
Abstract
Rheumatoid arthritis (RA) is an autoimmune disease affecting ∼1 % of the population. Although major advances have been made in the treatment of RA, relatively little is known about disease pathogenesis. Autoantibodies, present in approximately 60 % of the patients with early disease, might provide indications for immunological mechanisms underlying RA. Among the RA-associated autoantibodies, especially anti-citrullinated protein antibodies (ACPAs) have been studied intensively in the last decade. The discovery of ACPAs resulted into novel insight in RA pathogenesis and allowed division of the heterogeneous entity of RA into an ACPA-positive and ACPA-negative subset of disease. Other autoantibodies discovered in the serum of RA patients, including rheumatoid factors (RFs) targeting human IgG and anti-peptidylarginine deiminase (PAD)3/4 antibodies reactive against and activating the enzyme involved in citrullination, might contribute in collaboration with ACPAs to a feed-forward loop to aggravate erosive outcome of disease. Recently, a novel autoantibody system associated with RA was identified. These autoantibodies recognize carbamylated proteins (anti-CarP antibodies) and are detected in approximately 20 % of ACPA-negative patients, suggesting another parameter to sub-classify RA. In this review, the implication of autoantibodies in RA pathogenesis, diagnosis, prognosis and as biomarker for personalized medicine is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic autoimmune disease affecting 0.5–1 % of the population in industrialized countries. RA is characterized by persistent synovitis, systemic inflammation and the presence of several autoantibodies. The clinical course is extremely variable, showing a wide spectrum of clinical manifestations ranging from mild and self-limiting disease to rapidly progressive inflammation with joint destruction and severe physical disability [1, 2]. Because of the heterogeneous character of the disease, classification criteria have been developed to provide a basis for disease definition and to standardize recruitment in clinical trials and comparison of the results of multi-centred studies. Since 1987, RA has been classified based on the ACR 1987 criteria defined by a regression analysis of disease characteristics of “classic cases”. This resulted in the inclusion of a heterogeneous set of patients with conceivably different “pathogenic” backgrounds [3]. The ACR 1987 criteria are well accepted for disease definition, but have a significant limitation in identification of patients with recent-onset RA. Especially, this latter subset of patients would benefit from early effective intervention to avoid progress to the chronic, erosive state of RA exemplified in the 1987 criteria. Therefore, ACR/EULAR 2010 criteria were developed [4], which display a higher sensitivity but a lower specificity for RA to be able to diagnose RA and start treatment early in disease.
The heterogeneous character of RA is also reflected by a wide range of responses to therapies. Patients do not respond uniformly to therapeutics, conceivably because the specific biological pathway targeted by the drug may not be active in the particular patient subgroup. Currently, it is difficult to predict effectiveness of a specific therapy in individual patients because we lack knowledge to sub-classify RA based on biological pathways involved in pathogenesis. For the development of a personalized medicine approach or even more ambitious, curative therapeutics, it is essential to understand the immunological mechanisms underlying RA. Although RA is a considerable health problem, still relatively little is known on its immunopathology. Refinement of the understanding of molecular pathways involved in disease pathogenesis could be achieved by combined knowledge on RA-associated genes, environmental factors and the presence of serological factors.
Genetic and environmental contributions to RA
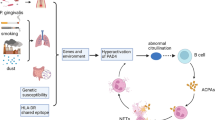
Both genetic and environmental risk factors contribute the development of RA. Smoking is the most prominent environmental factor, with its largest effect in autoantibody-positive patients [5, 6]. Although the prevalence of RA in the general population is <1 %, among monozygotic twins with one of them affected, for the unaffected sibling, the prevalence increases to 12.3 to 15.4 %. This implicates that genetic factors have a substantial impact on susceptibility to RA, with the genetic contribution estimated around 50 to 60 % [7]. The identification of disease-associated genes provided valuable insight into biological pathways that might contribute to RA pathogenesis. The strongest predisposing variants of the genetic risk factors are found in the human leukocyte antigen (HLA) alleles, accounting for 30 to 50 % of overall genetic susceptibility to RA [8, 9]. Already in 1969, it was demonstrated that in mixed lymphocyte cultures, peripheral blood lymphocytes from RA patients were non-reactive to each other [10] and that this non-reactivity was due to the sharing of genes in the HLA region [11]. Subsequently, it was discovered that multiple RA risk alleles within the HLA-DRB1 gene share a conserved amino acid sequence, leading to the “shared epitope” (SE) hypothesis [12]. The odds ratio to develop RA for one HLA SE allele is around 4, whereas two SE copies increase the odds ratio to approximately 12 [13]. The underlying mechanism by which HLA SE alleles predispose to the development of RA is still not understood, despite much progress in perception of the structure and function of HLA-DRB1 molecules. HLA-DR molecules are anchored in the membrane of antigen-presenting cells to present antigenic peptides to T lymphocytes. The T cell receptor recognizes residues from both the peptide as well as the HLA-DR molecule itself. The SE motif is situated at the part of the HLA-DR molecule that binds to the peptide. At this position, the SE influences peptide binding and presentation to T cells (Fig. 1). So far, no specific arthritogenic peptides have been identified that bind to SE HLA-DR molecules and contribute to RA.

Antigen-activated B cells require help from antigen-specific T cells to secrete isotype-switched antibodies. B cells recognizing citrullinated or carbamylated human proteins internalize antigen with their B cell receptor. After processing, antigen is presented to T cells on HLA-DR molecules anchored in the B cell membrane. The SE motif is situated at the part of the HLA-DR molecule that binds to the peptide and influences at this position peptide binding and subsequent presentation to T cells
It was not until 2001, almost 30 years after the identification of HLA-alleles as a risk factor for RA, that non-HLA loci associated with RA were revealed [14]. Interestingly, one of these loci encode the gene for PADI4, an enzyme that citrullinates proteins and hereby creating targets for anti-citrullinated protein antibodies (ACPAs) [15]. This directly implicates citrullination as important mechanism for RA development. After 2004, the identification of new genetic risk factors underwent an unprecedented acceleration. Genome-wide association studies rapidly expanded the number of genes associated with ACPA-positive RA. Currently, more than 30 genetic risk factors have been identified [16]. Most of them are located near genes that are linked to T or B cell activation or differentiation, and cytokine signalling, implicating—not surprisingly—immune-related events as an important component of RA-disease pathogenesis [17].
In addition to genes predisposing for RA, also, protective genes that are more often present in healthy controls compared to RA patients have been identified. A meta-analysis was performed to investigate which HLA-DRB1 alleles are associated with protection in ACPA-positive RA and ACPA-negative RA, stratified for SE alleles. In ACPA-positive RA, the alleles that conveyed protection after stratification for SE were HLA-DRB1*13 alleles, containing the amino acid DERAA. In ACPA-negative RA, no robust associations were found with protective HLA-DRB1 alleles [18].
RA-associated autoantibodies
The presence of autoantibodies in serum of RA patients has been recognized already for over 70 years. Rheumatoid factor (RFs), targeting the Fc part of human IgG were the first group of autoantibodies discovered [19]. Unfortunately, RFs do not have high specificity for RA and can be detected in up to 15 % of the healthy individuals. In 1964 and 1979, additional serum factors present in RA patients were described, anti-perinuclear factor antibodies and anti-keratin antibodies, respectively [20, 21]. Although their specificity for RA was very high, it was difficult to visualize the presence of these autoantibodies. Therefore, in daily practice, RF was still used to aid RA diagnosis, and consequently, the presence of RF was taken up in the ACR 1987 criteria for RA. It was not until 1995 when it was realized that anti-perinuclear factor antibodies and anti-keratin antibodies are the same autoantibodies [22] and that the epitopes they both target are generated by deimination of argine residues resulting in citrulline residues [23]. In 2002, the first commercial test to visualize ACPA as biomarker for RA to allow routine testing became available by the development of the cyclic citrullinated peptide (CCP)-2 assay. Extensive research into this unique autoantibody system boosted the understanding and classification of RA considerably, with both RF and ACPA being part of the ACR/EULAR 2010 classification criteria [4]. Recently, a novel subset of autoantibodies in sera of RA patients recognizing carbamylated proteins (anti-CarP antibodies) was reported [24]. This antibody system is independent from ACPA because antibodies from sera of RA patients can discriminate between citrullinated and carbamylated antigens. Correspondingly, a substantial part of ACPA-negative patients harbour anti-CarP antibodies [24]. Recently, anti-PAD antibodies targeting the enzyme involved in citrullination of proteins received much attention by the discovery that these antibodies not only target but also activate PAD [25]. The recent identification of a novel autoantibody system present in RA, together with the recent finding of autoantibodies in formerly defined ANCA-negative vasculitis [26], make the discovery of novel disease-specific autoantibodies in presumably autoantibody negative RA plausible.
Posttranslational modifications
Antibodies are important tools to protect the body from pathogenic intruders by neutralization of the pathogen and activation of the immune system. Unfortunately, individuals may produce antibodies directed against self-tissues, causing unwanted tissue damage observed in autoimmune diseases. Autoantibodies can be directed against chemical or enzymatic altered self-tissue, such as posttranslationally modified proteins. Although these modifications might occur in response to stress, also, under normal and pathological conditions, posttranslational modifications take please. However, they are generally invisible for the immune system. Apparently, in RA, the immune system is triggered to respond to modified self-proteins by the induction of an autoimmune response.
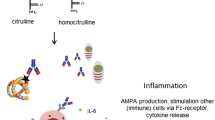
Over the past few years, important insight has been gained into the occurrence and aetiophathology of RA by the discovery of RA-specific autoantibodies directed against citrullinated proteins (ACPA) and carbamylated proteins (anti-CarP antibodies). Carbamylation and citrullination are both posttranslational modifications, resulting in carbamylated and citrullinated (self-) proteins, respectively, in which a positively charged amino acid is replaced by a neutral amino acid. Citrullinated proteins are generated by PAD enzymes, whereas carbamylation of proteins occurs when the amino acid lysine is converted to homocitrulline by a chemical reaction. The chemical structure of citrulline resembles homocitrulline, but is located at different positions of proteins as arginine and lysine are, by definition, located at different positions. Furthermore, citrulline is one methylene group longer (Fig. 2). Although some autoantibodies are reactive to both structures, also anti-CarP antibodies have been found that do not react to citrulline and vice versa [24].

Schematic illustration of citrullination and carbamylation. Citrullination and carbamylation occur on distinct amino acids by different mechanisms to generate similar posttranslational modifications
The posttranslational modification of arginine to citrulline by PAD enzymes is essential for the generation of the epitope recognized by ACPA. Citrullination is a physiological process occurring under different conditions, including inflammation [27]. It is tempting to speculate that presentation of peptides bearing posttranslational modifications can elicit highly specific T and B cell responses that might contribute to autoimmune disease. This hypothesis was reinforced by the demonstration that presentation of citrullinated peptides by B cells was not detected under normal culture conditions [28]. However, B cell stimulation by engagement of the B cell receptor resulted in autophagy associated with the presentation of citrullinated peptides to CD4 T cells [29]. These findings are in line with the notion that a cascade of immunological events precedes autoimmunity, starting with “inflammation-induced citrullination” and B cell receptor triggering followed by (autophagy-facilitated) HLA-restricted citrullinated antigen presentation to T cells, ultimately resulting in breach of T cell tolerance and development of ACPA responses (Fig. 1). To dissect the events that occur in the break of tolerance against citrullinated proteins, animal models of arthritis could be used. However, it is unclear at the moment whether a genuine ACPA response can be induced in animal models of arthritis [30, 31].
Carbamylated proteins contain a homocitrulline residue that has been generated through a chemical reaction in which mainly lysine is converted into a homocitrulline through the action of cyanate. Cyanate is naturally present in the body and in equilibrium with urea [32]. Under steady-state physiological conditions, the cyanate concentration might be too low to allow extensive carbamylation. Indeed, in renal failure, the urea concentration increases, resulting in extensive carbamylation of proteins [33]. Interestingly, smoking, the most prominent environmental risk factor for RA, increases the cyanate concentration and might consequently enhance carbamylation. However, most carbamylation is believed to be provoked under inflammatory conditions, when MPO is released from neutrophils. MPO converts thiocyanate to cyanate, allowing extensive carbamylation [34]. RA is characterized by inflamed joints; therefore, it is conceivable that carbamylation is a naturally occurring process in the inflamed synovium.
In addition to posttranslational modifications themselves, also, autoantibodies against the enzymes involved in generating posttranslational modifications are described in RA [35–38]. It was recently discovered that a subset of anti-PAD4 autoantibodies increase the catalytic efficiency of PAD4 by decreasing the enzyme’s requirement for calcium [25]. This implies that anti-PAD autoantibodies might be involved in the generation of the ligands for ACPA.
Characteristics of RA-associated autoantibodies
ACPA antigen recognition
ACPAs recognize posttranslationally modified proteins with the “non-encoded”, amino acid citrulline generated after enzymatic conversion of arginine. The first enzyme-linked immunosorbent assay (ELISA) to detect ACPA based upon the molecular identity of the antigens recognized by ACPA was developed in 1998, using citrullinated peptides derived from several filaggrin epitopes [39]. To allow routine testing, in 2002, the first commercial ELISA test was available with CCP to detect ACPA as biomarker in RA This anti-CCP ELISA proved to be extremely specific (98 %) for RA and had a significantly higher specificity in comparison with the IgM RF ELISA [40]. To confirm that the antibodies detected by CCP ELISA are in fact ACPA, it was shown that affinity-purified anti-CCP antibodies were able to recognize citrullinated proteins on Western blot. This indicates that anti-CCP antibodies recognize multiple citrullinated proteins and are a collection of ACPA. To determine to what extent antibody responses to different citrullinated antigens are cross-reactive, a method was established in which citrullinated antigen-specific antibodies were eluted from ELISA plates and then used for detection of other citrullinated antigens with Western blot or in ELISA, with citrullinated or control peptides as inhibitor. These experiments showed that antibodies specific for citrullinated fibrinogen and citrullinated myelin basic protein were cross-reactive. This cross-reactivity is not complete, as distinct non-cross-reactive responses can also be detected in RA patients [41]. Correspondingly, murine monoclonal ACPA cross-reacted with variable degrees to citrullinated epitopes on different peptide backbones [42]. Furthermore, by single B cell-based cloning technology it was demonstrated that human citrulline-reactive monoclonal antibodies reacted with more than one citrullinated antigen [43], confirming the cross-reactive nature of ACPA. This cross-reactivity towards different citrullinated proteins indicates that citrulline is recognized in a hapten-like manner to elicit immune responses towards several (self-) proteins.
Epitope spreading, an increase or shift in the antigen recognition profile, can have important pathophysiological consequences in autoimmune diseases. It was demonstrated that ACPA epitope spreading occurs several years prior to the onset of clinical RA [44]. At later stages of the disease course, the ACPA fine specificity does not change anymore [45]. Therefore, it is suggested that the initial autoimmune response is directed towards a limited, but not always the same set of autoantigens. At a later stage in the development of the autoimmune response, an increasing number of epitopes are recognized. This is paralleled by an increase in ACPA levels which also occurs before onset of symptoms; after which, both the ACPA-epitope recognition profile and the level of ACPA stabilize to relatively high levels at onset of disease [44] (Fig. 3). It is estimated that in ACPA-positive RA patients, up to 1 in 80 IgG molecules in serum are ACPA, indicating that absolute ACPA levels are high compared to other antibodies in established disease [46].

ACPA maturation in RA development. Before onset of arthritis, the epitope recognition profile, antibody levels, avidity for its antigen, glycosylation pattern and isotype usage of ACPA change
Within the ACPA repertoire, the presence and detection of different fine specificities offered hope for the possibility to use the ACPA epitope recognition profile as biomarker to predict disease development and progression. Although epitope spreading occurs on the group level before patients fulfill the ACR 1987 criteria for RA, current evidence indicated that this does not correlate with clinical outcome as measured by rate of joint destruction. For example, no statistically significant differences in clinical or in radiological outcomes were demonstrated between the ACPA recognizing citrullinated enolase peptide 1 (CEP1)-positive and negative subset of RA patients [47]. Since ACPA-positive patients display a heterogeneous ACPA recognition profile, cluster analyses have been performed in an attempt to identify subgroups of patients on the basis of their ACPA recognition profile. No apparent clustering of patients was found, and the recognition of specific citrullinated epitopes was not associated with baseline characteristics. Furthermore, patients with an extended fine-specificity repertoire did not display differences in baseline characteristics or joint damage after 7 years of follow-up compared to ACPA-positive patients recognizing few peptides [48], also indicating the limited use to detect ACPA fine specificities for diagnostic or prognostic purposes.
ACPA and the HLA system
Intriguingly, the strong association between SE-encoding HLA-DRB1 alleles and RA is only observed for ACPA-positive disease [13]. It was demonstrated that the HLA-SE-alleles influence not only the magnitude but also the specificity of the ACPA response by the finding that HLA-SE alleles predispose to the development of antibodies against citrullinated vimentin peptides, but not to the development of antibodies against citrullinated fibrinogen peptides [49]. However, although several ACPA fine specificities are more readily formed under the influence of SE alleles, again, no association between the presence of the HLA-SE alleles and radiological damage was observed in the ACPA-positive patient group [50]. These data indicate that although the HLA-SE alleles are instrumental in shaping the ACPA-repertoire, the “shape” of the ACPA-repertoire does not translate to meaningful clinical differences.
ACPA isotype switching
Antigen-activated B cells initially secrete antibodies of the IgM isotype, and after activation by T helper cells, they undergo isotype switching to produce IgG, IgA or IgE antibodies (Fig. 1). Isotype switching is a process in which the constant region of the antibody is changed, to affect the effector function of an antibody, whereas the variable region of the antibody, important for antigen recognition, is not affected. ACPA have been found in different forms, including IgG, IgA and IgM. The fine specificity and isotype usage of ACPA in health and disease differs. The ACPA-response present in healthy family members of patients with RA uses fewer ACPA isotypes than ACPA from RA patients [51]. Nonetheless, ACPA of both the IgG and IgA isotypes pre-date the onset of RA by years and predict the development of RA with the highest predictive value for IgG antibodies [52]. Although the usage of isotypes by ACPA increases during the development towards RA [45], the ACPA isotype distribution does not expand in established disease [53], indicating that most of the expansion of isotype usage by ACPA takes places before the onset of arthritis (Fig. 3). The number of isotypes used by ACPA does not only associate with RA development but also with RA progression, as the magnitude of the ACPA isotype profile at baseline reflects the risk of future radiographic damage [54].
ACPA avidity maturation
To evaluate if the evolution of the ACPA response differs intrinsically from the protective responses against pathogens, the avidity of ACPA in relation to the avidity of antibodies against recall antigens has been analysed. Intriguingly, the avidity of ACPA was significantly lower than the avidity of antibodies to the recall antigens tetanus toxoid and diphtheria toxoid. Moreover, ACPA did not show avidity maturation during longitudinal follow-up, even in patients who displayed extensive isotype switching [55]. These observations indicate that the evolution of ACPA differs from the development of antibodies against recall antigens as isotype-switching and avidity maturation are apparently uncoupled. The reason for low avidity of the ACPA response compared to “conventional” antibody responses against recall antigens in the same patients is unknown. However, it is conceivable that these observations are related to the notion that citrullinated antigens are abundantly expressed in the human body, including B cell follicles in lymph nodes. This avoids competition for only the strongest antigen-binding B cell receptors to bind these citrullinated antigens to receive survival signals. Abundant expression of citrullinated antigens could also explain the presence of high levels of IgM ACPA in RA patients [46]. Often, the presence of IgG prevents priming of new IgM-positive B cells, as a consequence of higher avidity for the same antigen. This principle is, for example, used in the context of Rhesus-antigen immunity, where a mother of a Rhesus-positive child is injected with anti-Rhesus IgG shortly after delivery, to prevent the formation of a new anti-Rhesus response by low-binding anti-Rhesus IgM from the mother. Probably, IgG ACPA is not able to capture the citrullinated antigens preventing them from binding to IgM-producing B cells, because of the low avidity of the IgG ACPA and/or the high concentration of citrullinated antigens. Interestingly, the detection of IgM ACPA also indicates that the presence of a continuous ongoing ACPA-immune response as IgM has a short half-life and is highly expressed in the synovial compartment [46].
Glycosylation of ACPA
Already in 1985, the importance of antibody glycosylation in RA was acknowledged. To obtain more information on autoantigenic reactivity of RF, Parekh et al. compared in detail the N-glycosylation pattern of serum IgG isolated from normal individuals and from patients with RA. The results of this study indicated that disease is associated with changes in the relative extent of galactosylation of IgG antibodies. The investigators proposed that RA might be a “glycosylation disease”, reflecting changes in the intracellular processing or postsecretory degradation of N-linked oligosaccharides [56]. After the discovery of ACPA, IgG ACPA glycosylation was determined, to address the occurrence of specific glycosylation features of antigen-specific subpopulations of antibodies. Glycosylation patterns of ACPA-Fc were analysed because glycosylation of the Fc part can underlie pro- versus anti-inflammatory characteristics of antibodies [57]. It was demonstrated that Fc glycosylation of IgG1 ACPA varies considerably from total serum IgG1 of patients with respect to galactosylation patterns [58]. Since RA is characterized by inflammation of the synovium, Fc glycosylation of IgG1 ACPA derived from serum and synovial fluid was compared. It was demonstrated that Fc glycosylation of ACPA differs considerably between synovial fluid and serum [59]. This might indicate that an inflammatory environment contributes to antibody glycosylation profiles. Indeed, environmental factors, including retinoic acid, TLR ligands and cytokines have specific impact on IgG1 Fc-glycosylation patterns, whereas no altered glycosylation patterns were observed for total cellular glycan proteins [60]. Because ACPA is present years before onset of disease and some molecular characteristics of ACPA change before disease onset, it was investigated whether ACPA exhibit specific changes in Fc glycosylation prior to the onset of arthritis. ACPA-IgG1 from arthralgia patients display Fc glycosylation patterns comparable to that of asymptomatic blood donors. However, around 3 months before disease onset, ACPA display significant changes in Fc galactosylation and fucosylation [61], again, indicating that specific changes in the molecular composition of ACPA occur before clinical precipitation of disease (Fig. 3).
Anti-CarP antibodies
The chemical structure of homocitulline highly resembles citrulline (Fig. 2). Therefore, it was hypothesized that ACPA might recognize homocitrulline present on proteins (carbamylated proteins) as well. Surprisingly, it was shown that most ACPA did not recognize peptides in which the citrulline residue was replaced by a homocitrulline residue, indicating that ACPA can discriminate between citrulline and homocitrulline. Intriguingly, reactivity towards homocitrulline containing antigens was noted in some patients in these studies. Therefore, an assay was developed to detect antibodies against carbamylated proteins. Interestingly, it was demonstrated that 45 % of the RA patients harbour IgG antibodies recognizing carbamylated proteins. Of note, not only ACPA-positive RA patients showed anti-CarP reactivity but also 16–30 % of the ACPA-negative patients harboured anti-CarP antibodies (Fig. 4) [24]. This observation demonstrated the identification of a novel group of autoantibodies present in RA. Since the discovery of this unique novel autoantibody system in RA was only a few years ago, the molecular characteristics of anti-CarP antibodies are to be defined. Likewise, it is currently not known whether carbamylated proteins are present in (inflamed) joints. The possibility to induce anti-CarP immunity by vaccination with carbamylated and citrullinated proteins [62, 63] and the emerge of anti-CarP antibodies upon arthritis induction in animal models (Stoop et al., submitted for publication) might allow to study the driving mechanisms underlying the anti-CarP immune response and their contribution to arthritis.

Percentage of RA patients positive for IgG ACPA and anti-CarP antibodies and IgA ACPA and anti-CarP antibodies
Pathogenic potential of RA-associated autoantibodies
The strong association with RA suggests a prominent role for ACPA in disease pathogenesis. The presence of ACPA in patients already diagnosed with RA predicts severe joint damage [64], whereas the presence of ACPA predicts progression towards RA in patients with undifferentiated arthritis [65]. Furthermore, the presence and levels of ACPA are indicative for arthritis development in arthralgia patients [66, 67]. ACPA-positive arthralgia patients show local subclinical inflammation in small joints already in the pre-clinical phase [68]. Likewise, it was demonstrated that most ACPA-positive RA patients appear to be ACPA positive already years before onset of disease [69]. In addition, polymorphisms in genes encoding the PAD enzymes associate with RA [15], indicating that the enzymes generating antigens for ACPA are involved in pathogenesis. Nonetheless, although suggestive, these observations do not show direct evidence of ACPA implication in disease pathogenesis.
Effector functions of ACPA
Intriguingly, shortly before the start of clinical symptoms, the epitope recognition profile, isotype usage, avidity maturation and glycosylation pattern of ACPA change [44, 45, 52, 61, 70] (Fig. 3), indicating that maturation of the ACPA response is required for its pathogenicity. Likewise, the magnitude of the ACPA isotype profile at baseline is associated with radiographic damage, showing an odds ratio of 1.4-fold increase for every additional isotype [54]. The mechanism behind elevated disease severity in association with isotype usage is still unknown; however, isotype usage might be implicated in the effector functions recruited by ACPA. These include complement activation [71], activation of Fc-receptor positive cells [72] and osteoclast activation. The presence of ACPA was associated with serum markers for osteoclast-mediated bone degradation. Interestingly, ACPA bind to osteoclast surfaces, resulting in osteoclastgenesis and bone degradation. This was confirmed by adoptive transfer of human ACPA into mice, leading to osteopenia and increased osteoclastgenesis [73].
Indications for a pathogenic role of ACPA were found in the synovium of ACPA-positive RA patients as well. Histological differences, mainly with regard to infiltrating lymphocytes, in the inflamed joint of ACPA-positive and ACPA-negative RA patients have been visualized [74]. Indeed, targets for ACPA are present in joints of RA patients, demonstrated by the detection of many citrullinated proteins in synovial fluid samples from the inflamed joints of RA patients [75]. Because ACPA display lower avidity to its antigen compared to antibodies against recall antigens [55], ACPA avidity in relation to biological activity and clinical outcome was analysed. Unexpectedly, patients with low-avidity ACPA display a higher rate of joint destruction [76]. The mechanisms behind this remain elusive, but could be due to a higher ability of low-avidity antibodies to penetrate tissues.
ACPA and neutrophil extracellular traps
The strong connection of HLA with (ACPA-positive) disease [13] elicits the focus of RA research on the adaptive immune system. Recently, a connection between the innate and adaptive immune system in RA was proposed by the observation that PAD4 is required for neutrophil extracellular trap (NET) formation by neutrophils [77]. NETs are highly decondensed chromatin structures that are formed by neutrophils to trap and kill bacteria. The observation that PAD4 is required for NETosis is interesting, because enhanced NET formation by neutrophils has already been shown to be related to autoimmunity, potentially because autoantigens together with immunostimulatory molecules are exposed to trigger the immune system. NETosis was directly linked to RA by the demonstration that citrullinated histone 4 from NETs is a target for RA sera [78]. Furthermore, enhanced NETosis in neutrophils derived from peripheral blood and synovial fluid of RA patients correlates with ACPA levels. Interestingly, immunoglobulin fractions from RA patients with high levels of ACPA and/or RF are able to significantly enhance NETosis in neutrophils from RA patients and these NETs induced by antibodies from RA serum harbour citrullinated proteins. Intriguingly, also, purified human ACPAs have been reported to induce potent NETosis in RA neutrophils [79]. This indicates that ACPA can enhance the formation of NETs by neutrophils, resulting in expel of immunostimulatory molecules together with its own target, citrullinated autoantigens. These observations suggest a mechanism that may promote and perpetuate disease (Fig. 5).

ACPA have been reported to enhance the formation of NETs by neutrophils, resulting in expel of immunostimulatory molecules together with citrullinated autoantigens, the targets of ACPA
Pathogenic potential of anti-PAD3/4 antibodies
Similar to ACPA and anti-CarP antibodies, anti-PAD4 antibodies have been associated with severe disease in established RA [35, 36]. Like RF, anti-PAD antibodies are associated with the presence of ACPA. Although anti-PAD4 antibodies have been detected with a mean duration of 4.7 years prior to the clinical diagnosis of RA in a small subset of patients, in the majority of patients, anti-PAD4 antibodies were detected after the appearance of ACPA [80]. To assess anti-PAD antibodies in health and disease, the presence of anti-PAD4 antibodies was tested in first-degree relatives of indigenous North American people with RA who are characterized by a high prevalence of ACPA. Interestingly, in contrast to ACPA, anti-PAD4 antibodies were almost exclusively found in people with established RA [81].
Recently, it was discovered that a subset of anti-PAD4 antibodies increases the catalytic capacity of PAD4 by decreasing the calcium requirement of this citrullination enzyme. The identification of autoantibodies that activate an enzyme that itself generates antigens for another group of autoantibodies associated with RA identifies an important feed-forward loop. This might drive worse outcome of disease and therefore potentially identify patient subsets that require aggressive treatment. The predictive value of anti-PAD antibodies for severe disease was confirmed by the finding that PAD3/PAD4 cross-reactive autoantibody-positive RA patients showed increased radiographic damage compared to PAD3/PAD4 cross-reactive autoantibody-negative individuals [25].
Autoreactive B cells
The introduction of biologicals for RA treatment provided valuable insight into the contribution of specific immunological mediators in pathogenesis. The effectiveness of biologicals depleting B cells, such as rituximab, demonstrates evidence for the involvement of B cells and/or (auto) antibodies in disease pathogenesis. In conventional B cell activation, which is intensively studied for vaccination strategies, antigen binding to the B cell receptor initiates signalling pathways for somatic hypermutation. After somatic hypermutation, B cells with the highest affinity receptors for their antigen are selected for survival. This process continues during evolution of the immune response, resulting in competitive survival and proliferation of B cells with high-affinity receptors. In contrast, low-affinity B cells die by apoptosis and eventually disappear from the population. Interestingly, ACPA-producing B cells behave differently, demonstrated by the identification of the avidity of ACPA for its antigen significantly lower than the avidity of antibodies to the recall antigens tetanus toxoid and diphtheria toxoid [55]. The avidity of ACPA is even low after extensive isotype switching [51], indicating that in ACPA-specific B cells, affinity maturation and isotype switching, in contrast to general B cell biology, is uncoupled.
Recently, a culture method has been developed to detect the presence of ACPA-producing B cells in different compartments of the human body and to study the characteristics and phenotype of these cells derived from RA patients. B cells were isolated from peripheral blood or synovial fluid from ACPA-positive RA patients and cultured with or without stimulating factors to assess the presence of ACPA-producing B cells in the different compartments. It was estimated that around 1 in 20,000 B cells in blood of RA patients secrete ACPA [82] versus around 10–20 % of the B cell population present in the synovial fluid compartment (Kerkman et al., unpublished observations). Likewise, it was demonstrated that ∼25 % of the synovial IgG-expressing B cells from ACPA-positive RA patients recognize citrullinated autoantigens [43]. Furthermore, it was shown that IgM-ACPA is the most abundantly present isotype of ACPA in synovial fluid, with the highest enrichment in the range of one IgM-ACPA for every eight IgM-antibodies [46]. Analysis of the BCR repertoire in blood and synovium showed multiple dominant clones in inflamed synovium and hardly any in blood. Interestingly, the fraction of IgM clones was higher in established RA compared to early RA [83]. Together, this indicates that the synovium is a niche for expanded autoantibody-producing B cells and is the décor of an ongoing induction of anti-citrulline immune responses in RA patients.
Autoantibodies as biomarkers for early disease and disease sub-classification
To prevent development towards the classical picture of RA, with massive erosions and deformities of inflamed joints, early treatment intervention is very important. Currently, it is hard to predict whether arthralgia and undifferentiated arthritis (UA) patients will go in remission or will develop RA. To decide which patients would benefit from aggressive intervention, robust biomarkers are required to diagnose RA early in disease.
RA is characterized by a wide spectrum of clinical manifestations, ranging from mild and self-limiting disease to rapidly progressive inflammation, joint destruction and severe physical disability. This heterogeneous character of RA is reflected by a broad range of responses to therapies. Currently, we are unable to predict the effectiveness of a specific therapy in individual patients, because we lack robust biomarkers for RA sub-classification. The discovery of the serological marker ACPA was of outstanding impact because for the first time, it was possible to sub-classify the heterogeneous entity of RA based on serological markers. ACPA-positive and ACPA-negative RA patients display major differences regarding genetic and environmental determinants of disease, molecular features of the affected joint, remission rates and, importantly, response to treatment.
Already before RA establishment, it could be considered to treat ACPA-positive UA patients differently than ACPA-negative UA patients. It was demonstrated that in ACPA-positive UA patients treated with methotrexaat, RA progression was circumvented or delayed. In contrast, for the ACPA-negative subset of UA patients, no effect of methotrexaat was observed [84]. Furthermore, ACPA-positive UA methotrexaat responders had lower levels of IgG ACPA before start of the treatment compared to ACPA-positive UA non-responders [85]. This suggests not only that different treatment strategies for ACPA-positive versus ACPA-negative patients might be required but also that in UA patients with high ACPA levels, only methotrexaat treatment might not be sufficient. In established RA, different treatments for ACPA-positive and ACPA-negative RA could be considered as well. This is supported by a study demonstrating that ACPA-positive RA patients initially treated with DMARD mono-therapy displayed increased radiographic joint destruction after 2 years compared to ACPA-negative patients. In contrast, in patients treated with initial combination therapy, no difference between ACPA-positive and ACPA-negative patients with respect to joint destruction was observed [86].
Assuming that ACPA is pathogenic, it is conceivable to hypothesize that ACPA-positive RA patients would benefit from therapies reducing B cells from the circulation. Indeed, it was demonstrated that in refractory RA patients ACPA positivity was predictive for a EULAR response at 24 weeks to rituximab, a therapy to deplete anti-CD20 positive cells. The investigators suggest that ACPA might be used as biomarker to predict responses to rituximab therapy [87]. Since ACPA consist of a collection of citrulline-recognizing antibodies with different reactivities, it was examined if the different fine specificities within the ACPA repertoire could predict response to therapies. Unfortunately, at least for anti-TNF agents, only the presence, but not individual ACPA specificities was associated with response to treatment [88]. Instead, intrinsic differences between the ACPA response and conventional antibody responses, including the disconnection of ACPA avidity maturation and isotype switching [55], and the detection of spontaneous ACPA production by circulating unstimulated plasmablasts/cells [82] might represent targets for novel therapeutic interventions.
Although taking a personalized medicine approach to treat the subgroup of ACPA-positive patients sounds promising, in the large subset of ACPA-negative RA patients, targets to distinguish different subsets are limited, mainly by the absence of robust biomarkers characterizing this manifestation of RA. Therefore, especially in the ACPA-negative group of patients, the identification of anti-CarP antibodies might be clinically useful. Possibly, the population of RA patients is more heterogeneous than only ACPA positive or negative, and anti-CarP-positive RA might represent an additional disease entity with its own genetic and environmental contributions and responses to specific therapies.
Conclusions
Several RA-associated autoantibody systems have been identified. Among these autoantibodies, ACPA exhibit a unique sensitivity for RA with the highest predictive value for RA development and severity. The importance of ACPA in RA diagnosis is emphasized by the inclusion of ACPA status in the 2010 criteria for RA. For the first time, ACPA allowed division of the heterogeneous entity of RA in an ACPA-positive and an ACPA-negative subset, with different genetic and environmental contribution factors. Importantly, ACPA status predicts response to therapies, indicating a biomarker for a personalized medicine approach. Over the past few years, important insight has been gained into the occurrence and aetiophathology of RA by the discovery of anti-citrullinated protein immunity, although the pathogenic potential of ACPA remains elusive. Major alterations in the biomolecular composition of ACPA in health and disease might be the key to discover the mechanisms underlying pathogenicity of ACPA. Recent evidence suggest a role for ACPA in exacerbating and perpetuating disease by the demonstration that ACPA enhances the formation of citrullinated autoantigens and immunostimulatory molecules containing NETs by neutrophils. Another feed-forward loop driving worse outcome of disease was demonstrated by another group of RA-associated autoantibodies, activating the enzymes that generate targets for ACPA. Although the discovery of ACPA induced a breakthrough in the understanding immunological events underlying ACPA-positive RA, these factors are still poorly understood for ACPA-negative RA. Recently, the identification of anti-CarP antibodies, also present in serum of ACPA-negative RA patients, indicated that even more subgroups of RA in addition to ACPA-positive and ACPA-negative disease might exist. Therefore, it is plausible that in the future, novel autoantibodies systems in RA will be identified, hopefully resulting in unravelling the immunological pathways underlying RA physiopathology, potentially of importance for the development of diagnostic and/or prognostic tools.
References
Scott DL, Wolfe F, Huizinga TW (2010) Rheumatoid arthritis. Lancet 376:1094–1108. doi:10.1016/S0140-6736(10)60826-4
Lee DM, Weinblatt ME (2001) Rheumatoid arthritis. Lancet 358:903–911. doi:10.1016/S0140-6736(01)06075-5
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, III, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G (2010) 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 69: doi:10.1136/ard.2010.138461
Klareskog L, Padyukov L, Alfredsson L (2007) Smoking as a trigger for inflammatory rheumatic diseases. Curr Opin Rheumatol 19:49–54. doi:10.1097/BOR.0b013e32801127c8
Silman AJ, Pearson JE (2002) Epidemiology and genetics of rheumatoid arthritis. Arthritis Res 4(Suppl 3):S265–S272
MacGregor AJ, Snieder H, Rigby AS, Koskenvuo M, Kaprio J, Aho K, Silman AJ (2000) Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 43:30–37. doi:10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B
Bowes J, Barton A (2008) Recent advances in the genetics of RA susceptibility. Rheumatology (Oxford) 47:399–402. doi:10.1093/rheumatology/ken005
Imboden JB (2009) The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol 4:417–434. doi:10.1146/annurev.pathol.4.110807.092254
Astorga GP, Williams RC Jr (1969) Altered reactivity in mixed lymphocyte culture of lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum 12:547–554
Stastny P (1976) Mixed lymphocyte cultures in rheumatoid arthritis. J Clin Invest 57:1148–1157. doi:10.1172/JCI108382
Gregersen PK, Silver J, Winchester RJ (1987) The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 30:1205–1213
Huizinga TW, Amos CI, van der Helm-van Mil AH, Chen W, van Gaalen FA, Jawaheer D, Schreuder GM, Wener M, Breedveld FC, Ahmad N, Lum RF, de Vries RR, Gregersen PK, Toes RE, Criswell LA (2005) Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum 52:3433–3438. doi:10.1002/art.21385
Jawaheer D, Seldin MF, Amos CI, Chen WV, Shigeta R, Monteiro J, Kern M, Criswell LA, Albani S, Nelson JL, Clegg DO, Pope R, Schroeder HW Jr, Bridges SL Jr, Pisetsky DS, Ward R, Kastner DL, Wilder RL, Pincus T, Callahan LF, Flemming D, Wener MH, Gregersen PK (2001) A genomewide screen in multiplex rheumatoid arthritis families suggests genetic overlap with other autoimmune diseases. Am J Hum Genet 68:927–936
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K (2003) Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet 34:395–402. doi:10.1038/ng1206
Bax M, van Heemst J, Huizinga TW, Toes RE (2011) Genetics of rheumatoid arthritis: what have we learned? Immunogenetics 63:459–466. doi:10.1007/s00251-011-0528-6
Zhernakova A, van Diemen CC, Wijmenga C (2009) Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat Rev Genet 10:43–55. doi:10.1038/nrg2489
van der Woude D, Lie BA, Lundstrom E, Balsa A, Feitsma AL, Houwing-Duistermaat JJ, Verduijn W, Nordang GB, Alfredsson L, Klareskog L, Pascual-Salcedo D, Gonzalez-Gay MA, Lopez-Nevot MA, Valero F, Roep BO, Huizinga TW, Kvien TK, Martin J, Padyukov L, de Vries RR, Toes RE (2010) Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: a meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum 62:1236–1245. doi:10.1002/art.27366
Frankline EC, Holman HR, Muller-Eberhard HJ, Kunkel HG (1957) An unusual protein component of high molecular weight in the serum of certain patients with rheumatoid arthritis. J Exp Med 105:425–438
Nienhuis RL, Mandema E (1964) A new serum factor in patients with rheumatoid arthritis; the antiperinuclear factor. Ann Rheum Dis 23:302–305
Young BJ, Mallya RK, Leslie RD, Clark CJ, Hamblin TJ (1979) Anti-keratin antibodies in rheumatoid arthritis. Br Med J 2:97–99
Sebbag M, Simon M, Vincent C, Masson-Bessiere C, Girbal E, Durieux JJ, Serre G (1995) The antiperinuclear factor and the so-called antikeratin antibodies are the same rheumatoid arthritis-specific autoantibodies. J Clin Invest 95:2672–2679. doi:10.1172/JCI117969
Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, Simon M, Senshu T, Masson-Bessiere C, Jolivet-Reynaud C, Jolivet M, Serre G (1999) The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol 162:585–594
Shi J, Knevel R, Suwannalai P, van der Linden MP, Janssen GM, van Veelen PA, Levarht NE, van der Helm-van Mil AH, Cerami A, Huizinga TW, Toes RE, Trouw LA (2011) Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci U S A 108:17372–17377. doi:10.1073/pnas.1114465108
Darrah E, Giles JT, Ols ML, Bull HG, Andrade F, Rosen A (2013) Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci Transl Med 5:186ra65. doi:10.1126/scitranslmed.3005370
Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, McGregor J, Burkart M, Hogan SL, Hu Y, Winnik W, Nachman PH, Stegeman CA, Niles J, Heeringa P, Kitching AR, Holdsworth S, Jennette JC, Preston GA, Falk RJ (2013) Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. J Clin Invest 123:1773–1783. doi:10.1172/JCI65292
Makrygiannakis D, af Klint E, Lundberg IE, Lofberg R, Ulfgren AK, Klareskog L, Catrina AI (2006) Citrullination is an inflammation-dependent process. Ann Rheum Dis 65:1219–1222. doi:10.1136/ard.2005.049403
Ireland J, Herzog J, Unanue ER (2006) Cutting edge: unique T cells that recognize citrullinated peptides are a feature of protein immunization. J Immunol 177:1421–1425
Ireland JM, Unanue ER (2011) Autophagy in antigen-presenting cells results in presentation of citrullinated peptides to CD4 T cells. J Exp Med 208:2625–2632. doi:10.1084/jem.20110640
Cantaert T, Teitsma C, Tak PP, Baeten D (2013) Presence and role of anti-citrullinated protein antibodies in experimental arthritis models. Arthritis Rheum 65:939–948. doi:10.1002/art.37839
Vossenaar ER, van Boekel MA, van Venrooij WJ, Lopez-Hoyoz M, Merino J, Merino R, Joosten LA (2004) Absence of citrulline-specific autoantibodies in animal models of autoimmunity. Arthritis Rheum 50:2370–2372. doi:10.1002/art.20296
Jaisson S, Pietrement C, Gillery P (2011) Carbamylation-derived products: bioactive compounds and potential biomarkers in chronic renal failure and atherosclerosis. Clin Chem 57:1499–1505. doi:10.1373/clinchem.2011.163188
Wynckel A, Randoux C, Millart H, Desroches C, Gillery P, Canivet E, Chanard J (2000) Kinetics of carbamylated haemoglobin in acute renal failure. Nephrol Dial Transplant 15:1183–1188
Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Horkko S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA, Hazen SL (2007) Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med 13:1176–1184. doi:10.1038/nm1637
Halvorsen EH, Haavardsholm EA, Pollmann S, Boonen A, van der Heijde D, Kvien TK, Molberg O (2009) Serum IgG antibodies to peptidylarginine deiminase 4 predict radiographic progression in patients with rheumatoid arthritis treated with tumour necrosis factor-alpha blocking agents. Ann Rheum Dis 68:249–252. doi:10.1136/ard.2008.094490
Harris ML, Darrah E, Lam GK, Bartlett SJ, Giles JT, Grant AV, Gao P, Scott WW Jr, El-Gabalawy H, Casciola-Rosen L, Barnes KC, Bathon JM, Rosen A (2008) Association of autoimmunity to peptidyl arginine deiminase type 4 with genotype and disease severity in rheumatoid arthritis. Arthritis Rheum 58:1958–1967. doi:10.1002/art.23596
Takizawa Y, Sawada T, Suzuki A, Yamada R, Inoue T, Yamamoto K (2005) Peptidylarginine deiminase 4 (PADI4) identified as a conformation-dependent autoantigen in rheumatoid arthritis. Scand J Rheumatol 34:212–215
Zhao J, Zhao Y, He J, Jia R, Li Z (2008) Prevalence and significance of anti-peptidylarginine deiminase 4 antibodies in rheumatoid arthritis. J Rheumatol 35:969–974
Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ (1998) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest 101:273–281. doi:10.1172/JCI1316
Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ (2000) The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum 43:155–163. doi:10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3
Ioan-Facsinay A, el Bannoudi H, Scherer HU, van der Woude D, Menard HA, Lora M, Trouw LA, Huizinga TW, Toes RE (2011) Anti-cyclic citrullinated peptide antibodies are a collection of anti-citrullinated protein antibodies and contain overlapping and non-overlapping reactivities. Ann Rheum Dis 70:188–193. doi:10.1136/ard.2010.131102
Uysal H, Bockermann R, Nandakumar KS, Sehnert B, Bajtner E, Engstrom A, Serre G, Burkhardt H, Thunnissen MM, Holmdahl R (2009) Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med 206:449–462. doi:10.1084/jem.20081862
Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, Engstrom M, Snir O, Israelsson L, Catrina AI, Wardemann H, Corti D, Meffre E, Klareskog L, Malmstrom V (2013) Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. J Exp Med 210:445–455. doi:10.1084/jem.20121486
van de Stadt LA, de Koning MH, van de Stadt RJ, Wolbink G, Dijkmans BA, Hamann D, van Schaardenburg D (2011) Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum 63:3226–3233. doi:10.1002/art.30537
van der Woude D, Rantapaa-Dahlqvist S, Ioan-Facsinay A, Onnekink C, Schwarte CM, Verpoort KN, Drijfhout JW, Huizinga TW, Toes RE, Pruijn GJ (2010) Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis 69:1554–1561. doi:10.1136/ard.2009.124537
Willemze A, Shi J, Mulder M, Stoeken-Rijsbergen G, Drijfhout JW, Huizinga TW, Trouw LA, Toes RE (2013) The concentration of anticitrullinated protein antibodies in serum and synovial fluid in relation to total immunoglobulin concentrations. Ann Rheum Dis 72:1059–1063. doi:10.1136/annrheumdis-2012-202747
Fisher BA, Plant D, Brode M, van Vollenhoven RF, Mathsson L, Symmons D, Lundberg K, Ronnelid J, Venables PJ (2011) Antibodies to citrullinated alpha-enolase peptide 1 and clinical and radiological outcomes in rheumatoid arthritis. Ann Rheum Dis 70:1095–1098. doi:10.1136/ard.2010.138909
Willemze A, Bohringer S, Knevel R, Levarht EW, Stoeken-Rijsbergen G, Houwing-Duistermaat JJ, van der Helm-van Mil AH, Huizinga TW, Toes RE, Trouw LA (2012) The ACPA recognition profile and subgrouping of ACPA-positive RA patients. Ann Rheum Dis 71:268–274. doi:10.1136/annrheumdis-2011-200421
Verpoort KN, van Gaalen FA, van der Helm-van Mil AH, Schreuder GM, Breedveld FC, Huizinga TW, de Vries RR, Toes RE (2005) Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum 52:3058–3062. doi:10.1002/art.21302
Scherer HU, van der Woude D, Willemze A, Trouw LA, Knevel R, Syversen SW, van der Linden MP, Lie B, Huizinga TW, van der Heijde DM, van der Helm-van Mil AH, Kvien TK, Toes RE (2011) Distinct ACPA fine specificities, formed under the influence of HLA shared epitope alleles, have no effect on radiographic joint damage in rheumatoid arthritis. Ann Rheum Dis 70:1461–1464. doi:10.1136/ard.2010.146506
Ioan-Facsinay A, Willemze A, Robinson DB, Peschken CA, Markland J, van der Woude D, Elias B, Menard HA, Newkirk M, Fritzler MJ, Toes RE, Huizinga TW, El-Gabalawy HS (2008) Marked differences in fine specificity and isotype usage of the anti-citrullinated protein antibody in health and disease. Arthritis Rheum 58:3000–3008. doi:10.1002/art.23763
Kokkonen H, Mullazehi M, Berglin E, Hallmans G, Wadell G, Ronnelid J, Rantapaa-Dahlqvist S (2011) Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res Ther 13:R13. doi:10.1186/ar3237
Verpoort KN, Jol-van der Zijde CM, Papendrecht-van der Voort EA, Ioan-Facsinay A, Drijfhout JW, van Tol MJ, Breedveld FC, Huizinga TW, Toes RE (2006) Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum 54:3799–3808. doi:10.1002/art.22279
van der Woude D, Syversen SW, van der Voort EI, Verpoort KN, Goll GL, van der Linden MP, van der Helm-van Mil AH, van der Heijde DM, Huizinga TW, Kvien TK, Toes RE (2010) The ACPA isotype profile reflects long-term radiographic progression in rheumatoid arthritis. Ann Rheum Dis 69:1110–1116. doi:10.1136/ard.2009.116384
Suwannalai P, Scherer HU, van der Woude D, Ioan-Facsinay A, Jol-van der Zijde CM, van Tol MJ, Drijfhout JW, Huizinga TW, Toes RE, Trouw LA (2011) Anti-citrullinated protein antibodies have a low avidity compared with antibodies against recall antigens. Ann Rheum Dis 70:373–379. doi:10.1136/ard.2010.135509
Parekh RB, Dwek RA, Sutton BJ, Fernandes DL, Leung A, Stanworth D, Rademacher TW, Mizuochi T, Taniguchi T, Matsuta K (1985) Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 316:452–457
Kaneko Y, Nimmerjahn F, Ravetch JV (2006) Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313:670–673. doi:10.1126/science.1129594
Scherer HU, Wang J, Toes RE, van der Woude D, Koeleman CA, de Boer AR, Huizinga TW, Deelder AM, Wuhrer M (2009) Immunoglobulin 1 (IgG1) Fc-glycosylation profiling of anti-citrullinated peptide antibodies from human serum. Proteomics Clin Appl 3:106–115. doi:10.1002/prca.200800098
Scherer HU, van der Woude D, Ioan-Facsinay A, el Bannoudi H, Trouw LA, Wang J, Haupl T, Burmester GR, Deelder AM, Huizinga TW, Wuhrer M, Toes RE (2010) Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum 62:1620–1629. doi:10.1002/art.27414
Wang J, Balog CI, Stavenhagen K, Koeleman CA, Scherer HU, Selman MH, Deelder AM, Huizinga TW, Toes RE, Wuhrer M (2011) Fc-glycosylation of IgG1 is modulated by B-cell stimuli. Mol Cell Proteomics 10:M110. doi:10.1074/mcp.M110.004655
Rombouts Y, Ewing E, van de Stadt LA, Selman MH, Trouw LA, Deelder AM, Huizinga TW, Wuhrer M, van Schaardenburg D, Toes RE, Scherer HU (2013) Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/annrheumdis-2013-203565
Mydel P, Wang Z, Brisslert M, Hellvard A, Dahlberg LE, Hazen SL, Bokarewa M (2010) Carbamylation-dependent activation of T cells: a novel mechanism in the pathogenesis of autoimmune arthritis. J Immunol 184:6882–6890. doi:10.4049/jimmunol.1000075
Turunen S, Koivula MK, Risteli L, Risteli J (2010) Anticitrulline antibodies can be caused by homocitrulline-containing proteins in rabbits. Arthritis Rheum 62:3345–3352. doi:10.1002/art.27644
Lindqvist E, Eberhardt K, Bendtzen K, Heinegard D, Saxne T (2005) Prognostic laboratory markers of joint damage in rheumatoid arthritis. Ann Rheum Dis 64:196–201. doi:10.1136/ard.2003.019992
van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, Toes RE, Huizinga TW (2004) Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum 50:709–715. doi:10.1002/art.20044
Bos WH, Wolbink GJ, Boers M, Tijhuis GJ, de Vries N, van der Horst-Bruinsma IE, Tak PP, van de Stadt RJ, van der Laken CJ, Dijkmans BA, van Schaardenburg D (2010) Arthritis development in patients with arthralgia is strongly associated with anti-citrullinated protein antibody status: a prospective cohort study. Ann Rheum Dis 69:490–494. doi:10.1136/ard.2008.105759
van de Stadt LA, van der Horst AR, de Koning MH, Bos WH, Wolbink GJ, van de Stadt RJ, Pruijn GJ, Dijkmans BA, van Schaardenburg D, Hamann D (2011) The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann Rheum Dis 70:128–133. doi:10.1136/ard.2010.132662
Krabben A, Stomp W, van der Heijde DM, van Nies JA, Bloem JL, Huizinga TW, Reijnierse M, van der Helm-van Mil AH (2013) MRI of hand and foot joints of patients with anticitrullinated peptide antibody positive arthralgia without clinical arthritis. Ann Rheum Dis 72:1540–1544. doi:10.1136/annrheumdis-2012-202628
Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, Van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA (2004) Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 50:380–386. doi:10.1002/art.20018
Suwannalai P, van de Stadt LA, Radner H, Steiner G, El-Gabalawy HS, Zijde CM, van Tol MJ, van Schaardenburg D, Huizinga TW, Toes RE, Trouw LA (2012) Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum 64:1323–1328. doi:10.1002/art.33489
Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, Huizinga TW, Toes RE (2009) Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum 60:1923–1931. doi:10.1002/art.24622
Clavel C, Nogueira L, Laurent L, Iobagiu C, Vincent C, Sebbag M, Serre G (2008) Induction of macrophage secretion of tumor necrosis factor alpha through Fcgamma receptor IIa engagement by rheumatoid arthritis-specific autoantibodies to citrullinated proteins complexed with fibrinogen. Arthritis Rheum 58:678–688. doi:10.1002/art.23284
Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, Jakobsson PJ, Baum W, Nimmerjahn F, Szarka E, Sarmay G, Krumbholz G, Neumann E, Toes R, Scherer HU, Catrina AI, Klareskog L, Jurdic P, Schett G (2012) Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest 122:1791–1802. doi:10.1172/JCI60975
van Oosterhout M, Sont JK, Bajema IM, Breedveld FC, van Laar JM (2006) Comparison of efficacy of arthroscopic lavage plus administration of corticosteroids, arthroscopic lavage plus administration of placebo, and joint aspiration plus administration of corticosteroids in arthritis of the knee: a randomized controlled trial. Arthritis Rheum 55:964–970. doi:10.1002/art.22340
van Beers JJ, Schwarte CM, Stammen-Vogelzangs J, Oosterink E, Bozic B, Pruijn GJ (2013) The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis Rheum 65:69–80. doi:10.1002/art.37720
Suwannalai P, Britsemmer K, Knevel R, Scherer HU, Levarht EW, van der Helm-van Mil AH, van Schaardenburg D, Huizinga TW, Toes RE, Trouw LA (2013) Low-avidity anticitrullinated protein antibodies (ACPA) are associated with a higher rate of joint destruction in rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202615
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207:1853–1862. doi:10.1084/jem.20100239
Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, Boscaro F, Panza F, Puxeddu I, Rovero P, Migliorini P (2013) Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202765
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P, Chen P, Fox DA, Pennathur S, Kaplan MJ (2013) NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 5:178ra40. doi:10.1126/scitranslmed.3005580
Kolfenbach JR, Deane KD, Derber LA, O’Donnell CI, Gilliland WR, Edison JD, Rosen A, Darrah E, Norris JM, Holers VM (2010) Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum 62:2633–2639. doi:10.1002/art.27570
Ferucci ED, Darrah E, Smolik I, Choromanski TL, Robinson DB, Newkirk MM, Fritzler MJ, Rosen A, El-Gabalawy HS (2013) Prevalence of anti-peptidylarginine deiminase type 4 antibodies in rheumatoid arthritis and unaffected first-degree relatives in indigenous north american populations. J Rheumatol 40:1523–1528. doi:10.3899/jrheum.130293
Kerkman PF, Rombouts Y, van der Voort EI, Trouw LA, Huizinga TW, Toes RE, Scherer HU (2013) Circulating plasmablasts/plasmacells as a source of anticitrullinated protein antibodies in patients with rheumatoid arthritis. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202893
Doorenspleet ME, Klarenbeek PL, de Hair MJ, van Schaik BD, Esveldt RE, van Kampen AH, Gerlag DM, Musters A, Baas F, Tak PP, de Vries N (2013) Rheumatoid arthritis synovial tissue harbours dominant B-cell and plasma-cell clones associated with autoreactivity. Ann Rheum Dis. doi:10.1136/annrheumdis-2012-202861
van Dongen H, van Aken J, Lard LR, Visser K, Ronday HK, Hulsmans HM, Speyer I, Westedt ML, Peeters AJ, Allaart CF, Toes RE, Breedveld FC, Huizinga TW (2007) Efficacy of methotrexate treatment in patients with probable rheumatoid arthritis: a double-blind, randomized, placebo-controlled trial. Arthritis Rheum 56:1424–1432. doi:10.1002/art.22525
Visser K, Verpoort KN, van Dongen H, van der Kooij SM, Allaart CF, Toes RE, Huizinga TW, van der Helm-van Mil AH (2008) Pretreatment serum levels of anti-cyclic citrullinated peptide antibodies are associated with the response to methotrexate in recent-onset arthritis. Ann Rheum Dis 67:1194–1195. doi:10.1136/ard.2008.088070
de Vries-Bouwstra JK, Goekoop-Ruiterman YP, Verpoort KN, Schreuder GM, Ewals JA, Terwiel JP, Ronday HK, Kerstens PJ, Toes RE, de Vries RR, Breedveld FC, Dijkmans BA, Huizinga TW, Allaart CF (2008) Progression of joint damage in early rheumatoid arthritis: association with HLA-DRB1, rheumatoid factor, and anti-citrullinated protein antibodies in relation to different treatment strategies. Arthritis Rheum 58:1293–1298. doi:10.1002/art.23439
Sellam J, Hendel-Chavez H, Rouanet S, Abbed K, Combe B, Le LX, Tebib J, Sibilia J, Taoufik Y, Dougados M, Mariette X (2011) B cell activation biomarkers as predictive factors for the response to rituximab in rheumatoid arthritis: a six-month, national, multicenter, open-label study. Arthritis Rheum 63:933–938. doi:10.1002/art.30233
Fisher BA, Plant D, Lundberg K, Charles P, Barton A, Venables PJ (2012) Heterogeneity of anticitrullinated peptide antibodies and response to anti-tumor necrosis factor agents in rheumatoid arthritis. J Rheumatol 39:929–932. doi:10.3899/jrheum.111315
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on B cell-mediated autoimmune diseases - Guest Editors: Thomas Winkler and Reinhard Voll
Rights and permissions
About this article

Cite this article
Bax, M., Huizinga, T.W.J. & Toes, R.E.M. The pathogenic potential of autoreactive antibodies in rheumatoid arthritis. Semin Immunopathol 36, 313–325 (2014). https://doi.org/10.1007/s00281-014-0429-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-014-0429-5