophia (37.5 % of cases), and ocular complications (50 % of cases) [41, 42].

Toxic epidermal necrolysis after the intake of allopurinol due to gout. a, b Maculopapular rash and skin detachment with erosions at the trunk. Please note the involvement of the lips and conjunctivae. c, d Full-blown TEN with massive, detached and detachable apoptotic skin (greyish color), erosions, and hemorrhagic crusts on the trunk, arm, and face
Most TEN cases are strongly associated with drug intake: i) preceding exposure to medications is reported in over 95 % of patients with TEN and ii) a strong association between drug ingestion and development of the cutaneous eruption is observed in 80 % of cases [1]. Other rare causes include infections and immunizations. The link between drugs and SJS is less strong, as only 50 % of reported SJS cases are claimed to be drug-related [1]. This is most probably an underestimation, most likely due, in part, to the confusion as to the clinical distinction between SJS and erythema multiforme. Up to date, approx. 100 compounds have been identified as the most likely triggers of individual SJS/TEN cases. The most frequently incriminated are allopurinol, antibiotics, non-steroidal anti-inflammatory drugs, and anticonvulsants [1, 43].
Pathophysiology
To date, the precise molecular and cellular pathomechanisms leading to the development of SJS/TEN are only partially understood. The pathophysiology is considered to be initiated by an immune response to an antigenic drug-host tissue complex [37, 44–47]. Today, three different concepts relating to the formation of the antigenic complex exist: i) covalent binding of the drug to a cellular peptide (hapten/pro-hapten concept); ii) non-covalent, direct interaction of the drug with a specific MHC I allotype (p-i concept); and iii) presentation of an altered-self repertoire by direct drug-MHC I interaction (altered peptide concept). Whereas the well-known hapten model is less likely to be HLA-restricted, the other two concepts favor specific HLA phenotypes. According to these concepts, the allergenic, pharmacological agent would directly bind to specific HLA molecules and/or T cell receptors without being processed beforehand in the antigen-presenting cell. In the case of the p-i concept, the mere pharmacological interaction of certain drugs with immune receptors is sufficient to elicit a drug hypersensitivity reaction [48]. However, recent publications have shown modifications of the HLA peptide repertoire through abacavir and carbamazepine, resulting in enhanced presentation of self-peptides and autoimmune reactivity (altered peptide model) [49, 50]. In line with the concepts of HLA-restricted drug presentation are reports on the genetic susceptibility, as shown by the identification of specific drug-related HLA alleles which strongly increase the susceptibility for the development of SJS or TEN [51–53]. This is of clinical importance as HLA-B*1502 screening in patients with Asian origin prior to drug intake could probably identify persons at risk from developing carbamazepine-induced SJS [54]. Several lines of evidence suggest that immune activation by the drug-tissue complex leads to a strong expression of the cytolytic molecule FasL on keratinocytes as well as granulysin and annexin A1 secretion from CTLs, NK cells, NKT cells, and monocytes [55–60] (Fig. 2). FasL- and granulysin-mediated apoptosis and/or annexin-dependent necroptosis of keratinocytes with subsequent epidermal necrosis and detachment follow. The role of CD8+ cytotoxic T cells has been discussed in fixed drug eruptions besides CD4+ helper T cells, which are a source of IL-10 [61]. This indicates that in skin inflammation, a balance between pro-inflammatory and immunomodulatory mechanisms may critically determine the clinical outcome. Interestingly, in the blister fluid of SJS/TEN patients, but not in EEM patients, Th17 cells were found alongside CD8+ T cells, the former being a source of IL-17, a cytokine-recruiting neutrophil [62]. The proportion of Th17 cells has been reported to decrease in the periphery upon treatment-related improvement of disease, suggesting a role for possible skin homing Th17 cells. In the light of the recent finding that Th17 cells may functionally trans-differentiate to T regulatory cells [63], the decrease of Th17 cells in improving SJS/TEN might be associated with a simultaneous rise of Tregs and should be monitored in future studies. In accordance, neutropenia is generally associated with a bad prognosis in SJS/TEN patients [64].
Management
Diagnostic workup should include rapid histological examination including direct immunofluorescence analysis of the skin biopsy. This approach helps rule out differential diagnoses such as autoimmune blistering diseases, bullous fixed drug eruption, acute generalized exanthematic pustulosis, and staphylococcal scalded skin syndrome, which can clinically mimic SJS/TEN. To date, prospective, controlled clinical trials showing efficacy of specific therapies in TEN are lacking. Cyclosporine, cyclophosphamide, plasmapheresis, N-acetylcysteine, TNF-α antagonists (e.g., etanercept, infliximab), systemic corticosteroids, thalidomide, and IVIg have been reported to have shown patient benefit in case reports and case series (reviewed in [1]). Regarding IVIg, early administration of high doses (≤2 g/kg) is recommended in TEN, even though the mechanism of action is still unclear [65–68]. Alternatively, a recent study has shown excellent efficacy of cyclosporine in the treatment of TEN [69, 70].
Acute generalized exanthematous pustulosis
The term pustulose exanthématique aiguë généralisée (PEAG) was first introduced by Beylot et al. in 1980 [71]. The disease is now called acute generalized exanthematous pustulosis (AGEP). The incidence of AGEP is estimated between 1 and 5 cases per million inhabitants per year. Genetic predisposition appears here also to play a role as HLA B51, DR11, and DQ3 were found to be more frequently associated with AGEP than observed in the average population [72]. The hallmark of AGEP is an edematous diffuse erythema with the rapid appearance of multiple, sterile non-follicular pustules [73] (Fig. 5). The pustules subsequently often merge together to form large areas of pustulosis. The large body flexures are classical sites of predilection. The acute phase of the disease is characterized by fever (>38 °C) and leukocytosis (neutrophil counts above 7 × 109/l) [74]. Lymphadenopathy, a slightly reduced creatinine clearance, or a mild elevation of liver enzymes may be present, but visceral organ involvement is rare [75]. AGEP usually resolves rapidly within 1–3 days after withdrawal of the causative agent leaving a characteristic collaret-shaped desquamation pattern. Differential diagnoses for AGEP include other cutaneous pustuloses, such as generalized acute pustular psoriasis; pustular vasculitis; subcorneal pustular dermatosis (Sneddon-Wilkinson); or other cutaneous adverse drug reactions like SJS, TEN, and DRESS. The vast majority of AGEP cases are drug-induced, although some viral infections have also been associated with the disease. Exposure to beta-lactam antibiotics and non-steroidal anti-inflammatory medications are the most frequent causes of AGEP.

AGEP after the intake of terbinafine
Pathophysiology
So far, the pathophysiology has remained largely unclear. The neutrophilic process may be orchestrated by T cells that release CXCL8 (IL-8) or IL-17 [76] (Fig. 2). Recent findings suggest an involvement of IL-36. Indeed, a defect in IL36Ra has been related to pustular forms of psoriasis [77]. Furthermore, Navarini et al. showed that out of a cohort of 96 patients having developed AGEP, 4 had mutations in the gene coding IL-36 [78]. Mutations in IL36RN may lead to uncontrolled IL-36 signaling and enhanced production of IL-6, IL-8, and IL-1, driving neutrophilic infiltration of the skin characteristic of the pustular eruptions of AGEP. In rare cases, a localized form of AGEP can occur [79].
Management
Rapid withdrawal of the culprit drug is the most important therapeutic action to be taken. Short-term application of topical or systemic corticosteroids may help to clear inflammation more quickly.
Drug reaction with eosinophilia and systemic symptoms
Drug reaction with eosinophilia with systemic symptoms (DRESS), also referred to as drug-induced hypersensitivity syndrome (DIHS), is a life-threatening systemic reaction affecting multiple organs and can be caused by a limited number of drugs [80]. DRESS is a rare adverse drug reaction with population-based studies in Japan reporting an incidence of 10 per million person-years. Upon drug intake, DRESS can manifest itself as late as 2–3 months after the initial contact with the causative agent, with symptoms including fever, rash, lymphadenopathy, hepatitis, and leukocytosis with eosinophilia. The cutaneous lesions are typically erythematous papules and patchy erythematous macules, which may be pruritic and confluent, sometimes resembling benign MPR and sometimes targetoid. The individual lesions are often hemorrhagic and are symmetrically distributed on the face, trunk, and extremities (Fig. 6a). Fever usually precedes the rash by 1–2 days. The most characteristic cutaneous lesions during the earliest phase of the disease are periorbital and facial edema and erythema with pinhead-sized pustules. Mucosal surfaces can show a few lesions, particularly lips and oral mucous membranes although mucous involvement is much less severe than in SJS/TEN. Cervical, axillary, and inguinal lymphadenopathy can be found in over 70 % of patients during the early course of the illness. Blood samples usually show a marked leukocytosis with atypical lymphocytosis and/or eosinophilia of various degrees which may lead to an erroneous leukemia diagnosis. Visceral involvement is common, with degree and patterns determining disease severity and prognosis (Fig. 6b). The liver (70 %), kidneys (11 %), and lungs [81] are the organs most frequently involved. Drugs causing DRESS are limited and often associated with the intake of carbamazepine, dapsone, phenytoin, salazosulfapyridine, phenobarbital, allopurinol, and zonisamide [79, 82].

a DRESS after the intake of trimethoprim/sulfamethoxazole. b Schematic diagram of the course of disease in DRESS patients
Pathophysiology
So far, the pathogenesis of DRESS has not been completely clarified. It has long been known that activated T cells play an important role [83] (Fig. 2). The second pathophysiological mechanism involves viral reactivation. Studies done in the past few years have shown that systemic manifestations of DRESS are related to human herpes virus (HHV) reactivation and to host immune response against the virus [84–86]. As the HHV can be detected in the blood of approximately 60–80 % of patients with DRESS at one time point during the course of the disease, HHV-6 reactivation has been included in the diagnostic criteria for DRESS syndrome developed by Japanese experts. In Japan, DRESS syndrome is known as drug-induced hypersensitivity syndrome or DIHS [87]. Furthermore, studies have shown that reactivation of other herpes viruses, namely, EBV, CMV, and HHV-7, is associated with systemic manifestations and flares of DRESS [16, 88, 89]. Two pathophysiological explanations have been put forward to explain viral involvement: i) an immune response against the drug with secondary viral reactivation related to a cytokine storm and ii) early viral reactivation responsible for most of the manifestations of DRESS syndrome. While there is evidence that certain drugs able to trigger DRESS can directly induce viral replication in T cells in vitro, the latency between drug intake and first appearance of DRESS symptoms remains to be explained [90]. It has been suggested that expansion of regulatory T cells might be of significance [83]; however, most experts favor the hypothesis that virus reactivation is a simple by-stander effect. While the detection of HHV reactivation might be useful in the diagnosis of DRESS, the significance of virus activation in the pathogenesis of DRESS remains unclear.
Management
In most cases, patients with DRESS are treated with systemic corticosteroids until complete disease control is achieved [91]. Care should be taken not to withdraw corticosteroids too early as this might result in reoccurrence of DRESS. In some situations, topical steroid therapy is sufficient without systemic therapy.
Conclusion
Luckily, most ADR follow a benign course. However, as approx. 2 % of all ADR are severe and potentially life-threatening, particular attention should be given to certain clinical symptoms which are to be considered red flags. These include facial edema, marked eosinophilia, mucous or conjunctival lesions, painful eyes or skin, greyish skin lesions, and epidermal detachment/erosions and indicate the increased possibility of a severe drug eruption (Table 1). Rapid identification and withdrawal of the causative drug are critical, although it is often difficult to determine the culprit drug in patients with polymedication. Therefore, the correct diagnosis of the type of skin reaction is important, as it helps to better define the likely latency and subsequently the culprit drug. Therapeutic options include topical corticosteroids as well as oral antihistamines for symptom relief, as well as systemic corticosteroids in more severe cases. In the absence of evidence supporting efficacy of other therapeutic options, high-dose IVIg treatment should be considered for cases of TEN.
References
French LE, Prins C (2013) Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis. In: Bolognia JL, Jorrizo JL, Schaffer JV (eds) Dermatology, vol 3, 3rd edn. Elsevier, New York, pp 319–334
Bircher AJ, Scherer K (2010) Delayed cutaneous manifestations of drug hypersensitivity. Med Clin North Am 94(4):711–725. doi:10.1016/j.mcna.2010.04.001
Brockow K, Garvey LH, Aberer W, Atanaskovic-Markovic M, Barbaud A, Bilo MB, Bircher A, Blanca M, Bonadonna B, Campi P, Castro E, Cernadas JR, Chiriac AM, Demoly P, Grosber M, Gooi J, Lombardo C, Mertes PM, Mosbech H, Nasser S, Pagani M, Ring J, Romano A, Scherer K, Schnyder B, Testi S, Torres M, Trautmann A, Terreehorst I, Group EEDAI (2013) Skin test concentrations for systemically administered drugs—an ENDA/EAACI Drug Allergy Interest Group position paper. Allergy 68(6):702–712. doi:10.1111/all.12142
Hoffmann HJ, Santos AF, Mayorga C, Nopp A, Eberlein B, Ferrer M, Rouzaire P, Ebo D, Sabato V, Sanz ML, Pecaric-Petkovic T, Patil SU, Hausmann OV, Shreffler WG, Korosec P, Knol EF (2015) The clinical utility of basophil activation testing in diagnosis and monitoring of allergic disease. Allergy. doi:10.1111/all.12698
Sicherer SH, Leung DY (2015) Advances in allergic skin disease, anaphylaxis, and hypersensitivity reactions to foods, drugs, and insects in 2014. J Allergy Clin Immunol 135(2):357–367. doi:10.1016/j.jaci.2014.12.1906
Porebski G, Pecaric-Petkovic T, Groux-Keller M, Bosak M, Kawabata TT, Pichler WJ (2013) In vitro drug causality assessment in Stevens-Johnson syndrome—alternatives for lymphocyte transformation test. Clin Exp Allergy 43(9):1027–1037. doi:10.1111/cea.12145
Polak ME, Belgi G, McGuire C, Pickard C, Healy E, Friedmann PS, Ardern-Jones MR (2013) In vitro diagnostic assays are effective during the acute phase of delayed-type drug hypersensitivity reactions. Br J Dermatol 168(3):539–549. doi:10.1111/bjd.12109
Rozieres A, Hennino A, Rodet K, Gutowski MC, Gunera-Saad N, Berard F, Cozon G, Bienvenu J, Nicolas JF (2009) Detection and quantification of drug-specific T cells in penicillin allergy. Allergy 64(4):534–542. doi:10.1111/j.1398-9995.2008.01674.x
Demoly P, Adkinson NF, Brockow K, Castells M, Chiriac AM, Greenberger PA, Khan DA, Lang DM, Park HS, Pichler W, Sanchez-Borges M, Shiohara T, Thong BY (2014) International consensus on drug allergy. Allergy 69(4):420–437. doi:10.1111/all.12350
Bigby M, Jick S, Jick H, Arndt K (1986) Drug-induced cutaneous reactions. A report from the Boston Collaborative Drug Surveillance Program on 15,438 consecutive inpatients, 1975 to 1982. Jama 256(24):3358–3363
Bigby M (2001) Rates of cutaneous reactions to drugs. Arch Dermatol 137(6):765–770
Nigen S, Knowles SR, Shear NH (2003) Drug eruptions: approaching the diagnosis of drug-induced skin diseases. J Drugs Dermatol: JDD 2(3):278–299
Hausermann P, Harr T, Bircher AJ (2004) Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis 51(5-6):297–310. doi:10.1111/j.0105-1873.2004.00445.x
Gell PGH, Coombs RRA (1963) Clinical aspects of immunology. Blackwell, Oxford
Scherer K, Spoerl D, Bircher AJ (2010) Adverse drug reactions to biologics. J Dtsch Dermatol Gesellschaft = J German Soc Dermatol: JDDG 8(6):411–426. doi:10.1111/j.1610-0387.2010.07339.x
Tohyama M, Hashimoto K, Yasukawa M, Kimura H, Horikawa T, Nakajima K, Urano Y, Matsumoto K, Iijima M, Shear NH (2007) Association of human herpesvirus 6 reactivation with the flaring and severity of drug-induced hypersensitivity syndrome. Br J Dermatol 157(5):934–940. doi:10.1111/j.1365-2133.2007.08167.x
Campos-Fernandez Mdel M, Ponce-De-Leon-Rosales S, Archer-Dubon C, Orozco-Topete R (2005) Incidence and risk factors for cutaneous adverse drug reactions in an intensive care unit. Revista Investig Clin; Organo Hosp Enferm Nutr 57(6):770–774
Schnyder B, Brockow K (2015) Pathogenesis of drug allergy, current concepts and recent insights. Clin Exp Allergy. doi:10.1111/cea.12591
Korkij W, Soltani K (1984) Fixed drug eruption. A brief review. Arch Dermatol 120(4):520–524
Shiohara T (2009) Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol 9(4):316–321. doi:10.1097/ACI.0b013e32832cda4c
Mizukawa Y, Yamazaki Y, Teraki Y, Hayakawa J, Hayakawa K, Nuriya H, Kohara M, Shiohara T (2002) Direct evidence for interferon-gamma production by effector-memory-type intraepidermal T cells residing at an effector site of immunopathology in fixed drug eruption. Am J Pathol 161(4):1337–1347
Mizukawa Y, Shiohara T (2010) Nonpigmenting fixed drug eruption as a possible abortive variant of toxic epidermal necrolysis: immunohistochemical and serum cytokine analyses. Clin Exp Dermatol 35(5):493–497. doi:10.1111/j.1365-2230.2009.03622.x
Mizukawa Y, Yamazaki Y, Shiohara T (2008) In vivo dynamics of intraepidermal CD8+ T cells and CD4+ T cells during the evolution of fixed drug eruption. Br J Dermatol 158(6):1230–1238. doi:10.1111/j.1365-2133.2008.08516.x
Park CO, Kupper TS (2015) The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 21(7):688–697. doi:10.1038/nm.3883
Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, Eidsmo L (2014) Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 192(7):3111–3120. doi:10.4049/jimmunol.1302313
Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA (2015) Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Trans Med 7(279):279ra239. doi:10.1126/scitranslmed.3010302
Shiohara T, Ushigome Y, Kano Y, Takahashi R (2014) Crucial role of viral reactivation in the development of severe drug eruptions: a comprehensive review. Clin Rev Allergy Immunol. doi:10.1007/s12016-014-8421-3
Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS (2012) Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483(7388):227–231. doi:10.1038/nature10851
Liu L, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS (2010) Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat Med 16(2):224–227. doi:10.1038/nm.2078
Liu L, Fuhlbrigge RC, Karibian K, Tian T, Kupper TS (2006) Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity 25(3):511–520. doi:10.1016/j.immuni.2006.06.019
Lyell A (1967) A review of toxic epidermal necrolysis in Britain. Br J Dermatol 79(12):662–671
Rzany B, Correia O, Kelly JP, Naldi L, Auquier A, Stern R (1999) Risk of Stevens-Johnson syndrome and toxic epidermal necrolysis during first weeks of antiepileptic therapy: a case-control study. Study Group of the International Case Control Study on Severe Cutaneous Adverse Reactions. Lancet 353(9171):2190–2194
La Grenade L, Lee L, Weaver J, Bonnel R, Karwoski C, Governale L, Brinker A (2005) Comparison of reporting of Stevens-Johnson syndrome and toxic epidermal necrolysis in association with selective COX-2 inhibitors. Drug Saf 28(10):917–924
Aguiar D, Pazo R, Duran I, Terrasa J, Arrivi A, Manzano H, Martin J, Rifa J (2004) Toxic epidermal necrolysis in patients receiving anticonvulsants and cranial irradiation: a risk to consider. J Neuro-oncol 66(3):345–350
Aydin F, Cokluk C, Senturk N, Aydin K, Canturk MT, Turanli AY (2006) Stevens-Johnson syndrome in two patients treated with cranial irradiation and phenytoin. J Eur Acad Dermatol Venereol:JEADV 20(5):588–590. doi:10.1111/j.1468-3083.2006.01510.x
Lebargy F, Wolkenstein P, Gisselbrecht M, Lange F, Fleury-Feith J, Delclaux C, Roupie E, Revuz J, Roujeau JC (1997) Pulmonary complications in toxic epidermal necrolysis: a prospective clinical study. Intensive Care Med 23(12):1237–1244
Revuz J, Penso D, Roujeau JC, Guillaume JC, Payne CR, Wechsler J, Touraine R (1987) Toxic epidermal necrolysis. Clinical findings and prognosis factors in 87 patients. Arch Dermatol 123(9):1160–1165
Chang YS, Huang FC, Tseng SH, Hsu CK, Ho CL, Sheu HM (2007) Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis: acute ocular manifestations, causes, and management. Cornea 26(2):123–129. doi:10.1097/ICO.0b013e31802eb264
Sotozono C, Ueta M, Koizumi N, Inatomi T, Shirakata Y, Ikezawa Z, Hashimoto K, Kinoshita S (2009) Diagnosis and treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis with ocular complications. Ophthalmology 116(4):685–690. doi:10.1016/j.ophtha.2008.12.048
Bastuji-Garin S, Rzany B, Stern RS, Shear NH, Naldi L, Roujeau J-C (1993) Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol 129:92–96
Yip LW, Thong BY, Lim J, Tan AW, Wong HB, Handa S, Heng WJ (2007) Ocular manifestations and complications of Stevens-Johnson syndrome and toxic epidermal necrolysis: an Asian series. Allergy 62(5):527–531. doi:10.1111/j.1398-9995.2006.01295.x
Magina S, Lisboa C, Leal V, Palmares J, Mesquita-Guimaraes J (2003) Dermatological and ophthalmological sequels in toxic epidermal necrolysis. Dermatology 207(1):33–36
Bentele-Jaberg N, Guenova E, Mehra T, Nageli M, Chang YT, Cozzio A, French LE, Hoetzenecker W (2015) The phytotherapeutic fenugreek as trigger of toxic epidermal necrolysis. Dermatology. doi:10.1159/000433423
Spielberg SP, Gordon GB, Blake DA, Goldstein DA, Herlong HF (1981) Predisposition to phenytoin hepatotoxicity assessed in vitro. N Engl J Med 305(13):722–727. doi:10.1056/NEJM198109243051302
Shear NH, Spielberg SP, Grant DM, Tang BK, Kalow W (1986) Differences in metabolism of sulfonamides predisposing to idiosyncratic toxicity. Ann Intern Med 105(2):179–184
Wolkenstein P, Carriere V, Charue D, Bastuji-Garin S, Revuz J, Roujeau JC, Beaune P, Bagot M (1995) A slow acetylator genotype is a risk factor for sulphonamide-induced toxic epidermal necrolysis and Stevens-Johnson syndrome. Pharmacogenetics 5(4):255–258
Dietrich A, Kawakubo Y, Rzany B, Mockenhaupt M, Simon JC, Schopf E (1995) Low N-acetylating capacity in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Exp Dermatol 4(5):313–316
Yun J, Marcaida MJ, Eriksson KK, Jamin H, Fontana S, Pichler WJ, Yerly D (2014) Oxypurinol directly and immediately activates the drug-specific T cells via the preferential use of HLA-B*58:01. J Immunol 192(7):2984–2993. doi:10.4049/jimmunol.1302306
Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, Miles JJ, Kjer-Nielsen L, Gras S, Williamson NA, Burrows SR, Purcell AW, Rossjohn J, McCluskey J (2012) Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 486(7404):554–558. doi:10.1038/nature11147
Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, Oseroff C, Lu S, Jakoncic J, de Oliveira CA, Yang L, Mei H, Shi L, Shabanowitz J, English AM, Wriston A, Lucas A, Phillips E, Mallal S, Grey HM, Sette A, Hunt DF, Buus S, Peters B (2012) Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A 109(25):9959–9964. doi:10.1073/pnas.1207934109
Hung SI, Chung WH, Liou LB, Chu CC, Lin M, Huang HP, Lin YL, Lan JL, Yang LC, Hong HS, Chen MJ, Lai PC, Wu MS, Chu CY, Wang KH, Chen CH, Fann CS, Wu JY, Chen YT (2005) HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A 102(11):4134–4139
Chung WH, Hung SI, Chen YT (2007) Human leukocyte antigens and drug hypersensitivity. Curr Opin Allergy Clin Immunol 7(4):317–323. doi:10.1097/ACI.0b013e3282370c5f
Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, Wu JY, Chen YT (2004) Medical genetics: a marker for Stevens-Johnson syndrome. Nature 428(6982):486
Chen P, Lin JJ, Lu CS, Ong CT, Hsieh PF, Yang CC, Tai CT, Wu SL, Lu CH, Hsu YC, Yu HY, Ro LS, Lu CT, Chu CC, Tsai JJ, Su YH, Lan SH, Sung SF, Lin SY, Chuang HP, Huang LC, Chen YJ, Tsai PJ, Liao HT, Lin YH, Chen CH, Chung WH, Hung SI, Wu JY, Chang CF, Chen L, Chen YT, Shen CY, Taiwan SJSC (2011) Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med 364(12):1126–1133. doi:10.1056/NEJMoa1009717
Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D, Hunziker T, Saurat JH, Tschopp J, French LE (1998) Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 282(5388):490–493
Wehrli P, Viard I, Bullani R, Tschopp J, French LE (2000) Death receptors in cutaneous biology and disease. J Investig Dermatol 115(2):141–148. doi:10.1046/j.1523-1747.2000.00037.x
Ito K, Hara H, Okada T, Shimojima H, Suzuki H (2004) Toxic epidermal necrolysis treated with low-dose intravenous immunoglobulin: immunohistochemical study of Fas and Fas-ligand expression. Clin Exp Dermatol 29(6):679–680
Chung WH, Hung SI, Yang JY, Su SC, Huang SP, Wei CY, Chin SW, Chiou CC, Chu SC, Ho HC, Yang CH, Lu CF, Wu JY, Liao YD, Chen YT (2008) Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med 14(12):1343–1350. doi:10.1038/nm.1884
Saito N, Qiao H, Yanagi T, Shinkuma S, Nishimura K, Suto A, Fujita Y, Suzuki S, Nomura T, Nakamura H, Nagao K, Obuse C, Shimizu H, Abe R (2014) An annexin A1-FPR1 interaction contributes to necroptosis of keratinocytes in severe cutaneous adverse drug reactions. Sci Transl Med 6(245):245
Viard-Leveugle I, Bullani RR, Meda P, Micheau O, Limat A, Saurat JH, Tschopp J, French LE (2003) Intracellular localization of keratinocyte Fas ligand explains lack of cytolytic activity under physiological conditions. J Biol Chem 278(18):16183–16188
Teraki Y, Shiohara T (2003) IFN-gamma-producing effector CD8+ T cells and IL-10-producing regulatory CD4+ T cells in fixed drug eruption. J Allergy Clin Immunol 112(3):609–615
Teraki Y, Kawabe M, Izaki S (2013) Possible role of TH17 cells in the pathogenesis of Stevens-Johnson syndrome and toxic epidermal necrolysis. J Allergy Clin Immunol 131(3):907–909. doi:10.1016/j.jaci.2012.08.042
Gagliani N, Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limon P, Paiva RS, Ching T, Weaver C, Zi X, Pan X, Fan R, Garmire LX, Cotton MJ, Drier Y, Bernstein B, Geginat J, Stockinger B, Esplugues E, Huber S, Flavell RA (2015) Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523(7559):221–225. doi:10.1038/nature14452
Ang CC, Tay YK (2011) Hematological abnormalities and the use of granulocyte-colony-stimulating factor in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Int J Dermatol 50(12):1570–1578. doi:10.1111/j.1365-4632.2011.05007.x
Barron SJ, Del Vecchio MT, Aronoff SC (2014) Intravenous immunoglobulin in the treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis: a meta-analysis with meta-regression of observational studies. Int J Dermatol. doi:10.1111/ijd.12423
Barron SJ, Del Vecchio MT, Aronoff SC (2015) Intravenous immunoglobulin in the treatment of Stevens-Johnson syndrome and toxic epidermal necrolysis: a meta-analysis with meta-regression of observational studies. Int J Dermatol 54(1):108–115. doi:10.1111/ijd.12423
Huang YC, Li YC, Chen TJ (2012) The efficacy of intravenous immunoglobulin for the treatment of toxic epidermal necrolysis: a systematic review and meta-analysis. Br J Dermatol 167(2):424–432. doi:10.1111/j.1365-2133.2012.10965.x
Prins C, Kerdel FA, Padilla RS, Hunziker T, Chimenti S, Viard I, Mauri DN, Flynn K, Trent J, Margolis DJ, Saurat JH, French LE (2003) Treatment of toxic epidermal necrolysis with high-dose intravenous immunoglobulins: multicenter retrospective analysis of 48 consecutive cases. Arch Dermatol 139(1):26–32
Valeyrie-Allanore L, Wolkenstein P, Brochard L, Ortonne N, Maitre B, Revuz J, Bagot M, Roujeau JC (2010) Open trial of ciclosporin treatment for Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 163(4):847–853. doi:10.1111/j.1365-2133.2010.09863.x
Kirchhof MG, Miliszewski MA, Sikora S, Papp A, Dutz JP (2014) Retrospective review of Stevens-Johnson syndrome/toxic epidermal necrolysis treatment comparing intravenous immunoglobulin with cyclosporine. J Am Acad Dermatol 71(5):941–947. doi:10.1016/j.jaad.2014.07.016
Beylot C, Bioulac P, Doutre MS (1980) Acute generalized exanthematic pustuloses (four cases) (author’s transl). Ann Dermatol Venereol 107(1-2):37–48
Bernard P, Lizeaux-Parneix V, Miossec V et al (1995) HLA et prédisposition génétique dans les pustuloses exanthématiques (PEAG) et les exanthémes maculo-papuleux (EMP). Ann Dermatol Venerol 122:S38–S39
Sidoroff A, Halevy S, Bavinck JN, Vaillant L, Roujeau JC (2001) Acute generalized exanthematous pustulosis (AGEP)—a clinical reaction pattern. J Cutan Pathol 28(3):113–119
Roujeau JC, Bioulac-Sage P, Bourseau C, Guillaume JC, Bernard P, Lok C, Plantin P, Claudy A, Delavierre C, Vaillant L et al (1991) Acute generalized exanthematous pustulosis. Analysis of 63 cases. Arch Dermatol 127(9):1333–1338
Leclair MA, Maynard B, St-Pierre C (2009) Acute generalized exanthematous pustulosis with severe organ dysfunction. CMAJ 181(6-7):393–396. doi:10.1503/cmaj.090137
Britschgi M, Pichler WJ (2002) Acute generalized exanthematous pustulosis, a clue to neutrophil-mediated inflammatory processes orchestrated by T cells. Curr Opin Allergy Clin Immunol 2(4):325–331
Stenerson M, Dufendach K, Aksentijevich I, Brady J, Austin J, Reed AM (2011) The first reported case of compound heterozygous IL1RN mutations causing deficiency of the interleukin-1 receptor antagonist. Arthritis Rheum 63(12):4018–4022. doi:10.1002/art.30565
Navarini AA, Valeyrie-Allanore L, Setta-Kaffetzi N, Barker JN, Capon F, Creamer D, Roujeau JC, Sekula P, Simpson MA, Trembath RC, Mockenhaupt M, Smith CH (2013) Rare variations in IL36RN in severe adverse drug reactions manifesting as acute generalized exanthematous pustulosis. J Invest Dermatol 133(7):1904–1907. doi:10.1038/jid.2013.44
Tresch S, Cozzio A, Kamarashev J, Harr T, Schmid-Grendelmeier P, French LE, Feldmeyer L (2012) T cell-mediated acute localized exanthematous pustulosis caused by finasteride. J Allergy Clin Immunol 129(2):589–594. doi:10.1016/j.jaci.2011.07.033
Tetsuo Shiohara YK, Ryo Takahashi, Tadashi Ishida and Yoshiko Mizukawa (2012) Adverse cutaneous drug eruptions. In: LE F (ed) Drug-induced hypersensitivity syndrome (DIHS/DRESS): Recent advances in the diagnosis, pathogenesis and management, vol 1. Karger, Basel, pp 122-135
Shiohara TTR, Kano Y (2007) Drug hypersensitivity. In: Pichler W (ed) Drug-induced hypersensitivity syndrome and viral reactivation, vol 1. Karger, Basel, pp pp 251–266
Cacoub P, Musette P, Descamps V, Meyer O, Speirs C, Finzi L, Roujeau JC (2011) The DRESS syndrome: a literature review. Am J Med 124(7):588–597. doi:10.1016/j.amjmed.2011.01.017
Takahashi R, Kano Y, Yamazaki Y, Kimishima M, Mizukawa Y, Shiohara T (2009) Defective regulatory T cells in patients with severe drug eruptions: timing of the dysfunction is associated with the pathological phenotype and outcome. J Immunol 182(12):8071–8079. doi:10.4049/jimmunol.0804002
Picard D, Janela B, Descamps V, D’Incan M, Courville P, Jacquot S, Rogez S, Mardivirin L, Moins-Teisserenc H, Toubert A, Benichou J, Joly P, Musette P (2010) Drug reaction with eosinophilia and systemic symptoms (DRESS): a multiorgan antiviral T cell response. Sci Transl Med 2:46–46ra62. doi:10.1126/scitranslmed.3001116
Descamps V, Valance A, Edlinger C, Fillet AM, Grossin M, Lebrun-Vignes B, Belaich S, Crickx B (2001) Association of human herpesvirus 6 infection with drug reaction with eosinophilia and systemic symptoms. Arch Dermatol 137(3):301–304
Suzuki Y, Inagi R, Aono T, Yamanishi K, Shiohara T (1998) Human herpesvirus 6 infection as a risk factor for the development of severe drug-induced hypersensitivity syndrome. Arch Dermatol 134(9):1108–1112
Shiohara T, Iijima M, Ikezawa Z, Hashimoto K (2007) The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations. Br J Dermatol 156(5):1083–1084. doi:10.1111/j.1365-2133.2007.07807.x
Kano Y, Hiraharas K, Sakuma K, Shiohara T (2006) Several herpesviruses can reactivate in a severe drug-induced multiorgan reaction in the same sequential order as in graft-versus-host disease. Br J Dermatol 155(2):301–306. doi:10.1111/j.1365-2133.2006.07238.x
Descamps V, Mahe E, Houhou N, Abramowitz L, Rozenberg F, Ranger-Rogez S, Crickx B (2003) Drug-induced hypersensitivity syndrome associated with Epstein-Barr virus infection. Br J Dermatol 148(5):1032–1034
Mardivirin L, Descamps V, Lacroix A, Delebassee S, Ranger-Rogez S (2009) Early effects of drugs responsible for DRESS on HHV-6 replication in vitro. J Clin Virol: Off Publ Pan Am Soc Clin Virol 46(3):300–302. doi:10.1016/j.jcv.2009.08.006
Descamps V, Ranger-Rogez S (2014) DRESS syndrome. Joint Bone Spine: Revue Rhum 81(1):15–21. doi:10.1016/j.jbspin.2013.05.002
Bircher AJ (2011) Uncomplicated drug-induced disseminated exanthemas. In: French L (ed) Adverse cutaneous drug eruptions, vol 1. Karger, Basel, pp 79–96
Funding
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Informed consent
Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Conflict of interest
The authors declare that they have no competing interests.
Additional information
This article is a contribution to the Special Issue on Immunodermatology - Guest Editors: Lars French and Alexander Navarini
Rights and permissions
About this article

Cite this article
Hoetzenecker, W., Nägeli, M., Mehra, E.T. et al. Adverse cutaneous drug eruptions: current understanding. Semin Immunopathol 38, 75–86 (2016). https://doi.org/10.1007/s00281-015-0540-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-015-0540-2