
Abstract
Neutrophils have long been viewed as short-lived cells crucial for the elimination of extracellular pathogens, possessing a limited role in the orchestration of the immune response. This dogma has been challenged by recent lines of evidence demonstrating the expression of an increasing number of cytokines and effector molecules by neutrophils. Moreover, in analogy with their “big brother” macrophages, neutrophils integrate the environmental signals and can be polarized towards an antitumoural or protumoural phenotype. Neutrophils are a major source of humoral fluid phase pattern recognition molecules and thus contribute to the humoral arm of innate immunity. Neutrophils cross talk and shape the maturation and effector functions of other leukocytes in a direct or indirect manner, through cell–cell contact or cytokine production, respectively. Therefore, neutrophils are integrated in the activation and regulation of the innate and adaptive immune system and play an important role in the resolution or exacerbation of diverse pathologies, including infections, chronic inflammation, autoimmunity and cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The role played by neutrophils in immunity has long been viewed as restricted to the acute phase of inflammation and to resistance against extracellular pathogens [1–3]. This view is consistent with the phagocytic theory of Metchnikoff who proposed more than 100 years ago that polymorphonuclear leukocytes patrol the bloodstream and migrate to the site of infection to phagocytose microbes. Several studies have recently challenged this dogma and placed the neutrophil as a key effector cell in the orchestration of adaptive immunity and in the resolution of inflammatory response [1–3]. Indeed, in addition to their phagocytic activity and the storage of a set of lytic enzymes and antimicrobial components in their intracellular granules, neutrophils are induced to express molecules (e.g. cytokines, chemokines) involved in the regulation of innate and adaptive response. Neutrophils have emerged as a major source of humoral pattern recognition molecules (PRMs) that recognise pathogen-associated molecular patterns and initiate the immune response in coordination with the cellular arm, therefore acting as functional ancestors of antibodies. Neutrophils directly interact with macrophages, dendritic cells, and lymphocyte subsets and modulate their effector functions. For instance, natural killer (NK) cell functions are impaired during neutropenia, and under steady state, neutrophils are crucial for NK cell development both in human and mouse [4]. Consequently, a bidirectional cross talk occurs between neutrophils and NK cells, which stimulates the production of IFN-γ by NK cells and promotes the survival and activation of neutrophils [5, 6]. In addition, polarized T helper (Th) 17 cells and innate IL17-producing cells rapidly activate neutrophilic inflammation through the production of granulopoietic factors and chemokines [7–9]. In this review, we will focus on the most recent findings related to the effector functions and plasticity of neutrophils as well as on their emerging role in regulating the innate and adaptive immune system. Finally, we will describe the relation between neutrophils and diverse pathologies.
Neutrophils in innate immunity
Pathogen recognition by neutrophils
Mammals are constantly in contact with microorganisms, and their ability to mount a protective immune response resides in their competence to identify potential pathogens. The life-threatening condition associated with acquired or congenital abnormalities in neutrophil life cycle or function underlines their essential role in innate immunity and resistance to pathogens [2]. Innate immune molecules involved in pathogen recognition are germline-encoded PRMs which belong to both the cellular and humoral arms of the innate immune system [10]. These receptors represent a class of sensors specialized in the discrimination of self versus non-self and modified self and participate in the initiation and regulation of the inflammatory process [11]. Neutrophils are endowed with a vast repertoire of cellular-associated PRMs which, upon recognition of pathogens or tissue damage, promote neutrophil effector functions (e.g. production of ROS, secretion of antimicrobial peptides) [12, 13]. These include all members of the Toll-like receptor (TLR) family, with the exception of TLR3 and a low or absent expression of TLR7 [14, 15]; the C-type lectin receptors Dectin-1 [16], CLEC-2 [17], Mincle [18] and CLECSF8 [19]; and functional cytoplasmic sensors, such as NOD-1, RIG1, MDA5 and IFI16 [20–22]. Dectin-1 (also known as CLEC7A) is the main β-glucan receptor on neutrophils and promotes phagocytosis and the killing of Candida albicans and Aspergillus fumigatus through the activation of a calcineurin signalling axis [16, 23]. Neutrophils express NOD-1, which primes the innate immune system [20], and the NLRP3/ASC/Caspase-1 inflammasome, which regulates IL-1β processing [24]. In addition, neutrophils express the NOD-like receptor family member NLRP6, a negative regulator of NF-kB and ERK activation after TLR engagement [25]. In contrast to other myeloid cells, the Myd88-independent pathway is not activated in human neutrophils stimulated by lipopolysaccharide (LPS) and neutrophils fail to produce IFN-β and, consequently, CXCL10 and other type I IFN-dependent genes after TLR4 engagement [26]. However, human neutrophils express a set of cytosolic DNA sensors, such as IFI16, MDA5, RIG1, LRRFIP1, DDX41 and STING [21, 22]. As a consequence, the expression of IFN-β and CXCL10 mRNAs was observed in human neutrophils transfected with plasmid DNA or infected by intracellular pathogens (e.g. Bartonella henselae, Listeria monocytogenes, Legionella pneumophila and adenovirus type 5) [21]. Formyl peptides, found in bacteria and mitochondria, activate neutrophils via the seven-transmembrane G-protein-coupled receptors FPR1 (high-affinity receptor for fMLF) and FPR2 (low-affinity receptor for fMLF), which have different effector functions [27, 28]. The activation of FPR1 or FPR2, depending on ligand concentration, promotes p38 or Erk activation, respectively, leading to the activation or inhibition of neutrophil chemotaxis [27]. Mitochondrial-derived formylated peptides induce the recruitment of neutrophils and inflammation and elicit neutrophil-mediated organ injury [29]. Since the production of formylated proteins is limited to bacteria and mitochondria, FPRs can be classified as PRMs recognising microbial moieties and tissue damage [29].
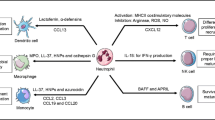
Fluid phase PRMs, including collectins, ficolins and pentraxins, are essential effectors and modulators of the innate resistance in animals and human and act as functional ancestors of antibodies [10]. Neutrophils have emerged as a source of humoral PRMs and notably serve as a ready-made reservoir of a set of PRMs, including long pentraxin PTX3, proteoglycan recognition receptor PGRP-S and M-ficolin (also known as ficolin 1), covering a temporal window preceding gene expression-dependent production (Fig. 1).

Neutrophils contribute to the humoral arm of innate immunity. Sensing microbial moieties or tissue damage by cellular receptors induces the release of humoral fluid phase pattern recognition receptors by innate immune cells (e.g. neutrophils, macrophages) and other cell types (e.g. endothelial cells, epithelial cells) with different tempos. Neutrophil granules contain a set of humoral fluid phase pattern recognition receptors (PGRP-S, PTX3, M-ficolin) rapidly released in minutes, covering a temporal window preceding gene expression-dependent production. These humoral sensors share fundamental mechanisms of effector function (e.g. opsonisation, agglutination, complement activation, regulation of inflammation) and participate in the initiation of immune response
PTX3 is a member of the long pentraxin family related to classic short pentraxins (e.g. C-reactive protein and serum amyloid P) and has served as a paradigm to study the humoral arm of the innate immune system [10]. PTX3 transcript expression is confined to immature myeloid cells, and mature neutrophils serve as a reservoir of preformed PTX3, ready for rapid release into neutrophil extracellular traps (NETs) [30]. Upon pathogen opsonisation (i.e. A. fumigatus, Pseudomonas aeruginosa), PTX3 interacts with Fcγ receptor IIA (FcγRIIA/CD32) and induces inside-out CR3 (CD11b/CD18) activation and amplification of C3b-opsonized pathogen phagocytosis [31, 32]. Accordingly, neutrophil-associated PTX3 is essential for resistance against A. fumigatus [30]. In addition, PTX3 is translocated from granules to the surface of apoptotic neutrophils and acts as a late “eat me” molecule involved in the recognition and engulfment of apoptotic neutrophils by macrophages [33]. Leukocyte-derived PTX3 also has a regulatory function on neutrophil recruitment and inflammation by interacting with P-selectin [34]. Thus, under conditions of full-blown neutrophilic inflammation, leukocyte-derived PTX3 acts as a negative feedback loop by binding to P-selectin and preventing further neutrophil recruitment [34].
PGRP-S and M-ficolin are stored in secondary and tertiary granules [35–37]. PGRP-S is also localized in NETs, binds to peptidoglycan and exerts bacteriostatic and bactericidal activities against selected microorganism (e.g. Micrococcus luteus, Staphylococcus aureus, Bacillus subtilis) [37, 38]. M-ficolin, which belongs to the lectin family, recognises selected Gram-positive and Gram-negative bacteria, activates the complement lectin pathway and exerts opsonic effects [10]. M-ficolin released from granules is also found associated with the neutrophil surface through a direct interaction with CD43 [36, 39]. This interaction enhances neutrophil aggregation and adhesion and activates complement on the neutrophil surface [39].
Collectively, these observations reveal that neutrophils participate in humoral innate immunity via the expression and release of fluid phase PRMs involved in the recognition and phagocytosis of non-self and modified self, complement activation and regulation of the inflammatory response.
Neutrophil extracellular traps
NETs are an extracellular fibrillary network formed by activated neutrophils [40] and composed of nuclear components (i.e. DNA, histones) [40] decorated by a set of proteins from primary (e.g. myeloperoxidase (MPO) and neutrophil elastase (NE)) [40], secondary (e.g. lactoferrin [40] and PTX3 [30]) and tertiary granules (e.g. MMP-9 [40] and PGRP-S [37]). In addition to genomic DNA, mitochondrial DNA has been reported in NETs [41]. These extracellular structures trap bacteria (e.g. Escherichia coli, Shigella flexneri, S. aureus); fungi (e.g. C. albicans, A. fumigatus); and human immunodeficiency virus-1 (HIV-1), favouring their interaction with effector molecules and their disposal (Fig. 2) [40, 42]. In addition to neutrophil-associated molecules, surfactant protein D present on many mucosal surfaces binds NETs and enhances microbial trapping [43]. However, direct microbicidal activity of NETs has recently been a matter of controversy and could require the presence of H2O2, chloride and the formation of HOCl catalysed by MPO [44, 45]. NET formation is a rapid active process called “NETosis” (Fig. 2) which occurs in vivo in animals and humans and prevents systemic bacterial dissemination [40, 46]. NET formation is induced by the Raf-MEK-ERK pathway, through the activation of the NADPH oxidase complex and upregulation of anti-apoptotic proteins, and by the mammalian target of rapamycin (mTOR), through the induction of hypoxia-inducible factor 1 alpha protein expression [47, 48].

Neutrophil extracellular traps in physiology and pathology. Neutrophil extracellular traps (NETs) are formed by activated neutrophils via an active process called NETosis. This extracellular network, composed of DNA and nuclear components decorated by a set of proteins from granules, traps microorganisms and facilitates their disposal by NET-associated effector molecules. NETs are also involved in a diverse set of pathological conditions
After stimulation, neutrophils lose their characteristic nuclear morphology. Chromatin decondensation is an essential event and requires the generation of ROS, the induction of neutrophil autophagy [49, 50], and the citrullination of histones by the peptidyl arginine deiminase 4 (PAD4) [51] and is favoured by NE and MPO [52]. Accordingly, genetic deficiency of these molecules, as observed for instance in chronic granulomatous disease or in MPO-deficient patients, results in defective NET formation, which is likely to contribute to the increased susceptibility to infections observed in these patients [49, 53].
NET formation is delayed in neutrophils isolated from preterm or healthy term neonates compared to neutrophils isolated from healthy adults [54]. Interestingly, this defect seems to be ROS-independent, which suggests additional mechanisms driving NET formation [55]. According to this observation, Leishmania donovani induces NET formation in a ROS-independent mechanism [56].
Microorganisms have acquired a set of mechanisms allowing them to escape NET trapping/killing and, thus, enhancing their virulence. For instance, the cell wall structure of Streptococcus pneumoniae and L. donovani is modified by a d-alanylation of the cytoplasmic membrane-anchored lipoteichoic acid and by the expression of a surface glycolipid lipophosphoglycan, respectively, which protect them from the antimicrobial activity of NETs [56, 57]. Expression of DNase by the M1 serotype strains of the pathogen group A Streptococcus (Streptococcus pyogenes or GAS) and S. pneumoniae inhibits their extracellular killing mediated by neutrophils and enhances their virulence in vivo [58, 59]. In addition, strains of P. aeruginosa isolated from cystic fibrosis patients have developed resistance to NET-mediated killing within the cystic fibrosis airway [60]. Finally, HIV-1 is also able to counteract the antiviral activity of NETs through DC-SIGN (CD209)-dependent IL-10 production, which inhibits NADPH oxidase-dependent NET formation [42]. Thus, as ancient Roman gladiators, neutrophils throw noxious NETs, whilst escape from NETs is an evolutionary strategy adopted by bacteria and HIV-1.
The growing number of neutrophil-derived cytokines
Beyond their classical preformed and rapidly secreted mediators, neutrophils have recently emerged as key regulators in innate and adaptive immunity through cytokine production and secretion. Table 1 summarizes the growing number of neutrophil-derived cytokines. Here, we describe the most recent evidence and refer the reader to previous reviews for background [2, 61].
A recent report has demonstrated that murine neutrophils constitutively express the NLRP3 inflammasome complex and that LPS-pretreated murine neutrophils are the major source of IL-1β in response to classical inflammasome activators (ATP, silica crystals) [24]. Previous results indicated that murine neutrophils are able to produce IL-1β and process it in a proteinase-3- and elastase-dependent manner [62]. However, Mankan and colleagues [24] showed that neutrophils isolated from proteinase 3/elastase double knockout mice are still able to produce functional IL-1β, whilst neutrophils isolated from NLRP, ASC or caspase-1 knockout mice are not, demonstrating that the NLRP3/ASC/caspase-1 pathway plays a major role in IL-1β production by murine neutrophils. IL-27 expressed by human neutrophils during sepsis suppresses the production of ROS and therefore reduces neutrophil bactericidal activity in vitro, suggesting that IL-27 exerts regulatory effects on neutrophils [63]. Human neutrophils are unable to activate the MyD88-independent/TRIF-dependent pathway upon TLR4 engagement and thereby fail to produce IFN-β upon stimulation by LPS [26]. Tamassia and colleagues [21] recently found that DNA-transfected human neutrophils express IFN-β and related genes via direct activation of IRF3 mediated by intracellular DNA sensors. Moreover, murine neutrophils also express IFN-β and related genes upon infection with encephalo-myocarditis virus via MDA5 engagement [22]. These results highlight the role of neutrophils in recognising intracellular pathogens and modulating innate and adaptive immune response.
Other neutrophil-derived cytokines, such as TNF-related leukocyte-expressed ligand (TRAIL), CCL20, CXCL8, B cell-activating factor BAFF) or IL-1 receptor antagonist, are stored in intracellular pools and rapidly secreted upon stimulation by pro-inflammatory stimuli [64].
Human neutrophils represent an important source of BAFF and A proliferation-inducing ligand (APRIL), two cytokines crucial for the survival, maturation and differentiation of B cells, suggesting a role of neutrophil-derived cytokines in autoimmune and neoplastic B cell-dependent disorders [2]. Under homeostatic conditions, Puga and colleagues [65] described a subpopulation of neutrophils presenting a singular phenotype characterized by high levels of BAFF, APRIL, CD40L and IL-21 production. These neutrophils activate B cells from the marginal zone of the spleen and promote the diversification and production of immunoglobulins, and are therefore called “B-cell helper neutrophils” (NBH) [65].
Neutrophil-derived proteases regulate the biological activity of cytokines in the inflammatory microenvironment. For instance, human and murine neutrophil-derived elastase and cathepsin G cleave full-length IL-33 into mature forms [66]. In a mouse model of P. aeruginosa-induced pneumonia, neutrophil-derived elastase (NE) induces the production of TNF-α, MIP-2/CXCL2 and IL-6 in the lungs, and in particular by macrophages, via a TLR4-dependent mechanism [67].
It has been proposed that both human and murine neutrophils infiltrating psoriatic skin lesions and inflamed synovia of rheumatoid arthritis patients express IL-17A [68, 69]. However, human and murine neutrophils show differences in cytokine expression. In particular, the expression of IFN-γ and IL-10 by human neutrophils remains controversial [2, 61, 70]. Surprisingly, and despite previous negative findings [70], human neutrophils have been reported to produce remarkable amounts of IL-10 upon stimulation with serum amyloid A and LPS [71]. These latter findings have not been reproduced in other laboratories, thus raising the issue of the need for stringent purification and control of monocyte contamination [72]. In addition, the IL-10 genomic locus is in an inactive state in human neutrophils, supporting their incapacity to produce IL-10 [76]. Concerning the murine counterpart, several studies have demonstrated that mouse neutrophils produce IL-10, for instance during disseminated Candida infection [23], methicillin-resistant S. aureus infection [73], pneumonia [74] or Trypanosoma cruzi infection [75].
Immune cell cross talk
Within tissues, neutrophils engage in an intricate cross talk with stromal elements, macrophages, dendritic cells (DC) and lymphocyte subsets. Integration of signals received by neutrophils during the migration process (i.e. cytokines, adhesion, transmigration, microbial products) is a critical step to increase their life span, thereby allowing these cross talks [77]. Activated tissue-resident mesenchymal stem cells and bone marrow-derived mesenchymal cells promote neutrophil recruitment, increase their life span and function in vitro [78, 79].
Activated neutrophils were shown to promote the maturation of human monocyte-derived DC (moDC), through interaction between CD18 and CEACAM1 expressed by neutrophil and DC-SIGN on moDC, and mouse bone marrow-derived DC, through the production of TNF-α [80–82]. However, the cross talk between neutrophils and DC may also inhibit or reduce their maturation and immunostimulatory activity through the secretion of ectosomes and neutrophil elastase [83, 84].
Neutrophils also communicate with T cells, B cells and NK cells. For example, activated neutrophils induce Th1 and Th17 cell chemotaxis through the production of CCL2, and CXCL10 or CCL2 and CCL20, respectively [9]. Accordingly, neutrophils and Th17 cells co-localize in the gut from Crohn’s disease and synovial fluid from rheumatoid arthritis patients [9]. In turn, Th17, γδ T cell or Treg, but not Th1, produce the neutrophil chemoattractant CXCL8 and activated T cells modulate the neutrophil life span and activation through the secretion of selected cytokines (e.g. IFN-γ, GM-CSF and TNF-α) [9, 85–87].
Neutrophils migrate to the lymph node in a CCR7-dependent manner and act as antigen-presenting cells [88–92]. Moreover, neutrophils take up and present exogenous antigens on MHC-I complex in vivo and promote the differentiation of naive CD8+ T cells into cytotoxic T cells, suggesting a direct interaction between neutrophils and CD8+ T cells [91]. In contrast, neutrophils may also interfere with professional antigen-presenting cells via a competition for the antigen and, therefore, reduce the T CD4+ response [93].
A “ménage a trois” composed by neutrophils, dendritic cells and T cells can enhance the development of the immune protective response. DC have the capacity to internalize live and UV-irradiated neutrophils in a CD18-dependent fashion [94]. Therefore, infected neutrophils sustain the maturation, activation and migration to the lymph node of DC through a cell contact-dependent fashion, which in turn cross-present antigens and elicit T cells to produce IFN-γ [94–96]. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to a slow down of the bacterial acquisition by DC, a slower migration to the lung-draining lymph node and a delayed activation of CD4+ T cells in vivo [97].
The interaction of neutrophils with DC can also result in NK cell activation. A cross talk between human neutrophils and 6-sulfo LacNAc+ myeloid DC (slanDC) increases the release of IL-12p70 by slanDC, which in turn potentiates the production of IFN-γ by NK cells [5]. Moreover, IFN-γ potentiates the interaction between neutrophils and slanDC and the release of IL-12p70, creating a positive amplification loop [5]. This tripartite network is supported by direct interactions between neutrophils and slanDC, via CD18–ICAM1 interaction, and between neutrophils and NK cells, via ICAM3 and probably CD18–CD11d complex expressed by NK cells [5]. Co-localization of neutrophils, slanDC and NK cells has been shown in the colonic mucosa of Crohn’s disease patients as well as in skin lesions of psoriasis patients, suggesting a pathophysiological relevance for this tripartite network [5].
Using a novel form of neutropenia obtained from a point mutation in the transcriptional repressor Gfi1, Jaeger and colleagues have recently demonstrated that neutrophils are crucial for the development of NK cells [4, 98]. These mice, called Genista, have normal viability, but are neutropenic due to a blockage of terminal granulocytic differentiation just after the metamyelocytic stage [98]. Interestingly, neutropenia was associated with poor survival, hyperproliferation, impaired development and hyporeactivity of NK cells [4]. For instance, most of the splenic NK cells are blocked at the double-positive CD27+CD11b+ stage and a low percentage expresses CD107a and IFN-γ upon contact with tumour cells [4]. Moreover, the depletion of neutrophils in wild-type mice is sufficient to induce NK cell hyporeactivity and impair their maturation [4]. Interestingly, NK cells from patients suffering from severe congenital neutropenia display similar maturation and functional defects, suggesting that neutrophils contribute to NK cell development and function also in human [4]. In turn, NK cell-derived products (e.g. IFN-γ, GM-CSF) promote the survival and activation of neutrophils [6, 99, 100], whereas cell–cell contact between NK cells and neutrophils induces neutrophil apoptosis via the natural cytotoxicity receptor NKp46 and the Fas pathway [6, 101].
As discussed above, a population of neutrophils around the marginal zone of the spleen (MZ) has been recently identified and called “B-cell helper neutrophils” (NBH) [65]. These neutrophils colonize the MZ during foetal life and become more prominent after postnatal mucosal colonization by bacteria. Indeed, LPS-activated splenic sinusoidal endothelial cells produce neutrophil chemoattractant molecules (e.g. CXCL8, CXCL1, CXCL2, CXCL3) and IL-10, which reprogram neutrophils towards a NBH phenotype [65]. NBH express higher levels of B cell-stimulating molecules such as BAFF, APRIL, IL-21 and CD40L and B cell chemoattractant molecules such as CXCL12 and CXCL13 compared to circulating neutrophils [65]. Based on various parameters, including their expression levels of CD16 and CD15, NBH were divided into NBH1 (CD15int, CD16int) and NBH2 (CD15low, CD16low). Moreover, higher expression levels of CD27, CD40L, CD86, CD95 and HLA-II associated with a lower expression level of CD24 are found in NBH1 compared to NBH2, suggesting that NBH1 are more activated subsets. NBH, and in particular NBH2, via higher secretion levels of BAFF, APRIL and IL-21 compared to NBH1, activate B cells from the MZ and promote immunoglobulin class switching, somatic hypermutation and antibody production [65]. Interestingly, patients with neutrophil disorders present low levels of IgM, IgG and IgA antibodies against microbial T cell-independent antigens (e.g. LPS, peptidoglycan), whereas immunoglobulin levels against T cell-dependent antigens (e.g. diphtheria toxins, tetanus) are unmodified, suggesting that NBH regulate the immunoglobulin response to T cell-independent antigens in vivo [65]. Though this study suggests that neutrophils may regulate the immunoglobulin response, their implication in human immunoglobulin deficiency, such as in acquired IgA deficiency, has not been demonstrated.
Though invariant NKT (iNKT) cells are known to modulate inflammation and neutrophilic inflammation [102–104], neutrophil modulation of iNKT activity has not been previously reported. Recently, Weingender and colleagues [105] have demonstrated that cell–cell contact between neutrophils and iNKT cells occurs in vitro and impairs iNKT function. Accordingly, the expression levels of GATA3 and T-bet in iNKT as well as the levels of α-galactosyl ceramide-induced cytokines are reduced during neutrophilic inflammation in mice and humans [105].
All these data demonstrate that neutrophils are not isolated players that are quickly substituted by more specialized cells, but they guide, support and regulate the immune response throughout its development.
Neutrophils in resolution of inflammation
Neutrophils have long been viewed as final effector cells of the acute phase of the inflammatory response and as passive cells during the resolution of inflammation. However, the critical role played by neutrophils in the resolution phase of inflammation and to maintain tissue homeostasis is now demonstrated and accepted [2].
The cell death receptor ligand TRAIL is produced by neutrophils [106] and, in turn, accelerates neutrophil apoptosis in vitro and in vivo [107]. Accordingly, TRAIL deficiency has been associated with increased neutrophil number and inflammation during neutrophilic inflammation [107]. This phenotype is reversed by treatment with recombinant TRAIL, suggesting that neutrophil apoptosis driven by TRAIL could be a potential therapeutic target in neutrophilic inflammation [107]. Moreover, neutrophil-associated p40phox, a subunit of the NADPH oxidase complex involved in the generation of ROS, plays a crucial role in the resolution phase of intestinal inflammation [108]. Indeed, NADPH oxidase activity controls the resolution phase of inflammation via the downregulation of CCR1 expression in mouse neutrophils and the upregulation of enzymes involved in glycan modifications (e.g. fucosyl transferases, sialyl transferases), which are regulators of leukocyte trafficking through selectin ligand synthesis [108].
During the late phase of the inflammatory response, the biosynthesis of eicosanoids by neutrophils is shifted from leukotriene B4 (LTB4) to the lipoxin A4 (LXA4), reducing tissue neutrophil infiltration through a direct interaction between LXA4 and FPR2, also known as the G-protein-coupled receptor (GPCR) LXA4 receptor [109]. Neutrophils contribute to the biosynthesis of omega-3 essential polyunsaturated fatty acid-derived mediators, resolvins (Rv; i.e. RvE1, RvE2, RvD1, RvD2, RvD5), and protectin D1 (PD1), which inhibit neutrophil infiltration in several in vivo inflammatory models [109–111]. RvD1 and RvE2 have the capacity to interact with GPCRs and RvE1 with leukotriene B4 receptor 1 (BLT1), leading to the inhibition of subsequent migration induced by LTB4 [109, 112]. Accordingly, in a zymosan-induced peritonitis model, the anti-inflammatory effects observed after low-dose administration of RvE1 are lost in BLT1-deficient mice [113]. Moreover, RvE1 and RvD1 dampen leukocyte–endothelial interactions via the modulation of the expression of leukocyte adhesion receptors (e.g. CD11b, CD18) and the regulation of L-selectin (CD62L) shedding [109, 111, 112]. Other mediators, such as the recently described macrophage-derived compound maresin 1 and the new 18S series resolvins, inhibit neutrophil transendothelial migration and tissue infiltration in vivo [114, 115]. In addition to limiting neutrophil infiltration, these pro-resolving mediators enhance phagocytosis and the clearance of microorganisms by neutrophils [110]. For instance, in a model of E. coli-induced peritonitis, RvD5 and RvD1 increased the phagocytosis of E. coli by macrophages and neutrophils, reduced pro-inflammatory cytokine levels and bacterial burden, and enhanced survival, potentiating the effect of antibiotics [110].
Disposal of apoptotic neutrophils regulated by the expression of “eat me” signal on apoptotic cells and receptors on phagocytes is a fundamental step to trigger an anti-inflammatory response and the resolution of inflammation [116]. For instance, recognition and ingestion of apoptotic neutrophils by macrophages induce an IL-10high IL-12low M2-like phenotype that negatively regulates inflammation and stimulates tissue repair [117]. The biosynthesis of pro-resolving mediators is increased in neutrophils during apoptosis and in macrophages after engulfment of apoptotic neutrophils. In turn, these mediators enhance the phagocytosis and clearance of apoptotic cells [109, 118]. RvE1 shortens the life span of neutrophils in the presence of opsonized pathogens, probably through the activation of caspase-8 [119]. Accordingly, apoptotic neutrophils protected mice against LPS-induced septic shock, and in a model of pneumonia, treatment with RvE1 increased the percentage of apoptotic neutrophils and reduced neutrophil infiltration and lung injury [119, 120]. Moreover, the pro-resolving mediators LXA4, RvE1 and PD1 increased the expression of CCR5 on apoptotic neutrophils that assist in vivo in the sequestration and clearance of CCL3 and CCL5 from inflamed sites [121]. Live neutrophils also have the capacity to trap and scavenge chemokines and cytokines. For instance, neutrophils express the IL-1 receptor antagonist, a soluble molecule which binds to IL-1R without inducing any intracellular signal [122] and the type II IL-1 decoy receptor (IL-1RII), both in membrane-bound or released forms, which binds IL-1 and prevents its interaction with its signalling receptor complex IL-1R1 [123].
Neutrophils in pathology
Infection
Neutrophils have long been recognised for their ability to sense and eliminate extracellular pathogens. However, recent evidence demonstrating their recruitment during the IL-17/Th17 response and their involvement in the host response against intracellular pathogens challenges this view [8]. For instance, the granule-associated molecules cathepsin G and neutrophil elastase play a crucial role in pulmonary protective immunity against mycobacterial infection [124]. In a genome-wide transcriptional profile from the blood of patients with active tuberculosis (TB), latent TB and healthy controls, a signature revealed an overexpression of type I IFN-inducible transcripts in blood neutrophils from infected patients, thus supporting a role for these cells in the pathogenesis of TB [125]. Consistently, neutrophils express various cytosolic DNA sensors promoting IFN-β production upon infection by intracellular pathogens (e.g. Legionella pneumophilia, B. henselae, L. monocytogenes) [21]. Neutrophilic inflammation observed during M. tuberculosis infection is detrimental to the host and has been associated with increased infection [126]. IFN-γ controls neutrophilic inflammation via inhibition of the IL-17/Th17 response and may directly act on neutrophils to prevent their accumulation [126, 127].
Upon neutrophil activation via TLR engagement, G-protein-coupled receptor kinase-2 induces CXCR2 desensitization and internalization. Accordingly, TLR9 deficiency enhances neutrophil recruitment to the site of infection, increases bacterial clearance and improves sepsis outcome [128]. IL-33, a member of the IL-1 family, is involved in regulating the activity of neutrophils in infections. For instance, it inhibits the downregulation of CXCR2 expression induced by TLR4 ligation in mouse and human neutrophils. Interestingly, patients who do not recover from sepsis have high serum levels of soluble ST2 (ST2 is also known as IL-1RL1), the decoy receptor of IL-33, and their neutrophils have reduced expression of CXCR2, suggesting a therapeutic potential of IL-33 in sepsis [129]. Moreover, IL-33 increases the expression of CR3 in neutrophils via a mechanism involving both the TLR and dectin-1 signalling pathways [130]. Therefore, IL-33 enhances the phagocytosis and killing of opsonized C. albicans by neutrophils and the administration of IL-33 protects mice against C. albicans infection in a neutrophil-dependent mechanism [130].
Neutrophils in chronic inflammation and autoimmunity
Although neutrophils are generally linked to acute inflammation, recent reports have challenged the dogma and demonstrated their fundamental implication in the development and/or persistence of chronic inflammation. For instance, neutrophil infiltration in response to zymosan-induced peritonitis is enhanced in mice experiencing various chronic inflammatory conditions, probably via IL-17A produced during chronic inflammation [131].
In chronic obstructive pulmonary disease (COPD), immunoglobulin-free light chains (IgLC), found in serum and lung tissue of patients, bind to neutrophils, activating and inducing them to secrete CXCL8 [132]. Interestingly, in a murine model of lung emphysema induced by cigarette smoke, the administration of IgLC antagonist reduces neutrophil influx in the broncho-alveolar space and activation [132]. Moreover, selective neutrophil chemoattraction promoted by the tripeptide proline–glycine–proline (PGP) has been implicated in the persistence of COPD [133, 134]. The leukotriene A4 hydrolase, which promotes the synthesis of the chemotactic factor, LTB4, also has the capacity to degrade PGP via its aminopeptidase activity [133]. However, the aminopeptidase but not the hydrolase activity of LTAH4 is inhibited by cigarette smoke, thereby leading to sustained neutrophil recruitment and chronic lung inflammation by the combined action of LTB4 and PGP [133, 134]. Since the extracellular levels of chloride ions selectively activate the peptidase activity of LTAH4, a similar mechanism may be at play in cystic fibrosis, characterized by chronic neutrophilic inflammation [133]. Indeed, due to mutations in the cystic fibrosis transmembrane conductance regulator protein, the extracellular levels of chloride ions are reduced in cystic fibrosis and are likely to be responsible for the measurable levels of PGP in the sputum of these patients [135]. In addition, gamma-glutamyl transferase, observed in high levels in cystic fibrosis sputum and involved in the catabolism of the antioxidant and mucolytic glutathione, are found associated with the neutrophil secretory vesicles and released after stimulation [136]. Thus, neutrophils can directly contribute to the low concentration levels of glutathione found in cystic fibrosis airways and in worsening respiratory function [136].
The nature of the contribution of neutrophils in autoimmune disorders is not well defined, despite their recognised role in pathogenesis. Recent observations demonstrated the involvement of NETs in autoimmunity. Indeed, the degradation of NETs by DNase I, normally observed in healthy human serum, is compromised in a subset (36.1 %) of patients with systemic lupus erythematosus (SLE), a multi-organ autoimmune disease characterized by an interferon and granulopoiesis signature [137, 138]. NETs activate the classical complement pathway leading to C1q deposition, which inhibits DNase I and, thus, their degradation [139]. A positive correlation has been reported between undegraded NETs and the levels of antinuclear and anti-NET antibodies and with the frequency of lupus nephritis development [138]. Accordingly, netting neutrophils are found in the skin and kidney of lupus patients, and serum from lupus patients contains immune complexes composed of autoantibodies, notably anti-ribonucleoproteins (anti-RNP IgG), self-DNA and antimicrobial peptides, such as LL-37 or HNP [140–142]. Therefore, defective NET clearance leads to the expression of a set of autoantigens and danger-associated molecular patterns, known to trigger and promote inflammation [116]. In Felty’s syndrome, a variant of rheumatoid arthritis defined by an enlarged spleen and an abnormally low white blood cell count, circulating autoantibodies, in particular directed against PAD4-deiminated histones, are found associated with NETs [143]. These immune complexes are transported across pDC membranes via binding to CD32 (FcγRIIB), where self-DNA induces the production of IFN-α. In turn, IFN-α primes neutrophils for further NET formation [140–142]. Interestingly, a distinct subset of neutrophils (low-density granulocytes) found in SLE patients display an activated phenotype; secrete high levels of type I IFNs, TNF-α and IFN-γ; overexpress a set of immunostimulatory proteins and alarmins; have a high capacity to produce NETs; and induce significant endothelial cell cytotoxicity [140, 144].
Neutrophils are also linked to vascular diseases where the presence of anti-neutrophil cytoplasmic antibodies (ANCAs) is a hallmark of ANCA-associated vasculitis (e.g. Wegener’s granulomatosis, Churg–Strauss syndrome and microscopic polyangiitis) [145]. NETs promote injury of the endothelium and the surrounding tissue via the presence of extracellular histones, which have been involved in cytotoxic activity, organ failure and death [146, 147]. Accordingly, pre-incubation of NETs with antibodies against histones reduced NET-induced cytotoxicity in vitro [148]. NETs are produced by ANCA-stimulated neutrophils and found in kidney biopsies from patients with small-vessel vasculitis and SLE, where they contribute to endothelial damage [140, 149].
In a mouse model of transfusion-related acute lung injury (TRALI), neutrophils capture circulating platelets, which results in the production of ROS by neutrophils and subsequent vascular injury [150]. Moreover, activated platelets, found during vessel injury and sepsis, induce NET formation, causing endothelial and tissue damage [146, 151–153]. Disruption of NETs by intranasal DNase I treatment in TRALI improves blood arterial oxygenation and reduces lung oedema, lung vascular permeability and mortality [151, 152]. In humans, NETs were detected in plasma and lungs of patients with TRALI, suggesting that clinical trials to target platelet activation and platelet–neutrophil interactions should be considered in this condition [151].
Neutrophils, and in particular NET formation, are involved in the coagulation cascade and thrombus formation by supporting platelet adhesion and aggregation, thrombin-dependent fibrin formation and blood clot formation [153–155]. Accordingly, in an experimental model of deep vein thrombosis, NETs are found in the thrombus and the administration of DNaseI protects mice from thrombosis [154, 156]. Moreover, NET formation has been involved in cancer-associated thrombosis, one of the major causes of death in cancer patients [155].
Neutrophil recruitment into joints, induced by LTB4 and chemokines produced by stimulated synovial cells, is a hallmark of arthritis [157]. Their recruitment into joints requires the GPI-anchored protein Ly6G, which is closely associated with β2-integrins at the neutrophil surface [158]. Moreover, Syk-dependent signalling in neutrophils, required for FcγR-induced signalling, is essential to establish an immune complex-mediated arthritis [159].
Finally, CXCR2-dependent neutrophil activation and consequent inflammatory disease has been involved in susceptibility to murine multiple sclerosis, an inflammatory demyelinating disorder of the central nervous system [160, 161]. For instance, transfer of CXCR2-positive neutrophils into CXCR2-deficient resistant mice restores susceptibility to autoimmune encephalomyelitis [161]. Moreover, in a model of cuprizone-induced demyelination, CXCR2 knockout mice are resistant due to deficient neutrophil effector responses [160]. Thus, CXCR2 not only contributes to neutrophil migration into tissues but also promotes their effector functions.
Neutrophils and cancer
Genetic instability and inflammation, such as inflammatory cell infiltration, chemokine and cytokine expression in the microenvironment of most neoplastic tissues, have been proposed to represent hallmarks of cancer [162]. Among myeloid cell subsets infiltrating the tumour stroma, tumour-associated macrophages (TAM) were the most prominent and best-characterized cells implicated in tumour progression, stroma deposition and remodelling, angiogenesis and antitumour T cell-dependent immunity [163]. However, tumour-associated neutrophils (TAN) have recently emerged as key mediators in malignant transformation, tumour progression and in the regulation of antitumour immunity [2, 127].
Neutrophils and prognostic significance
The relationship between TAN infiltration and prognosis in human cancer has not been systematically investigated, as has been done for macrophages [163]. However, evidence based on epidemiological studies and animal models is consistent with the view that neutrophil infiltration and accumulation within neoplastic tissues may be associated with a poor clinical outcome, as observed in human patients with bronchoalveolar carcinoma, hepatocellular carcinoma, colorectal carcinomas, aggressive types of pancreatic tumours, or head and neck squamous cell carcinoma [164–169]. In contrast, neutrophil infiltration has been associated with a favourable prognosis in patients with gastric carcinoma [170]. Collectively, these results suggest that depending on the localisation, the prognostic significance of infiltrating neutrophils may differ, as observed for other leukocyte populations [163].
Neutrophil recruitment into the tumour and tumour promotion
The tumour-associated inflammation, a hallmark of cancer, triggers the production of CXC chemokines (CXCL8, CXCL1, CXCL2, CXCL3, CXCL5) by cells infiltrating or surrounding the tumour (e.g. TAM) and by tumour cells themselves [2, 171, 172]. This family of chemokines was linked to cancer progression and in particular to tumour angiogenesis and metastasis [173]. In animal models, a correlation was found between the expression levels of CXC chemokines and lung tumour progression, and CXCR2 inhibition reduced pancreatic ductal adenocarcinoma progression [174, 175]. Most recently, Jamieson and colleagues [176] reported that in models of inflammation-induced skin papillomas, colitis-associated tumour or spontaneous malignancy, CXCR2 deficiency or neutrophil depletion suppressed the inflammation-associated tumourigenesis and the spontaneous development of tumours.
More than 30 years ago, Clark and Klebanoff [177] suggested a neutrophil-dependent tumour cell cytotoxicity mediated by the peroxidase system. In contrast, in 1995, Pekarek et al. [178] found that granulocytes are required for the rapid growth of tumour cells and that their depletion inhibits tumour development. Evidence suggesting the involvement of neutrophils in the promotion and progression of cancer has followed these original observations [2, 127].
Neutrophil-associated proteins stored within granules and directed to defence towards pathogens are also involved in tumour promotion. Neutrophil elastase shows a dual role in tumour initiation. For instance, NE is taken up into a specific endosomal compartment of adjacent epithelial tumour cells and hydrolyses insulin receptor substrate-1, which normally interacts with a subunit of the PI3K and blocks its activity [179]. Therefore, upon NE activity, PI3K enhances the signalling of the platelet-derived growth factor receptor, thereby leading to tumour cell proliferation [179]. In contrast, NE taken up by breast cancer cells cleaves cyclin E into a truncated isoform, which is subsequently presented in the context of HLA-I molecule and promotes a T lymphocyte-mediated lysis of cancer cells [180].
Neutrophil-derived cytokines contribute also to tumour progression. The expression of GM-CSF by breast tumour cells promotes the production of oncostatin M by neutrophils, which in turn induces the production of vascular endothelial growth factor (VEGF) by breast cancer cells, reduces cancer cell adhesiveness and increases their invasive capacity [181]. Hepatocyte growth factor (HGF), a cytokine involved in cell proliferation and motility, is stored in secretory vesicles and granules of neutrophils [182]. Interestingly, neutrophils enhance the invasiveness of human cholangiocellular carcinoma and hepatocellular carcinoma cells in vitro via a HGF secretion-dependent mechanism [183]. Moreover, in patients with bronchoalveolar carcinoma, tumour cells express the HGF receptor and neutrophil-derived HGF promotes their migration [166]. Accordingly, the levels of HGF found in the bronchoalveolar lavage fluids of patients are correlated with the neutrophil counts and associated with poor prognosis [166].
A mechanism facilitating the occurrence of metastasis has been directly linked with the recruitment and activation of neutrophils. Indeed, melanoma cell-derived CXCL8 increases the expression of β2-integrin on neutrophils and promotes a neutrophil–melanoma cell interaction via the expression of ICAM-1 by melanoma cells [171]. In turn, this cross talk favours the transmigration of melanoma cells across the endothelium and facilitates the development of lung metastasis [171]. In contrast, in mice orthotopically implanted with breast cancer cells, TAN accumulate in the premetastatic lung and their depletion increases the metastatic burden. The authors suggested that tumour-educated neutrophils are recruited to the premetastatic lung and inhibit tumour cell seeding through an H2O2-dependent cytotoxic activity [184]. Neutrophils acquire this cytotoxic phenotype upon accumulation and sequestration in the lung mediated by granulocyte colony-stimulating factor (G-CSF) and a subsequent activation induced by CCL2 [184].
Angiogenic switch mediated by neutrophils
TAM and TAN are involved in tumour angiogenesis through the production of growth and matrix remodelling factors such as VEGF, basic fibroblast growth factor, platelet-derived growth factor, urokinase-type plasminogen activator and metalloproteinases (MMPs). For instance, neutrophil-derived VEGF has been identified as the major factor responsible for the in vivo angiogenesis activity induced by CXCL1 [185]. In addition, neutrophil-derived MMP-9 induces VEGF expression in the neoplastic tissue and therefore catalyses tumour angiogenesis [186]. In patients with hepatocellular carcinoma and head and neck cancer, neutrophils are the major source of MMP-9 in the peritumoural stroma and within the tumour, respectively [164, 187]. Interestingly, a microvascular architecture promoting blood-borne metastasis is observed in hepatoma samples presenting an elevated number of neutrophils [164]. Moreover, depletion of granulocytes reduces tumour angiogenesis and growth in murine hepatoma, demonstrating that neutrophils control the progression of tumour angiogenesis in vivo [164].
In a model of subcutaneous injection of melanoma or fibrosarcoma cells, neutrophils were identified as the major cells with angiogenic activity negatively controlled by IFN-β, a potential adjuvant in cancer vaccines. High expression levels of VEGF, CXCR4 and MMP-9, associated with better developed blood vessels, are found in neutrophils from IFN-β mice [188]. Interestingly, neutrophil depletion in IFN-β-deficient mice reduces tumour growth [188], suggesting the relevance of the interplay between INF-β and neutrophils in the early stages of cancer development.
Tumour-associated G-CSF enhances the expression of Bv8 (also known as prokineticin-2) by neutrophils, which promotes their mobilization and tumour angiogenesis [189, 190]. Interestingly, treatment of tumours refractory to anti-VEGF therapy with anti-G-CSF or anti-Bv8 reduces tumour growth and angiogenesis [191].
Finally, NET formation has been recently linked to cancer-associated thrombosis, a highly common cause of death in cancer patients [155]. Neutrophils isolated from leukaemic mice or tumour-bearing mice are primed to release NETs, most likely via the increased levels of G-CSF found in cancer, which thus promotes a prothrombotic milieu [155].
Neutrophilic inflammation is linked to genetic instability
More than 25 years ago, neutrophilic inflammation and, in particular, neutrophil-derived ROS were linked to genetic instability [192]. Today, several lines of evidence are consistent with the view that neutrophils are linked with the process of carcinogenesis through ROS-dependent and ROS-independent mechanisms. Accordingly, the expression of neutrophil-derived inducible nitric oxide synthase and ROS, notably the MPO-mediated formation of HOCl, have been linked with point mutations or DNA damages, and levels of TAN significantly correlated with DNA mutations [193–195]. In ulcerative colitis, neutrophils located in crypt abscesses are activated and induce DNA damage, as suggested by G2/M cell cycle checkpoint arrest in colon epithelial cells [196].
Plasticity of TAN
In 1989, the plasticity of neutrophils was suggested in the context of tumours in which neutrophils isolated from tumour-bearing animals significantly increased the metastatic potential of adenocarcinoma cells compared to neutrophils isolated from healthy animals [197]. More recently, reports have confirmed that neutrophils are endowed with unexpected plasticity [70, 172]. Therefore, mirroring the M1–M2 and Th1–Th2 paradigms, neutrophils can be polarized towards a pro-inflammatory phenotype with antitumour activity (N1) or towards a protumoural N2 phenotype [172]. TGF-β, which has a regulatory role on neutrophil functions, including chemotaxis or cytotoxicity, and plays a critical role in tumour initiation, progression and metastasis (e.g. suppressor or promoter depending on the context and stage of the tumour), is a crucial effector in this polarization [172]. In models of lung adenocarcinoma and mesothelioma, TGF-β drives neutrophils towards a N2 phenotype, whereas TGF-β blockade enhances the infiltration of N1 TAN, which are characterized by a cytotoxic activity against tumour and a pro-inflammatory phenotype (i.e. TNF-αhigh, CCL3high, ICAM-1high, arginaselow) [172]. The authors have demonstrated that TGF-β blockade is associated with the activation of a CD8+ T cell-dependent arm that involves neutrophils as effectors [172].
Since studies have indicated that neutrophils can also exert an antitumour immunity [184], in analogy with their “big brothers”, macrophages, and depending on environmental signals, neutrophils can exert a dual influence on tumour growth. Moreover, in parallel to the differences found between mouse and human macrophage polarization, the existence and functional properties of N1 and N2 in human have to be carefully investigated.
Relation between neutrophils, TAN and MDSC
Cancer has provided a paradigm for myeloid-derived suppressor cells (MDSC), which represent a heterogeneous group of myeloid cells composed of monocytic (M-MDSC) and granulocytic (G-MDSC) subsets, recognised for their immune-suppressive activity [198]. Neutrophils and TAN share phenotypic (e.g. cell markers and morphology) and functional properties (e.g. recruitment within tumours, production of arginase, promotion of tumour angiogenesis) with G-MDSC [198]. Moreover, G-MDSC and neutrophils were usually defined and isolated using the same phenotype markers (i.e. CD11b+ Ly6G+ Ly6Clow), creating confusion to discriminate these cells. Therefore, whether TAN activity can be attributed to MDSC remains a matter of debate. Reports have suggested that activated neutrophils present an immunosuppressive activity in cancer patients and could account for the arginase I-mediated suppression of lymphocytes in renal cell carcinoma [199, 200]. Recently, in a genetic model of lung adenocarcinoma, but also in patients with invasive cancer, splenic granulocyte/macrophage progenitors have been proposed as TAN progenitors [201]. Consistent with the hypothesis that a part of the MDSC activity can be attributed to TAN, a subset of MDSC accumulated within the spleen of tumour-bearing animals are indeed immature myeloid cells [201–203]. However, other studies have reported that MDSC and neutrophils are functionally and phenotypically different [202, 204]. For instance, transcriptomic analysis of mouse TAN, naive neutrophils and G-MDSC has suggested that TAN and G-MDSC are distinct populations of cells and that naive neutrophils and G-MDSC are more closely related to each other than to TAN [205]. Interestingly, upon stimulation by GM-CSF, G-MDSC acquire the same characteristics as neutrophils, suggesting that G-MDSC are immature neutrophils [204]. These immature neutrophilic MDSC have also been reported in the peripheral blood of patients with cancer, and high levels are associated with poor prognosis [202, 203]. Collectively, MDSC are a heterogeneous population of myeloid cells which includes immature granulocytes. Therefore, further investigations are required to identify new biomarkers and discriminate the distinct subpopulation of cells within MDSC.
Concluding remarks
Long viewed as ultimate short-lived effectors, several lines of evidence have demonstrated in vitro, but also in vivo, that neutrophils are endowed with unsuspected diversity and plasticity. Conforming to Metchnikoff’s model, neutrophils patrol the bloodstream to detect and phagocyte pathogens. However, this classical mechanism has been complemented by the discovery of NETs, which trap and kill extracellular pathogens. Their importance in innate immunity was notably put into evidence by evolutionary strategies adopted by pathogens to escape these poisonous NETs.
Neutrophils use a set of membrane and intracellular molecules to sense their local environment signals and switch their phenotype towards a pro-inflammatory and antitumour (N1) or anti-inflammatory and protumoural (N2) programs. In turn, neutrophils are involved in a bidirectional cross talk with most other types of leukocytes and can directly or indirectly modify their maturation, activation or effector functions, depending on the context. These new vistas shed new light on neutrophil function, but also raise new questions. For instance, the existence of neutrophil plasticity and diversity in humans together with their role in human pathologies require further studies.
References
Borregaard N (2010) Neutrophils, from marrow to microbes. Immunity 33(5):657–670
Mantovani A, Cassatella MA, Costantini C, Jaillon S (2011) Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol 11(8):519–531
Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489. doi:10.1146/annurev-immunol-020711-074942
Jaeger BN, Donadieu J, Cognet C, Bernat C, Ordonez-Rueda D et al (2012) Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med 209(3):565–580. doi:10.1084/jem.20111908
Costantini C, Calzetti F, Perbellini O, Micheletti A, Scarponi C et al (2011) Human neutrophils interact with both 6-sulfo LacNAc+ DC and NK cells to amplify NK-derived IFN{gamma}: role of CD18, ICAM-1, and ICAM-3. Blood 117(5):1677–1686. doi:10.1182/blood-2010-06-287243
Costantini C, Micheletti A, Calzetti F, Perbellini O, Pizzolo G et al (2010) Neutrophil activation and survival are modulated by interaction with NK cells. Int Immunol 22(10):827–838. doi:10.1093/intimm/dxq434
Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto-Garcia E et al (2012) IL-17 and TNF-alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol 188(12):6287–6299. doi:10.4049/jimmunol.1200385
Cua DJ, Tato CM (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 10(7):479–489
Pelletier M, Maggi L, Micheletti A, Lazzeri E, Tamassia N et al (2010) Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 115(2):335–343. doi:10.1182/blood-2009-04-216085
Bottazzi B, Doni A, Garlanda C, Mantovani A (2010) An integrated view of humoral innate immunity: pentraxins as a paradigm. Annual Rev Immunol 28:157–183
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820
Nathan C (2006) Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6(3):173–182
Segal AW (2005) How neutrophils kill microbes. Annu Rev Immunol 23:197–223
Hayashi F, Means TK, Luster AD (2003) Toll-like receptors stimulate human neutrophil function. Blood 102(7):2660–2669
Berger M, Hsieh CY, Bakele M, Marcos V, Rieber N et al (2012) Neutrophils express distinct RNA receptors in a non-canonical way. J Biol Chem 287(23):19409–19417. doi:10.1074/jbc.M112.353557
Kennedy AD, Willment JA, Dorward DW, Williams DL, Brown GD et al (2007) Dectin-1 promotes fungicidal activity of human neutrophils. Eur J Immunol 37(2):467–478
Kerrigan AM, Dennehy KM, Mourao-Sa D, Faro-Trindade I, Willment JA et al (2009) CLEC-2 is a phagocytic activation receptor expressed on murine peripheral blood neutrophils. J Immunol 182(7):4150–4157
Lee WB, Kang JS, Yan JJ, Lee MS, Jeon BY et al (2012) Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the mincle pathway. PLoS Pathog 8(4):e1002614. doi:10.1371/journal.ppat.1002614
Graham LM, Gupta V, Schafer G, Reid DM, Kimberg M et al (2012) The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J Biol Chem 287(31):25964–25974. doi:10.1074/jbc.M112.384164
Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y et al (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16(2):228–231
Tamassia N, Bazzoni F, Le Moigne V, Calzetti F, Masala C et al (2012) IFN-beta expression is directly activated in human neutrophils transfected with plasmid DNA and is further increased via TLR-4-mediated signaling. J Immunol 189(3):1500–1509. doi:10.4049/jimmunol.1102985
Tamassia N, Le Moigne V, Rossato M, Donini M, McCartney S et al (2008) Activation of an immunoregulatory and antiviral gene expression program in poly(I:C)-transfected human neutrophils. J Immunol 181(9):6563–6573
Greenblatt MB, Aliprantis A, Hu B, Glimcher LH (2010) Calcineurin regulates innate antifungal immunity in neutrophils. J Exp Med 207(5):923–931
Mankan AK, Dau T, Jenne D, Hornung V (2012) The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur J Immunol 42(3):710–715. doi:10.1002/eji.201141921
Anand PK, Malireddi RK, Lukens JR, Vogel P, Bertin J et al (2012) NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 488(7411):389–393. doi:10.1038/nature11250
Tamassia N, Le Moigne V, Calzetti F, Donini M, Gasperini S et al (2007) The MyD88-independent pathway is not mobilized in human neutrophils stimulated via TLR4. J Immunol 178(11):7344–7356
Liu X, Ma B, Malik AB, Tang H, Yang T et al (2012) Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol 13(5):457–464. doi:10.1038/ni.2258
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I et al (2010) Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (New York, NY) 330(6002):362–366. doi:10.1126/science.1195491
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T et al (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464(7285):104–107
Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A et al (2007) The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med 204(4):793–804
Moalli F, Jaillon S, Inforzato A, Sironi M, Bottazzi B et al (2011) Pathogen recognition by the long pentraxin PTX3. J Biomed Biotechnol 2011:830421
Moalli F, Doni A, Deban L, Zelante T, Zagarella S et al (2010) Role of complement and Fc{gamma} receptors in the protective activity of the long pentraxin PTX3 against Aspergillus fumigatus. Blood 116(24):5170–5180
Jaillon S, Jeannin P, Hamon Y, Fremaux I, Doni A et al (2009) Endogenous PTX3 translocates at the membrane of late apoptotic human neutrophils and is involved in their engulfment by macrophages. Cell Death Differ 16(3):465–474
Deban L, Russo RC, Sironi M, Moalli F, Scanziani M et al (2010) Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol 11(4):328–334
Dziarski R, Platt KA, Gelius E, Steiner H, Gupta D (2003) Defect in neutrophil killing and increased susceptibility to infection with nonpathogenic Gram-positive bacteria in peptidoglycan recognition protein-S (PGRP-S)-deficient mice. Blood 102(2):689–697
Rorvig S, Honore C, Larsson LI, Ohlsson S, Pedersen CC et al (2009) Ficolin-1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J Leukoc Biol 86(6):1439–1449
Cho JH, Fraser IP, Fukase K, Kusumoto S, Fujimoto Y et al (2005) Human peptidoglycan recognition protein S is an effector of neutrophil-mediated innate immunity. Blood 106(7):2551–2558
Kashyap DR, Wang M, Liu LH, Boons GJ, Gupta D et al (2011) Peptidoglycan recognition proteins kill bacteria by activating protein-sensing two-component systems. Nat Med 17(6):676–683. doi:10.1038/nm.2357
Moreno-Amaral AN, Gout E, Danella-Polli C, Tabarin F, Lesavre P et al (2012) M-ficolin and leukosialin (CD43): new partners in neutrophil adhesion. J Leukoc Biol 91(3):469–474. doi:10.1189/jlb.0911460
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y et al (2004) Neutrophil extracellular traps kill bacteria. Science (New York, NY) 303(5663):1532–1535
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16(11):1438–1444
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M et al (2012) Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 12(1):109–116. doi:10.1016/j.chom.2012.05.015
Douda DN, Jackson R, Grasemann H, Palaniyar N (2011) Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol 187(4):1856–1865. doi:10.4049/jimmunol.1004201
Menegazzi R, Decleva E, Dri P (2012) Killing by neutrophil extracellular traps: fact or folklore? Blood 119(5):1214–1216. doi:10.1182/blood-2011-07-364604
Parker H, Albrett AM, Kettle AJ, Winterbourn CC (2012) Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol 91(3):369–376. doi:10.1189/jlb.0711387
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN et al (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18:1386–1393. doi:10.1038/nm.2847
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H et al (2011) Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7(2):75–77
McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW et al (2012) Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood 120:3118–3125. doi:10.1182/blood-2012-01-405993
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I et al (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176(2):231–241
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E et al (2011) Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21(2):290–304
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N et al (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207(9):1853–1862
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191(3):677–691
Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J et al (2011) Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood 117(3):953–959
Marcos V, Nussbaum C, Vitkov L, Hector A, Wiedenbauer EM et al (2009) Delayed but functional neutrophil extracellular trap formation in neonates. Blood 114(23):4908–4911, author reply 4911–4902
Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM et al (2009) Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood 113(25):6419–6427
Gabriel C, McMaster WR, Girard D, Descoteaux A (2010) Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J Immunol 185(7):4319–4327
Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A et al (2007) Capsule and d-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 9(5):1162–1171
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A et al (2006) An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 16(4):401–407
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA et al (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16(4):396–400
Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR et al (2011) Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS One 6(9):e23637. doi:10.1371/journal.pone.0023637
Cassatella MA (1999) Neutrophil-derived proteins: selling cytokines by the pound. Adv Immunol 73:369–509
Guma M, Ronacher L, Liu-Bryan R, Takai S, Karin M et al (2009) Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum 60(12):3642–3650. doi:10.1002/art.24959
Rinchai D, Khaenam P, Kewcharoenwong C, Buddhisa S, Pankla R et al (2012) Production of interleukin-27 by human neutrophils regulates their function during bacterial infection. Eur J Immunol 42:3280–3290. doi:10.1002/eji.201242526
Scapini P, Bazzoni F, Cassatella MA (2008) Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunol Lett 116(1):1–6. doi:10.1016/j.imlet.2007.11.009
Puga I, Cols M, Barra CM, He B, Cassis L et al (2012) B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol 13(2):170–180. doi:10.1038/ni.2194
Lefrancais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B et al (2012) IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A 109(5):1673–1678. doi:10.1073/pnas.1115884109
Benabid R, Wartelle J, Malleret L, Guyot N, Gangloff S et al (2012) Neutrophil elastase modulates cytokine expression: contribution to host defense against Pseudomonas aeruginosa-induced pneumonia. J Biol Chem 287:34883–34894. doi:10.1074/jbc.M112.361352
Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M et al (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol 187(1):490–500. doi:10.4049/jimmunol.1100123
Moran EM, Heydrich R, Ng CT, Saber TP, McCormick J et al (2011) IL-17A expression is localised to both mononuclear and polymorphonuclear synovial cell infiltrates. PLoS One 6(8):e24048. doi:10.1371/journal.pone.0024048
Cassatella MA, Locati M, Mantovani A (2009) Never underestimate the power of a neutrophil. Immunity 31(5):698–700
De Santo C, Arscott R, Booth S, Karydis I, Jones M et al (2010) Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol 11(11):1039–1046
Davey MS, Tamassia N, Rossato M, Bazzoni F, Calzetti F et al (2011) Failure to detect production of IL-10 by activated human neutrophils. Nat Immunol 12(11):1017–1018. doi:10.1038/ni.2111, author reply 1018–1020
Tsuda Y, Takahashi H, Kobayashi M, Hanafusa T, Herndon DN et al (2004) Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity 21(2):215–226
Zhang X, Majlessi L, Deriaud E, Leclerc C, Lo-Man R (2009) Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity 31(5):761–771. doi:10.1016/j.immuni.2009.09.016
Tosello Boari J, Amezcua Vesely MC, Bermejo DA, Ramello MC, Montes CL et al (2012) IL-17RA signaling reduces inflammation and mortality during Trypanosoma cruzi infection by recruiting suppressive IL-10-producing neutrophils. PLoS Pathog 8(4):e1002658. doi:10.1371/journal.ppat.1002658
Tamassia N, Zimmermann M, Castellucci M, Ostuni R, Bruderek K et al (2013) Cutting edge: an inactive chromatin configuration at the IL-10 locus in human neutrophils. J Immunol 190:1921–1925. doi:10.4049/jimmunol.1203022
Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A (1992) Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 80(8):2012–2020
Brandau S, Jakob M, Hemeda H, Bruderek K, Janeschik S et al (2010) Tissue-resident mesenchymal stem cells attract peripheral blood neutrophils and enhance their inflammatory activity in response to microbial challenge. J Leukoc Biol 88(5):1005–1015. doi:10.1189/jlb.0410207
Cassatella MA, Mosna F, Micheletti A, Lisi V, Tamassia N et al. (2011) Toll-like receptor-3-activated human mesenchymal stromal cells significantly prolong the survival and function of neutrophils. Stem Cells 29:1001–1011
van Gisbergen KP, Ludwig IS, Geijtenbeek TB, van Kooyk Y (2005) Interactions of DC-SIGN with Mac-1 and CEACAM1 regulate contact between dendritic cells and neutrophils. FEBS Lett 579(27):6159–6168
van Gisbergen KP, Sanchez-Hernandez M, Geijtenbeek TB, van Kooyk Y (2005) Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J Exp Med 201(8):1281–1292
Bennouna S, Bliss SK, Curiel TJ, Denkers EY (2003) Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J Immunol 171(11):6052–6058
Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C et al (2008) Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol 180(2):817–824
Gasser O, Schifferli JA (2004) Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 104(8):2543–2548. doi:10.1182/blood-2004-01-0361
Davey MS, Lin CY, Roberts GW, Heuston S, Brown AC et al (2011) Human neutrophil clearance of bacterial pathogens triggers anti-microbial gammadelta T cell responses in early infection. PLoS Pathog 7(5):e1002040. doi:10.1371/journal.ppat.1002040
Himmel ME, Crome SQ, Ivison S, Piccirillo C, Steiner TS et al (2011) Human CD4+FOXP3+ regulatory T cells produce CXCL8 and recruit neutrophils. Eur J Immunol 41(2):306–312. doi:10.1002/eji.201040459
Pelletier M, Micheletti A, Cassatella MA (2010) Modulation of human neutrophil survival and antigen expression by activated CD4+ and CD8+ T cells. J Leukoc Biol 88(6):1163–1170. doi:10.1189/jlb.0310172
Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ et al (2005) Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood 106(5):1843–1850. doi:10.1182/blood-2005-03-1281
Abi Abdallah DS, Egan CE, Butcher BA, Denkers EY (2011) Mouse neutrophils are professional antigen-presenting cells programmed to instruct Th1 and Th17 T-cell differentiation. Int Immunol 23(5):317–326
Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S et al (2011) CCR7 is involved in the migration of neutrophils to lymph nodes. Blood 117(4):1196–1204. doi:10.1182/blood-2009-11-254490
Beauvillain C, Delneste Y, Scotet M, Peres A, Gascan H et al (2007) Neutrophils efficiently cross-prime naive T cells in vivo. Blood 110(8):2965–2973
Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M et al (2008) Dynamics of neutrophil migration in lymph nodes during infection. Immunity 29(3):487–496. doi:10.1016/j.immuni.2008.07.012
Yang CW, Strong BS, Miller MJ, Unanue ER (2010) Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. J Immunol 185(5):2927–2934. doi:10.4049/jimmunol.1001289
Alfaro C, Suarez N, Onate C, Perez-Gracia JL, Martinez-Forero I et al (2011) Dendritic cells take up and present antigens from viable and apoptotic polymorphonuclear leukocytes. PLoS One 6(12):e29300. doi:10.1371/journal.pone.0029300
Morel C, Badell E, Abadie V, Robledo M, Setterblad N et al (2008) Mycobacterium bovis BCG-infected neutrophils and dendritic cells cooperate to induce specific T cell responses in humans and mice. Eur J Immunol 38(2):437–447. doi:10.1002/eji.200737905
Blomgran R, Ernst JD (2011) Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol 186(12):7110–7119. doi:10.4049/jimmunol.1100001
Blomgran R, Desvignes L, Briken V, Ernst JD (2012) Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11(1):81–90. doi:10.1016/j.chom.2011.11.012
Ordonez-Rueda D, Jonsson F, Mancardi DA, Zhao W, Malzac A et al (2012) A hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia. Eur J Immunol 42(9):2395–2408. doi:10.1002/eji.201242589
Costantini C, Cassatella MA (2011) The defensive alliance between neutrophils and NK cells as a novel arm of innate immunity. J Leukoc Biol 89(2):221–233. doi:10.1189/jlb.0510250
Bhatnagar N, Hong HS, Krishnaswamy JK, Haghikia A, Behrens GM et al (2010) Cytokine-activated NK cells inhibit PMN apoptosis and preserve their functional capacity. Blood 116(8):1308–1316. doi:10.1182/blood-2010-01-264903
Thoren FB, Riise RE, Ousback J, Della Chiesa M, Alsterholm M et al (2012) Human NK cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J Immunol 188(4):1668–1674. doi:10.4049/jimmunol.1102002
Michel ML, Keller AC, Paget C, Fujio M, Trottein F et al (2007) Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med 204(5):995–1001. doi:10.1084/jem.20061551
Emoto M, Emoto Y, Yoshizawa I, Kita E, Shimizu T et al (2010) Alpha-GalCer ameliorates listeriosis by accelerating infiltration of Gr-1+ cells into the liver. Eur J Immunol 40(5):1328–1341. doi:10.1002/eji.200939594
Wintermeyer P, Cheng CW, Gehring S, Hoffman BL, Holub M et al (2009) Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology 136(3):1048–1059. doi:10.1053/j.gastro.2008.10.027
Wingender G, Hiss M, Engel I, Peukert K, Ley K et al (2012) Neutrophilic granulocytes modulate invariant NKT cell function in mice and humans. J Immunol 188(7):3000–3008. doi:10.4049/jimmunol.1101273
Cassatella MA (2006) On the production of TNF-related apoptosis-inducing ligand (TRAIL/Apo-2L) by human neutrophils. J Leukoc Biol 79(6):1140–1149. doi:10.1189/jlb.1005558
McGrath EE, Marriott HM, Lawrie A, Francis SE, Sabroe I et al (2011) TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol 90(5):855–865. doi:10.1189/jlb.0211062
Conway KL, Goel G, Sokol H, Manocha M, Mizoguchi E et al (2012) p40phox expression regulates neutrophil recruitment and function during the resolution phase of intestinal inflammation. J Immunol 189(7):3631–3640. doi:10.4049/jimmunol.1103746
Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8(5):349–361
Chiang N, Fredman G, Backhed F, Oh SF, Vickery T et al (2012) Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484(7395):524–528. doi:10.1038/nature11042
Spite M, Norling LV, Summers L, Yang R, Cooper D et al (2009) Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461(7268):1287–1291. doi:10.1038/nature08541
Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee CH et al (2010) Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci U S A 107(4):1660–1665
Arita M, Ohira T, Sun YP, Elangovan S, Chiang N et al (2007) Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J Immunol 178(6):3912–3917
Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS et al (2009) Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 206(1):15–23. doi:10.1084/jem.20081880
Oh SF, Pillai PS, Recchiuti A, Yang R, Serhan CN (2011) Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J Clin Invest 121(2):569–581. doi:10.1172/JCI42545
Jeannin P, Jaillon S, Delneste Y (2008) Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol 20(5):530–537
Filardy AA, Pires DR, Nunes MP, Takiya CM, Freire-de-Lima CG et al (2010) Proinflammatory clearance of apoptotic neutrophils induces an IL-12(low)IL-10(high) regulatory phenotype in macrophages. J Immunol 185(4):2044–2050
Dalli J, Serhan C (2012) Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 120:e60–e72. doi:10.1182/blood-2012-04-423525
El Kebir D, Gjorstrup P, Filep JG (2012) Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A 109(37):14983–14988. doi:10.1073/pnas.1206641109
Ren Y, Xie Y, Jiang G, Fan J, Yeung J et al (2008) Apoptotic cells protect mice against lipopolysaccharide-induced shock. J Immunol 180(7):4978–4985
Ariel A, Fredman G, Sun YP, Kantarci A, Van Dyke TE et al (2006) Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol 7(11):1209–1216
Cassatella MA, Meda L, Gasperini S, Calzetti F, Bonora S (1994) Interleukin 10 (IL-10) upregulates IL-1 receptor antagonist production from lipopolysaccharide-stimulated human polymorphonuclear leukocytes by delaying mRNA degradation. J Exp Med 179(5):1695–1699
Bourke E, Cassetti A, Villa A, Fadlon E, Colotta F et al (2003) IL-1 beta scavenging by the type II IL-1 decoy receptor in human neutrophils. J Immunol 170(12):5999–6005
Steinwede K, Maus R, Bohling J, Voedisch S, Braun A et al (2012) Cathepsin G and neutrophil elastase contribute to lung-protective immunity against mycobacterial infections in mice. J Immunol 188(9):4476–4487. doi:10.4049/jimmunol.1103346
Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA et al (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466(7309):973–977. doi:10.1038/nature09247
Nandi B, Behar SM (2011) Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med 208(11):2251–2262. doi:10.1084/jem.20110919
Fridlender ZG, Albelda SM (2012) Tumor-associated neutrophils: friend or foe? Carcinogenesis 33(5):949–955. doi:10.1093/carcin/bgs123
Trevelin SC, Alves-Filho JC, Sonego F, Turato W, Nascimento DC et al (2012) Toll-like receptor 9 activation in neutrophils impairs chemotaxis and reduces sepsis outcome. Crit Care Med 40(9):2631–2637. doi:10.1097/CCM.0b013e318258fb70
Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA Jr et al (2010) Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med 16(6):708–712
Le HT, Tran VG, Kim W, Kim J, Cho HR et al (2012) IL-33 priming regulates multiple steps of the neutrophil-mediated anti-Candida albicans response by modulating TLR and dectin-1 signals. J Immunol 189(1):287–295. doi:10.4049/jimmunol.1103564
Bian Z, Guo Y, Ha B, Zen K, Liu Y (2012) Regulation of the inflammatory response: enhancing neutrophil infiltration under chronic inflammatory conditions. J Immunol 188(2):844–853. doi:10.4049/jimmunol.1101736
Braber S, Thio M, Blokhuis BR, Henricks PA, Koelink PJ et al (2012) An association between neutrophils and immunoglobulin free light chains in the pathogenesis of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 185(8):817–824. doi:10.1164/rccm.201104-0761OC
Snelgrove RJ, Jackson PL, Hardison MT, Noerager BD, Kinloch A et al (2010) A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science (New York, NY) 330(6000):90–94. doi:10.1126/science.1190594
Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD et al (2006) A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12(3):317–323. doi:10.1038/nm1361
Gaggar A, Jackson PL, Noerager BD, O'Reilly PJ, McQuaid DB et al (2008) A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol 180(8):5662–5669
Corti A, Franzini M, Cianchetti S, Bergamini G, Lorenzini E et al (2012) Contribution by polymorphonucleate granulocytes to elevated gamma-glutamyltransferase in cystic fibrosis sputum. PLoS One 7(4):e34772. doi:10.1371/journal.pone.0034772
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J et al (2003) Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197(6):711–723
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA et al (2010) Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A 107(21):9813–9818
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C et al (2012) Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol 188(7):3522–3531. doi:10.4049/jimmunol.1102404
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R et al (2011) Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol 187(1):538–552. doi:10.4049/jimmunol.1100450
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F et al (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3(73):73ra20. doi:10.1126/scitranslmed.3001201
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C et al (2011) Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med 3(73):73ra19. doi:10.1126/scitranslmed.3001180
Dwivedi N, Upadhyay J, Neeli I, Khan S, Pattanaik D et al (2012) Felty’s syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum 64(4):982–992. doi:10.1002/art.33432
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M et al (2010) A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol 184(6):3284–3297. doi:10.4049/jimmunol.0902199
Gomez-Puerta JA, Bosch X (2009) Anti-neutrophil cytoplasmic antibody pathogenesis in small-vessel vasculitis: an update. Am J Pathol 175(5):1790–1798
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z et al (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13(4):463–469
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT et al (2009) Extracellular histones are major mediators of death in sepsis. Nat Med 15(11):1318–1321
4Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G et al (2012) Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7(2):e32366. doi:10.1371/journal.pone.0032366
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL et al (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 15(6):623–625
Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY et al (2009) Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med 15(4):384–391. doi:10.1038/nm.1939
Caudrillier A, Looney MR (2012) Platelet–neutrophil interactions as a target for prevention and treatment of transfusion-related acute lung injury. Curr Pharm Des 18(22):3260–3266
Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB et al (2012) Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 119(26):6335–6343. doi:10.1182/blood-2012-01-405183
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S et al (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16(8):887–896. doi:10.1038/nm.2184
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M et al (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107(36):15880–15885
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR et al (2012) Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A 109(32):13076–13081. doi:10.1073/pnas.1200419109
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K et al (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10(1):136–144. doi:10.1111/j.1538-7836.2011.04544.x
Chou RC, Kim ND, Sadik CD, Seung E, Lan Y et al (2010) Lipid–cytokine–chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 33(2):266–278. doi:10.1016/j.immuni.2010.07.018
Wang JX, Bair AM, King SL, Shnayder R, Huang YF et al (2012) Ly6G ligation blocks recruitment of neutrophils via a beta2-integrin-dependent mechanism. Blood 120(7):1489–1498. doi:10.1182/blood-2012-01-404046
Elliott ER, Van Ziffle JA, Scapini P, Sullivan BM, Locksley RM et al (2011) Deletion of Syk in neutrophils prevents immune complex arthritis. J Immunol 187(8):4319–4330. doi:10.4049/jimmunol.1100341
Liu L, Belkadi A, Darnall L, Hu T, Drescher C et al (2010) CXCR2-positive neutrophils are essential for cuprizone-induced demyelination: relevance to multiple sclerosis. Nat Neurosci 13(3):319–326. doi:10.1038/nn.2491
Carlson T, Kroenke M, Rao P, Lane TE, Segal B (2008) The Th17-ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J Exp Med 205(4):811–823. doi:10.1084/jem.20072404
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122(3):787–795. doi:10.1172/JCI59643
Kuang DM, Zhao Q, Wu Y, Peng C, Wang J et al. (2011) Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol 54:948–955
Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F et al (2009) Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol 27(28):4709–4717
Wislez M, Rabbe N, Marchal J, Milleron B, Crestani B et al (2003) Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res 63(6):1405–1412
Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B et al (1998) Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol 152(1):83–92
Rao HL, Chen JW, Li M, Xiao YB, Fu J et al (2012) Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS One 7(1):e30806. doi:10.1371/journal.pone.0030806
Trellakis S, Bruderek K, Dumitru CA, Gholaman H, Gu X et al (2011) Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int J Cancer 129(9):2183–2193. doi:10.1002/ijc.25892
Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L et al (2002) Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Mod Pathol 15(8):831–837. doi:10.1097/01.MP.0000020391.98998.6B
Huh SJ, Liang S, Sharma A, Dong C, Robertson GP (2010) Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res 70(14):6071–6082
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G et al (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16(3):183–194
Keeley EC, Mehrad B, Strieter RM (2010) CXC chemokines in cancer angiogenesis and metastases. Adv Cancer Res 106:91–111
Ijichi H, Chytil A, Gorska AE, Aakre ME, Bierie B et al (2011) Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest 121(10):4106–4117. doi:10.1172/JCI42754
Keane MP, Belperio JA, Xue YY, Burdick MD, Strieter RM (2004) Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol 172(5):2853–2860
Jamieson T, Clarke M, Steele CW, Samuel MS, Neumann J et al (2012) Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest 122(9):3127–3144. doi:10.1172/JCI61067
Clark RA, Klebanoff SJ (1975) Neutrophil-mediated tumor cell cytotoxicity: role of the peroxidase system. J Exp Med 141(6):1442–1447
Pekarek LA, Starr BA, Toledano AY, Schreiber H (1995) Inhibition of tumor growth by elimination of granulocytes. J Exp Med 181(1):435–440
Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE et al (2010) Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 16(2):219–223
Mittendorf EA, Alatrash G, Qiao N, Wu Y, Sukhumalchandra P et al (2012) Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res 72(13):3153–3162. doi:10.1158/0008-5472.CAN-11-4135
Queen MM, Ryan RE, Holzer RG, Keller-Peck CR, Jorcyk CL (2005) Breast cancer cells stimulate neutrophils to produce oncostatin M: potential implications for tumor progression. Cancer Res 65(19):8896–8904
Grenier A, Chollet-Martin S, Crestani B, Delarche C, El Benna J et al (2002) Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood 99(8):2997–3004
Imai Y, Kubota Y, Yamamoto S, Tsuji K, Shimatani M et al (2005) Neutrophils enhance invasion activity of human cholangiocellular carcinoma and hepatocellular carcinoma cells: an in vitro study. J Gastroenterol Hepatol 20(2):287–293
Granot Z, Henke E, Comen EA, King TA, Norton L et al (2011) Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20(3):300–314. doi:10.1016/j.ccr.2011.08.012
Scapini P, Morini M, Tecchio C, Minghelli S, Di Carlo E et al (2004) CXCL1/macrophage inflammatory protein-2-induced angiogenesis in vivo is mediated by neutrophil-derived vascular endothelial growth factor-A. J Immunol 172(8):5034–5040
Nozawa H, Chiu C, Hanahan D (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A 103(33):12493–12498
Dumitru CA, Fechner MK, Hoffmann TK, Lang S, Brandau S (2012) A novel p38-MAPK signaling axis modulates neutrophil biology in head and neck cancer. J Leukoc Biol 91(4):591–598. doi:10.1189/jlb.0411193
Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S (2010) Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest 120(4):1151–1164
Shojaei F, Singh M, Thompson JD, Ferrara N (2008) Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc Natl Acad Sci U S A 105(7):2640–2645
Shojaei F, Wu X, Zhong C, Yu L, Liang XH et al (2007) Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450(7171):825–831. doi:10.1038/nature06348
Shojaei F, Wu X, Qu X, Kowanetz M, Yu L et al (2009) G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A 106(16):6742–6747. doi:10.1073/pnas.0902280106
Weitzman SA, Weitberg AB, Clark EP, Stossel TP (1985) Phagocytes as carcinogens: malignant transformation produced by human neutrophils. Science (New York, NY) 227(4691):1231–1233
Sandhu JK, Privora HF, Wenckebach G, Birnboim HC (2000) Neutrophils, nitric oxide synthase, and mutations in the mutatect murine tumor model. Am J Pathol 156(2):509–518
Gungor N, Knaapen AM, Munnia A, Peluso M, Haenen GR et al (2010) Genotoxic effects of neutrophils and hypochlorous acid. Mutagenesis 25(2):149–154
Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y et al (2003) Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol 163(6):2221–2232
Campregher C, Luciani MG, Gasche C (2008) Activated neutrophils induce an hMSH2-dependent G2/M checkpoint arrest and replication errors at a (CA)13-repeat in colon epithelial cells. Gut 57(6):780–787
Welch DR, Schissel DJ, Howrey RP, Aeed PA (1989) Tumor-elicited polymorphonuclear cells, in contrast to “normal” circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proc Natl Acad Sci U S A 86(15):5859–5863
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12(4):253–268. doi:10.1038/nri3175
Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J et al (2009) Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res 69(4):1553–1560
Schmielau J, Finn OJ (2001) Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res 61(12):4756–4760
Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A et al (2012) Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A 109(7):2491–2496. doi:10.1073/pnas.1113744109
Brandau S, Trellakis S, Bruderek K, Schmaltz D, Steller G et al (2011) Myeloid-derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J Leukoc Biol 89(2):311–317. doi:10.1189/jlb.0310162
Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L et al (2011) A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 118(8):2254–2265. doi:10.1182/blood-2010-12-325753
Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI (2012) Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 91(1):167–181. doi:10.1189/jlb.0311177
Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G et al (2012) Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One 7(2):e31524. doi:10.1371/journal.pone.0031524
Acknowledgments
The contribution of the European Commission (FP7-HEALTH-F4-2008 “TOLERAGE” 202156, FP7-HEALTH-2011-ADITEC-280873), European Research Council (project HIIS), Fondazione CARIPLO (project Nobel and project 2009-2582), Ministero della Salute (Ricerca finalizzata), the Italian Association for Cancer Research (AIRC; special project 5 × 1000) and Regione Lombardia (project Metadistretti—SEPSIS) is gratefully acknowledged. S.J. is the recipient of a Mario e Valeria Rindi Fellowship from AIRC.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Neutrophils – Guest Editors: Paul Hasler and Sinuhe Hahn
Rights and permissions
About this article
Cite this article
Jaillon, S., Galdiero, M.R., Del Prete, D. et al. Neutrophils in innate and adaptive immunity. Semin Immunopathol 35, 377–394 (2013). https://doi.org/10.1007/s00281-013-0374-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-013-0374-8