
Abstract
Cancer cells, both in vivo and in vitro, have been demonstrated to release membranous structures, defined as microvesicles or exosomes, consisting of an array of macromolecules derived from the originating cells, including proteins, lipids, and nucleic acids. While only recently have the roles of these vesicular components in intercellular communication become elucidated, significant evidence has demonstrated that tumor exosomes can exert a broad array of detrimental effects on the immune system—ranging from apoptosis of activated cytotoxic T cells to impairment of monocyte differentiation into dendritic cells, to induction of myeloid-suppressive cells and T regulatory cells. Immunosuppressive exosomes of tumor origin can be found within neoplastic lesions and in biologic fluids from cancer patients, implying a potential role of these pathways in in vivo tumor progression and systemic paraneoplastic syndromes. Through the expression of molecules involved in angiogenesis promotion, stromal remodeling, signaling pathway activation through growth factor/receptor transfer, chemoresistance, and genetic intercellular exchange, tumor exosomes could represent a central mediator of the tumor microenvironment. By understanding the nature of these tumor-derived exosomes/microvesicles and their roles in mediating cancer progression and modulating the host immune response will significantly impact therapeutic approaches targeting exosomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The release of nano-sized membranous vesicles by viable tumors was initially described by our group over three decades ago [1] and has since been verified in multiple tumor and cell types. These membranous vesicles have been identified by various terms, from “high molecular weight complexes,” “membrane fragments,” “microvesicles,” “microparticles,” and “exosomes.” While restrictive definitions have been applied to these vesicular structures [2], considerable overlap exists between various circulating cell-derived vesicles isolated from cancer patients, suggesting the distinctions may not be clear-cut, and these different terms may include the same components.
The term “exosome” was coined by Trams et al. in 1981 [3] for “exfoliated membrane vesicles with 5′-nucleotidase activity.” The “exosome” term originated from a discovery of the secretion of neoplastic cell line-derived exfoliated vesicles, which mirrored the 5′-nucleotidase activity of the parent cells [3]. Subsequently, the canonical pathway of “vesicle” release following multivesicular endosome fusion with the cell surface was demonstrated in cultured sheep [4] and rat [5] reticulocytes. After purification by ultracentrifugation, the sedimented microvesicles were found to contain transferrin receptors, which were also found in native reticulocytes [6]. These microvesicles were redefined as “exosomes” [7].
The release of tumor-derived microvesicles was initially demonstrated in ovarian cancer patients [1, 8, 9]. Within this patient population, intact membrane fragments or vesicles from the peripheral circulation and malignant effusions (ascites and cyst fluids) were found to express molecular markers that were inherent to the tumor plasma membrane, 5′-nucleotidase, and placental-type alkaline phosphatase [10, 11]. In addition to tumor cells and embryonic cells, microvesicles/exosomes are released by a variety of cells, particularly activated cells of the immune system, including dendritic cells, macrophages, B cells, T cells, and NK cells [12–14]. We now recognize that these shed microvesicles are key intercellular communication vehicles, serving to regulate normal immune responses. Exosomes from activated dendritic cells can present antigens in the context of MHC II to T cells [15]. Exosomes from activated T cells can mediate “activation-induced cell death” in a cell-autonomous manner, defined by the nature of the initial T cell activation events and can play central roles in both central and peripheral deletion events involved in tolerance and homeostasis [16]. Exosomes released by tumors may elicit a tolerogenic response and participate in other immune mechanisms, such as platelet activation, mast cell degranulation, germinal center reaction, and potential engulfment of apoptotic cells. Since, under normal condition, microvesicles/exosomes are enriched in MHC class I and II antigens and costimulatory molecules, they are thought to be an alternative antigen delivery pathway mediated by cell surface molecules. The aberrant release of exosomes by tumors may allow them to circumvent these immunoregulatory antigen delivery pathways and evade immunosurveillance [10].
Microvesicle/exosome formation
There are multiple mechanisms leading to the release of cellular components into the extracellular space. Three mechanisms have been proposed for the release of membranous vesicles. These are exocytic fusion of multivesicular bodies (MVBs) resulting in exosomes, budding of vesicles directly from the plasma membranes resulting in shed “microvesicles,” and cell death leading to apoptotic bodies [17]. The first two mechanisms are properties of viable cells and are energy-requiring events. While most isolation protocols readily exclude apoptotic bodies, they do not differentiate exosomes from shed “microvesicles.” Thus, these extracellular vesicle populations may include a mixture of exosomes and microvesicles, which may confuse interpretation of biochemical data. In terms of their characteristics, exosomes/microvesicles derived from the extracellular environment of tumors, either in vitro or in vivo, exhibit overlapping similarities in size (defined by dynamic light scattering), morphology (defined by electron microscopy), density (defined by of sucrose gradient centrifugation), and protein markers of both the endosomes and plasma membranes (defined by western immunoblotting and mass spectrometry) [18, 19]. We have compared the vesicle populations obtained from biologic fluids of ovarian cancer patients by both the technique described to isolate exosomes and our original chromatographic method isolating “microvesicles” [18]. This comparative study demonstrated that these in vivo-derived vesicles from both techniques isolated cup-shaped vesicles, with a density between 1.13 and 1.17 g/ml, a diameter between 50 and 100 nm, and expressing CD63, Alix, VPS35, galectin 3, HSP90, fibronectin, and placental alkaline phosphatase (Fig. 1). Thus, these patient-derived circulating vesicles fall within the definition of exosomes. However, it is unclear whether this distinction between exosomes and shed microvesicles is critical to understand the biologic activities of these vesicles as they can interact with target cells of the host as a mixture. This review focuses on the vesicle populations actively released by viable cells.

Schematic diagram of a tumor-derived exosome, presenting protein components defined by ion trap mass spectrometry
The increased release of exosomes/microvesicles and their accumulation appear to be important in the malignant transformation process. Although extracellular shedding of exosomes occurs in other types of cells under specific physiological conditions, the accumulation of membranous vesicles from non-neoplastic cells is rarely observed, in vivo [20]. In contrast, exosomes released by tumor cells accumulate in biologic fluids, including sera, urine, ascites, and pleural fluids. Exosome release and its accumulation appear to be important features of the malignant transformation. Shed tumor-derived exosomes do not mirror the general composition of the plasma membrane of the originating tumor cells, but represent ‘micromaps,’ with enhanced expression of tumor antigens [21, 22].
While the exact mechanisms of exosome/microvesicle release remain unclear, this release is an energy-requiring phenomenon, modulated by extracellular signals. The most common process is the release of large biomolecules through the plasma membrane by a process termed exocytosis, which has regulatory and signaling functions. Exocytosis can be either constitutive (non-calcium-triggered) or regulated (calcium-triggered) [23]. Constitutive exocytosis occurs in all cells and serves to secrete extracellular matrix components or to incorporate newly synthesized proteins into the plasma membrane following fusion with transport vesicles. Regulated exocytosis is critical to events, such as neurologic signaling, as synaptic vesicles fuse with the membrane at the synaptic cleft. The formation of these endosomes is initiated cell surface-mediated invagination to generate endocytic vesicles that migrate and fuse with the early endosome [24]. Exosomes appear to form by invagination and budding from the limiting membrane of late endosomes, resulting in vesicles that contain cytosol and that expose the extracellular domain of transferring receptors at their surface. Using electron microscopy, studies have shown fusion profiles of multivesicular endosomes with the plasma membrane, leading to the secretion of the internal vesicles into the extracellular environment [24].
Since the formation of these membrane vesicles has an endocytic origin, this mechanism is a secretion process of the endosomal system, including endocytic vesicles, early endosomes, late endosomes, and lysosomes. These endocytic vesicles form through clathrin- or non-clathrin-mediated endocytosis at the plasma membrane and are transported to early endosomes [25]. The late endosomes develop from early endosomes via acidification, changes in their protein content, and their ability to fuse with vesicles or other cellular membranes. Early endosomes are localized near the outer margin of the cells and exhibit a tubular appearance, in contrast, late endosomes are localized proximal to the nucleus and are spherical in shape. A critical step in the formation of MVBs from late endosomes is reversed budding. In this step, limiting membranes of late endosomes “bud” into their lumen, resulting in a continuous enrichment of internal luminal vesicles [26] (http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T75-4M4KJ04-1&_user=134779&_coverDate=11%2F15%2F2006&_rdoc=1&_fmt=high&_orig=search&_origin=search&_sort=d&_docanchor=&view=c&_acct=C000011238&_version=1&_urlVersion=0&_userid=134779&md5=347321f447ba003d55a8380cc328666d&searchtype=a-bib6). MVBs have been demonstrated to be involved in the exocytic fusion of their external membrane with the plasma membrane of the cell, resulting in release of their segregated vesicles to the extracellular space.
The molecular composition of the cell surface can be modulated by endosomes, facilitating segregation of proteins destined for degradation from proteins destined to be recycled [27]. As a consequence of multiprotein complex, the Endosomal Sorting Complexes Required for Transport (ESCRTs), specific proteins and lipids of the MVB external membrane are enriched in the exosomes, while other components are excluded. The formation of MVBs relies on ubiquitin-binding proteins [28]. The protein-sorting mechanisms include ubiquitination of the target protein and preferential sorting. The ligation of one ubiquitin molecule (mono-ubiquitinylation) can serve as a signal for endocytosis and segregation into MVBs; however, the attachment of multiple ubiquitin molecules (poly-ubiquitinylation) can earmark proteins for degradation in the proteasome [29]. Some studies have also suggested that oligoubiquitination may also be a sorting signal for trafficking to MVBs, which may increase MVB sorting efficiency. ESCRT-I, -II, and -III recognize monoubiquitinated cargoes and promote their inclusion in MVBs [30]. The ESCRT complex recognizes the ubiquitinylated proteins via vacuolar protein sorting (VPS)-27 [31]. Subsequently, VPS27 recruits an additional ESCRT complex and TSG101, activating AIP/Alix. This complex initiates sorting specific proteins into the budding vesicles. Although mono-ubiquitinylation prompts their uptake into MVBs, not all proteins in exosomes are ubiquitinylated, potentially resulting from a passive mechanism involved in protein sorting to MVBs. It has been proposed that the signals responsible for the “passive” processes are, in some cases, the presence of tetraspanin-enriched [32] or cholesterol-enriched (i.e., lipid rafts) membrane microdomains [33]. A similar but unclear mechanism is potentially associated with the accumulation of specific cytosolic proteins in the exosome lumen.
Exosome/microvesicle characterization/composition
Exosomes have been described as microvesicles containing 5′-nucleotidase activity that are released from neoplastic cells [3]. These small nanovesicles, present within MVBs (endosomes), contain transferrin receptors, a marker used to identify endocytosis and recycling of internalized plasma membrane proteins [34]. Biophysically, exosomes are equivalent to cytoplasm enclosed in a lipid bilayer with the external domains of transmembrane proteins exposed to the extracellular environment. Exosome composition varies depending on the cell type of origin.
The lipid and protein content of exosomes has been extensively analyzed by various techniques including western immunoblotting, fluorescence-activated cell sorting, immuno-electron microscopy, and mass spectrometry.
Ultrastructural studies, western blot, and mass spectrometry analysis of exosomes from different sources have allowed the distinction between commonly expressed proteins, as well as cell type-specific proteins [22, 35]. Exosomes contain a number of common protein components. Based on their endosomal origin, exosomes, independent of the parental cell of origin, exhibit multiple proteins involved in MVB formation (annexins, Rab family GTPases, and ESCRT complex proteins (TSG101, Alix)) [36]. Additional protein markers linked with exosomes include tetraspanins (CD81, CD63, and CD9) and heat shock proteins (HSP60, HSP70, and HSP90) [22]. Exosome-associated HSP70 and HSP90 can facilitate peptide loading onto MHC class I and class II proteins. Exosome express cell type-specific markers, such as class I and II MHC, co-stimulatory proteins (CD80 and CD86) on antigen presenting cell-derived exosomes [37], integrin CD41a and Von Willebrand factor on platelet-derived exosomes [38], and perforin and granzyme on cytotoxic T lymphocyte-derived exosomes [39]. The cytosolic proteins present on exosomes include Rabs, which promote exosome docking and the membrane fusion events [40]. The annexins, including annexin I, II, V, and VI, may regulate membrane cytoskeleton dynamics and membrane fusion events [41]. Adhesion molecules, including intercellular adhesion molecule-1, CD146, CD9, EGFRvIII, CD18, CD11a, CD11b, CD11c, CD166, and LFA-3/CD58, have also been identified in exosomal preparations [42, 43]. One of the characteristic features of exosomes is the presence of tetraspanins, including CD9, CD63, CD81, and CD82 [44]. Exosomes are also enriched in proteins that participate in vesicle formation and trafficking, such as the lysobisphosphatidic acid (LBPA)-binding protein, Alix [45]. Other proteins demonstrated to be present exosomes include metabolic enzymes, such as peroxidases, pyruvate and lipid kinases, and enolase-1 [46].
Recently, we have analyzed the patient-derived exosomal proteome using ion trap mass spectrometry (Table 1). This study identified 232 unique proteins. These proteins were classified as percent of the identified total proteins into molecular chaperones (8.5%), vesicle fusion (8.5%), cytoskeletal proteins and proteins involved in the assembly/disassembly of the cytoskeletal networks (17.6%), anionic and cationic ion transport channels (3.7%), proteins involved in lipid (6.9%), carbohydrate (3.2%), and amino acids (2.1%) metabolisms, proteins involved in DNA replication (6.9%), messenger RNA (mRNA) splicing (5.3%), transcription/translation (5.3%), post-transcriptional protein modification (13.8%), and signal transduction (2.7%). Our studies demonstrated that cytosolic proteins were highly represented, and we observed a diverse array of cytoskeletal constituents (actin, α-actinin-1, cofilin, filamin-A-B-C, tubulins, gelsolin, profilin-1, spectrin, symplekin, talin, vinculin, and myosins). We identified that transmembrane proteins were also abundant, including multiple integrins (β1, α3, and αv), intercellular adhesion molecule 1 (ICAM-1), and mucin-4. A variety of channels were observed, such as the voltage-dependent anion-selective channel protein 2 and 3, chloride intracellular channel protein 1, sodium/potassium-transporting ATPase subunit β-3, long of sodium/potassium-transporting ATPase subunit α-1, and transitional endoplasmic reticulum ATPase. In line with their endocytic origin, exosomal proteins belonging to the ESCRT complex that are important protein complexes involved in ubiquitin-dependent exosome biogenesis have also been observed. These ESCRT-associated proteins include vacuolar protein sorting-associated protein 35 (VPS-35), Alix, ubiquitin-like modifier-activating enzyme, and ubiquitin carboxyl-terminal hydrolases. We demonstrated that proteins involved in membrane trafficking and fusion processes were enriched (annexin A2, A5, A6, clathrin heavy chain 1/2, coatomer subunit β, Rab1b, Rab2a, and Rab7a). A group of markers of endosomes and lysosomes were also detected (cathepsin-C, -D, EH domain-containing protein 1, and β-hexoaminidase), and several chaperonnes were identified (HSP70, HSP90, HSC70, HSPA4, -8, -9, HSPA1A/B, HSPB1, HSP47, HSPA5, HSPβ1, HSPD1, HSP90AB1, B1, AA1; T-complex protein 1, endoplasmin, and protein disulfide-isomerase A3, A4, and A6).
The initial identification of exosome release by tumor cells was envisioned as the discovery of a new cell-free source of tumor antigens for in vivo immune priming or tumor vaccine design [47]. Exosomes are close replicas of the originating cells in terms of selected protein content and express a large array of tumor antigens when secreted by neoplastic cells, including highly immunogenic antigens MelanA/Mart-1 and gp100; colon carcinoma cells express CEA and HER2. This antigenic content is not only a feature of in vitro-released exosomes but also can be found in microvesicles isolated from plasma of cancer patients as well, evidence that demonstrates the tumor origin of these organelles [48].
Exosomes as vehicles for intercellular communication
Tumor-secreted exosomes have recently gained increased attention as a “vehicle” for intercellular communication with extensive autocrine/paracrine functions. One of the most important functions of cell-derived microvesicles/exosomes appears to be intercellular communication. By exposing cell type-specific adhesion receptors or ligands, exosomes can interact with specific cells and deliver their “signals,” including bioactive lipids, cytokines, growth factors, receptors, and genetic materials. Thus, the microvesicle/exosomal pathway may constitute a mechanism for local and systemic intercellular transfer of information, with a complexity superior to that of secreted soluble factors, but similar to that observed with direct cell–cell contact.
Exosomes provide stable conformational conditions for their protein content (due to maintenance of their three-dimensional transmembrane structure), conserve bioactivity of their proteins (based on the protective membrane structure), improve bio-distribution (based on their capacity to circulate in biologic fluids and migrate to secondary sites), and support an efficient interaction with target cells (due to the fusogenic properties of exosomes) [49, 50]. Due to these features, tumor-derived exosomes are efficient platforms for the in vivo transfer of cross-talk signals. The multiplicity of bioactive molecules associated with exosomes suggests that they exhibit a central role in generating the tumor microenvironment [51, 52]. Exosomes have the ability to transfer specific proteins to homologous and heterologous target cells for the delivery of signaling pathways [53, 54]. The presence of tumor-derived exosomes can increase matrix metalloproteinase (MMP) secretion and VEGF expression in target cells through the expression of proangiogenic molecules, such as members of the tetraspanin family, thereby promoting neoangiogenesis even at secondary metastatic sites [55]. The released MMPs can digest the extracellular matrices where they arise. This degradation is enhanced when MMPs are co-released with exosome-associated extracellular matrix metalloproteinase inducer (EMMPRIN) [56].
Studies have shown that cancer ascites-derived exosomes carry extracellular matrix-remodeling enzymes, such as metalloproteinases 2 and 9 (MMP-2 and MMP-9) [57, 58], and urokinase plasminogen activator [59], leading to an increase in extracellular matrix degradation. The expression of matrix-remodeling enzymes increases the tumor's invasive phenotype and promotes metastasis. The presence of proangiogenic factors supports neovascularization of the developing tumor. A common cellular component of the tumor microenvironment is the monocyte/macrophage. Within the microenvironment, these tumor-associated macrophages have been shown to assist in tumor progression by angiogenesis, growth, metastasis, and immunosuppression [60].
When shed vesicles fuse with their target cells, they can transfer important membrane components, such as receptors and ligands. The transferring of receptors between exosomes and target cells was demonstrated by the observation that bystander B cells acquire antigen receptors from activated B cells by membrane transfer [61]. This transfer allows the amplified expansion of antigen-binding B cells with the ability to present a specific antigen to CD4 T cells. Exosomes can transfer the adhesion molecule CD41 from platelets to endothelial cells or to tumor cells, conferring pro-adhesive properties to the target cell [62]. Exosome-mediated transfer of Fas ligand from tumor cells induces apoptosis of activated T cells favoring tumor immune escape [63]. Exosomes can also be protective for cells that remove from their membranes to the extracellular compartment the potentially harmful molecules, such as Fas or the membrane attack complex.
Exosomes have also been postulated to contribute to the spread of infective agents, such as human immunodeficiency virus (HIV) type 1 [64]. In macrophages receiving chemokine receptors, this can induce an increased risk of HIV infection together with resistance to apoptosis. The transfer of the chemokine (CXC motif) receptor 4 and the chemokine (CC motif) receptor 5, chemokine co-receptors for HIV type I by released exosomes, can enhance the entry of the virus into cell types other than the lympho-hemopoietic lineage [65]. In addition to transferring receptors, exosomes can transfer viruses, contained within exosomes, by the “Trojan exosome hypothesis” involving direct delivery [66].
In human gliomas, only a fraction of the cells, exhibiting a transformed phenotype, expressed the truncated epidermal growth factor receptor, EGFRvIII, associated with dysregulated tumor growth [67]. Al-Nedawi et al. [68] demonstrated transfer of the oncogenic EGFRvIII from human glioma cancer cells expressing the receptor to glioma cells without the EGFRvIII via the fusion of exosomes. After transfer, the glioma cells, lacking the receptor, were transformed to express EGFRvIII-regulated genes, including VEGF, Bcl-xL, and p27 [69]. Subsequent studies demonstrated that the oncogenic EGFRvIII from human squamous cell carcinoma cells was transferred via exosomes to tumor-associated endothelial cells to activate MAPK and Akt cell signaling pathways and promote endothelial VEGF expression [69].
The occurrence of epigenetic changes has been frequently reported in cancer. Epigenetic regulation of gene transcription, mediating cell proliferation, differentiation, and survival are additional targets in tumor progression, resulting in genomic instability [70]. One explanation for this genomic instability lies in the mediation by microvesicular horizontal transfer [71]. Horizontal transfer via microvesicles has been validated in a number of tumor-associated cells including gliomas, monocytes, mast cells, and T cells [72]. One explanation of this phenomenon is the transfer of genetic information between cells. It has been shown that tumor-derived exosomes may transfer not only surface determinants but also mRNA of tumor cells to monocytes. Janowska-Wieczorek et al. [73] demonstrated that exosomes derived from murine embryonic stem cells (ESCs) could induce epigenetic reprogramming of target cells. ESC-derived exosomes were shown to improve survival of hematopoietic stem/progenitor cells, to induce upregulation of early pluripotent and early hematopoietic markers, and to induce phosphorylation of mitogen-activated protein kinase p42/44 and Akt. ESC-derived exosomes were shown to express mRNAs for several pluripotent transcription factors that can be delivered to target cells and translated to their corresponding proteins [74]. As RNase treatment inhibited their exosome-mediated biological effect, the involvement of mRNA in the observed biological effects was suggested. Yuan et al. [75] have shown that in addition to mRNA, exosomes can transfer microRNA to target cells. They demonstrated that exosomes derived from ESCs contain abundant microRNA and that they can transfer a subset of microRNAs to mouse embryonic fibroblasts in vitro. Since microRNAs are regulators of protein translation, this observation opens the possibility that stem cells can alter the expression of genes in neighboring cells by transferring exosomal microRNAs. When shed vesicles fuse with their target cells, the portion of cytosol segregated within their lumen is discharged to and integrates with the cytosol of the target cell. Because this transfer can also include transmission of specific mRNAs, it can ultimately contribute to the epigenetic and proteomic properties of target cells.
It has been suggested that tumor cell progression could use multiple forms of exosome/microvesicle-mediated communication to simultaneously affect multiple effector populations, based on release of tissue factors, immunoregulators, and oncogenic molecules. Thus, the signals transferred to neighboring cells via exosomes may mirror the transcriptional status of the parent cell, but due to the exosomal mRNA and microRNA being transferred, their consequences on the translational machinery of the target cells are extensive.
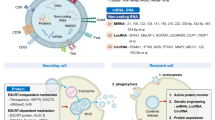
Exosomes/microvesicles as mediators immune regulation
Cancer cells are postulated to modulate components of the microenvironment and affect immune system function, primarily through pathways involving cell-to-cell contact and the release of suppressive soluble factors. However, a unique alternative mechanism has emerged that involves the active release of immunosuppressive microvesicles/exosomes by tumor cells [76, 77]. As tumor-derived microvesicles/exosomes are abundant in the blood and malignant effusions derived from cancer patients [78, 79], their release appears to be important features of intercellular communication. Evidence supports the concept that tumors constitutively shed exosomes with pleiotropic immunosuppressive effects [80, 81] that are protective and supportive of the tumor to facilitate escape from lymphocyte immunosurveillance [82]. Since released exosomes express molecules with biologic activity (such as Fas ligand, PD-1, MICA/B, mdr1, MMPs, CD44, and autoreactive antigens), the ability of exosomes to modulate lymphocyte and monocyte functions has been analyzed [83–85]. The immunological significance of exosomes, while far from clear, has been linked with their potential to modulate the host's immune system, which may be their major function. Supporting this view is that the topology of the macromolecules displayed on exosomes is analogous to that observed on the tumor's plasma membrane, making them well positioned for interactions with target cell surface receptors. This positioning can mediate signal transduction without the need for direct cell–cell contact. Further, these microvesicles can also fuse with the recipient cell, leading to the acquisition of novel molecules by the cells and the delivery of mRNA and miRNA through this route.
Tumor exosome release has been described as capable of modulating the evasion from anti-tumor immune responses [86]. It has been suggested that the anti-tumor immune response can be divided into three different phases, and tumor-derived microvesicles/exosomes can exhibit roles in each phase [87]. The first step includes the recognition of tumor cells by innate immune cells. The progression and development of tumors is coordinated by biochemical and biophysical signals from the tumor microenvironment [88]. After growing to a certain threshold size, solid tumors exceed their capacity to acquire oxygen and nutrients in this hypoxic environment [89, 90]. For tumor progression to occur, the “angiogenic switch” has been reported to be required [91]. The “angiogenic switch” promotes the transition of the tumor to a highly vascularized and progressive outgrowth [51]. This process further induces extracellular matrix remodeling and the production of a pro-inflammatory environment leading to the recruitment of innate immune cells, including NK, macrophages, and dendritic cells and the mediation of T cells into the tumor microenvironment [57–59].
Recent studies have demonstrated that the MHC class I-related chain (MIC) A and MICB ligands for the NK cell activating receptor NKG2D is released by tumor cells as a component of exosomes [92]. This shedding of MICA/B by tumors not only prevents recognition of MICA/B-expressing tumor cells but also results in the downregulation of NKG2D expression on circulating CD8 T cells, NK cells, and γδT cells, leading to impairment of the antitumor immune response [93]. Ashiru et al. [92] demonstrated that treatment of NK cells with MICA-expressing exosomes suppressed NK expression of NKG2D on the cell surface and also suppressed NK cytotoxicity, which is independent of NKG2D ligand expression. Thus, their findings demonstrated exosomal MICA/B expression as a mechanism of NK suppression, facilitating immune escape and progression.
The second phase of an anti-tumor immune response involves the maturation and migration of tissue dendritic cells and priming of naïve T cells. Blood-derived exosomes from melanoma patients have been shown to promote the generation of myeloid-derived suppressor cells (MDSCs) from peripheral blood monocytes [94], which acts as one of the major mechanisms used by tumors to escape immune recognition [95]. MDSCs have potent immunosuppressive functions that can suppress T cell immune responses by a variety of mechanisms [96–99]. Further, tumor exosomes have been shown to be involved in the regulation of the adaptive immune responses to cancer cells in animal models and cancer patients [12, 100] by impairing peripheral blood monocyte differentiation into dendritic cells [101]. As a result, the generation of tumor-specific T cells has been reported to be a very inefficient process. Most importantly, tumor exosomes have the ability to induce a series of functional defects in tumor reactive-effector T cells [102], through the expression of apoptosis-inducing ligands, such as FasL and TRAIL [103] or PDL-2, which directly stimulate their T cell targets to negatively regulate T cell activation [104]. For example, ovarian cancer patient-derived exosomes inhibit T cell functions by increasing expression of FasL (on the exosome surface) and suppressing CD3-zeta (on the T cells) to collectively induce T cell apoptosis [105]. An investigation of human prostate cancer exosomes added to activated T cells exposed a dose-dependent inhibition of CD8+ T cell proliferation stimulated by Fas–FasL interaction [106]. Additionally, tumor-derived exosomes block innate immune effector cell function as seen in NK cells via production of exosome-associated MICA/B to downregulate NKG2D expression, thereby decreasing NKG2D-mediated killing [107, 108]. Production of tumor exosome-associated MICA/B and FasL has been shown to decrease the effectiveness of not only the innate immune system but also the adaptive immune system to reject the tumor [109, 110]. It has been theorized that these released exosomes modulate lymphocyte functions by mimicking “activation induced cell death” (AICD) [111, 112]. Lymphoid cells appear to release exosomes following activation, and these appear to play an essential role in immunoregulation, by preventing excessive immune responses and the development of autoimmunity [113]. It was postulated that exosome release by tumor cells is a re-expression of the fetal cell exosomes and that both constituted pathways to circumvent immunosurveillance.
The third and final phase involved in the anti-tumor response is the generation and homing of tumor-specific T cells. Tumor-reactive CD4+ and CD8+ T cells homing to the primary tumor site is an important step in eradicating the tumor [114, 115]. Tumor exosomes express membrane bound ICAM-1 that efficiently blocks the interaction between lymphocytes and endothelial cells [42] and therefore decreases the recruitment of adaptive immune cells. Several studies have shown that tumor-infiltrating T cells are impaired by the tumor and display altered expression in intracellular signal transduction molecules such as CD3-zeta [108, 116]. Tumor exosomes co-incubation with T cells leads to a decrease in CD3-zeta, which suggests that tumor exosomes may be an additional mechanism used by tumors to evade immune recognition [82, 108]. The alteration of TCR-CD3-zeta has been observed in several types of tumors namely malignant melanoma [117], ovarian [118], and pancreatic [119]. Furthermore, studies have reported that cancer patients display a high frequency of suppressive peripheral blood regulatory T cells when compared to normal controls [120–122]. These cells have been shown to infiltrate the tumor and are involved in the induction of CD8+ tumor-reactive cytotoxic T lymphocyte (CTL) apoptosis [42, 123, 124]. A study from Szajnik et al. shows that tumor-derived microvesicles expand and promote the suppressive activities of human regulatory T cells (Treg) by upregulating the expression of FasL, interleukin (IL)-10, TGF-β1, CTLA-4, granzyme B, and perforin [125]. Therefore, the immunoregulatory properties attributed to tumor-derived exosomes might be essential in regulating peripheral tolerance and promoting immune evasion of tumors [53]. Collectively, these studies support a role for exosomes in adapting the host microenvironment to allow escape from immune surveillance via stimulation of angiogenesis and metastasis of tumors [126, 127], which suggests that tumors may use exosomes to keep the host immune system under control without a direct interaction with host immune cells.
Critical components of the immune response, such as antigen presenting cells, are significantly affected by interactions with tumor exosomes. These microvesicles not only impair the capacity of circulating monocytes to differentiate into functional DCs but they also skew the differentiation of these cells towards altered CD14+ monocytes expressing low or absent levels of HLA-DR [128]. These cells, which are present in relatively high numbers in PBMCs of melanoma patients, exert suppressive activity on lymphocyte proliferation and impair the expression of effector molecules (such as perforin and IFN-γ) in a TGF-β-mediated fashion. CD14+HLA-DR−/low cells behave as MSC, undergoing in vivo expansion upon administration of GM-CSF. These hallmark alterations induced by tumor microvesicles on target immune cells in vitro can also be detected on immune cells isolated from cancer patients, which supports the hypothesis that these suppressive pathways are present in vivo.
These exosomes activated a stronger pro-inflammatory response in the form of NF-κB activation and TNF-α release from untreated macrophages as compared to macrophages exposed to control exosomes. Parallel evaluations of the structural components of tumor-derived microvesicles demonstrated enhanced expression of tumor antigens on the vesicular surface [129, 130]. In murine B16 melanoma cultures, the expression of surface glycoproteins on the isolated microvesicles represented a profile similar to that found in the melanoma cell membrane [130]. Continued characterization of tumor-derived microvesicles was achieved through biochemical analyses, which identified molecules with immunologic and biologic activity. Microvesicles released by late stage tumor cells were found to exert a dose-dependent suppression of MHC II in monocytes/macrophages in comparison to early stage tumor cells [131, 132]. In addition, microvesicles suppressed lymphocyte activation induced by phytohemoglutinin, anti-CD3, concanavalin A [10, 11, 130], and IL-2 [133].
As evidence of the pleiotropic effect of tumor-secreted exosomes, tumor exosomes can interfere directly with T cell effector functions. Exosomal expression of bioactive FasL and TRAIL has been shown on exosomes derived from human tumors to induce apoptosis in activated tumor-specific T cells. This phenomenon highly resembles the one utilized under physiological conditions not only by T cells to downsize immune responses [134] but also by placenta cells that recently have been shown to promote a state of immune privilege by inducing FasL-mediated apoptosis and defects in the expression of crucial TCR signaling components (such as CD3-zeta and JAK3) in local T cells, which have been reported with microvesicles isolated from plasma of cancer patients and may help to explain the high frequency of apoptotic or CD3-zeta− lymphocytes that are often found in the peripheral circulation of these patients [135, 136]. Natural killer cells lose their cytolytic potential, through the suppression perforin expression, upon encounter with tumor-secreted microvesicles.
A pro-inflammatory microenvironment is associated with the development and progression of cancer. Macrophages are prominent in the development of the pro-inflammatory environment, with IL-1β serving as a “master” cytokine regulator. Macrophages are critical for the resolution of inflammation by producing anti-inflammatory cytokines and chemokines and by increasing phagocytic activity. Based on Th1/Th2 polarization, phenotypically polarized macrophages are termed pro-inflammatory M1 (classically activated) and anti-inflammatory M2 (alternatively activated) [137]. In vitro, macrophages can be polarized to the M1 state by treatment with IFN-γ and inducers of TNF-α, such as lipopolysaccharide (LPS) [138]. These M1 macrophages induce synthesis of pro-inflammatory cytokines and chemokines, including TNF-α, IL-12, IL-6, CCL2, and IL-1β, as well as increased production of reactive oxygen species [139, 140]. Elevated levels of IL-1β are present in M1 polarized macrophages due to activation of the NF-κB and MAPK pathways [141], while no IL-1β protein is found in M2 polarized macrophages [142]. Using cytochalasin D, a known inhibitor of actin polymerization, while a dramatic suppression of exosome internalization was observed, this internalization was not essential for the induction of IL-1β mRNA and protein, following the exposure to exosomes.
IL-1β exhibits profound effects on immune cell function during inflammation. Components of the extracellular matrices have been demonstrated to be capable of stimulating the expression of IL-1β [143]. Fibronectin is highly expressed in injured tissues [144] and appears to be positioned to modulate the expression of IL-1β in diseased tissues [143]. In vitro, fibronectin stimulates the expression of IL-1β mRNA and its translation into the 31-kDa intracellular precursor protein, along with secretion of the 17-kDa active form in human mononuclear cells [145]. This effect of fibronectin is mediated by specific cell surface α5β1 integrin receptor, which activates poorly understood intracellular signals to induce IL-1β expression [146]. Fibronectin contains a sequence, termed Arg-Gly-Asp (RGD), which promotes its attachment to integrin receptors [147]. Monocytes and macrophages have been shown to possess fibronectin receptors that recognize the RGD motif and mediate pro-inflammatory cytokine production. The effect of fibronectin has been shown to be dependent on binding of the RGD sequence of fibronectin to integrin receptors, as this effect could be inhibited by integrin receptor blocking peptides (anti-RGD sequence mimics) [148]. The use of RGD antagonists demonstrates that exosome-induced IL-1β production by macrophages is mediated by exosome-associated fibronectin (Fig. 2).

Schematic presenting exosome-associated fibronectin induction and release of IL1-β by macrophages
Shedding vesicles released by various cell types are now known to participate as well [149, 150]. Their role can vary depending on the stage of the process. At an early stage, the vesicles shed by neutrophils stimulate the release of anti-inflammatory factors such as TGFβ1 and IL-10 from macrophages with reduction of TNFα and IL-8 [151, 152]. At later time points, however, shed vesicles can become pro-inflammatory, mediating the transfer of chemokine receptors, such as CCR4 and CCR5, and stimulating release of other mediators, such as IL-6 and the monocyte chemotactic protein 1 (MCP1), which induce and strengthen inflammatory responses [153].
Conclusions
It has become increasingly clear, as new exosome studies are published, that these small bioactive vesicles are important in a number of biological functions. Exosomes exhibit important roles in intercellular communication, and under normal conditions, this communication mediates the activation of the immune response. However, in cancer, tumor exosomes can induce apoptosis of activated cytotoxic T cells, impairment of monocyte differentiation, induction of myeloid-suppressive cells and T regulatory cells, and suppression of lymphoid activation signaling molecules. Tumor-derived exosomes express molecules involved in angiogenesis promotion, stromal remodeling, signaling pathway activation through growth factor/receptor transfer, chemoresistance, and genetic intercellular exchange. Tumor exosomes induce a pro-inflammatory environment from macrophages due to expression of exosomal fibronectin. As a result of these exosomal effects, they can represent a central mediator of the tumor-supportive microenvironment. From the removal of unwanted proteins from maturing reticulocytes to their role in immune surveillance, the inventory of functions continues to grow.
References
Taylor DD, Doellgast GJ (1979) Quantitation of peroxidase-antibody binding to membrane fragments using column chromatography. Anal Biochem 98:53–59
Théry C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nature Rev Immunol 9:581–593
Trams EG, Lauter CJ, Salem C Jr, Heine U (1981) Exfoliation of membrane ecto-enzymes in the form of microvesicles. Biochim Biophys Acta 645:63–70
Pan BT, Blostein R, Johnstone RM (1983) Loss of the transferrin receptor during the maturation of sheep reticulocytes in vitro: an immunological approach. Biochem J 210:37–47
Harding C, Heuser J, Stahl P (1983) Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol 97:329–339
Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C (1987) Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem 262:9412–9420
van Niel G, Porto-Carreiro I, Simoes S, Raposo G (2006) Exosomes: a common pathway for a specialized function. J Biochem 140:13–21
Taylor DD, Homesley HD, Doellgast GJ (1980) Binding of specific peroxidase-labeled antibody to placental-type phosphatase on tumor-derived membrane fragments. Cancer Res 40:4064–4069
Taylor DD, Homesley HD, Doellgast GJ (1983) Membrane-associated immunoglobulins in cyst and ascites fluids of ovarian cancer patients. Am J Reprod Immunol 3:7–11
Taylor DD, Levy EM, Black PH (1985) Shed membrane vesicles: a mechanism for tumor induced immunosuppression. In: Mitchell MS, Reif AE (eds) Immunity to cancer. Academic Press, Orlando, pp 369–373
Taylor DD, Black PH (1986) Shedding of plasma membrane fragments: neoplastic and developmental importance. In: Steinberg M (ed) Developmental Biology. Plenum Press, New York, vol 3., pp 33–57
Xie Y, Zhang H, Li W, Deng Y, Munegowda MA, Chibbar R, Qureshi M, Xiang J (2010) Dendritic cells recruit T cell exosomes via exosomal LFA-1 leading to inhibition of CD8+ CTL responses through downregulation of peptide/MHC class I and Fas ligand-mediated cytotoxicity. J Immunol 185:5268–5278
Zumaquero E, Muñoz P, Cobo M, Lucena G, Pavón EJ, Martín A, Navarro P, García-Pérez A, Ariza-Veguillas A, Malavasi F, Sancho J, Zubiaur M (2010) Exosomes from human lymphoblastoid B cells express enzymatically active CD38 that is associated with signaling complexes containing CD81, Hsc-70, and Lyn. Exp Cell Res 316:2692–2706
Anand PK (2010) Exosomal membrane molecules are potent immune response modulators. Commun Integr Biol 3:405–408
Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang Z, Divito SJ, Papworth GD, Watkins SC, Robbins PD, Larregina AT, Morelli AE (2008) Exosomes as a short-range mechanism to spread alloantigen between dendritic cells during T cell allorecognition. J Immunol 180:3081–3090
Prado N, Cañamero M, Villalba M, Rodríguez R, Batanero E (2010) Bystander suppression to unrelated allergen sensitization through intranasal administration of tolerogenic exosomes in mouse. Mol Immunol 47:2148–2151
Rak J (2010) Microparticles in cancer. Semin Thromb Hemost 36:888–906
Taylor DD, Gercel-Taylor C (2005) Tumor-derived exosomes as mediates of T-cell signaling defects. Brit J Cancer 92:305–311
Kesimer M, Scull M, Brighton B, DeMaria G, Burns K, O'Neal W, Pickles RJ, Sheehan JK (2009) Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: a possible role in innate defense. FASEB J 23:1858–1868
Taylor DD, Bender DP, Gercel-Taylor C, Stanson J, Whiteside TL (2001) Modulation of TcR/CD3-zeta chain expression by a circulating factor derived from ovarian cancer patients. Br J Cancer 84:1624–1629
Mathivanan S, Ji H, Simpson RJ (2010) Exosomes: extracellular organelles important in intercellular communication. J Proteomics 73:1907–1920
Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, Bigner DD (2009) Proteomic and immunologic analyses of brain tumor exosomes. FASEB J 23:1541–1557
Parsons TD, Lenzi D, Almers W, Roberts WM (1994) Calcium-triggered exocytosis and endocytosis in an isolated presynaptic cell: capacitance measurements in saccular hair cells. Neuron 13:875–883
Simons K, Gerl MJ (2010) Revitalizing membrane rafts: new tools and insights. Nature Rev Mol Cell Biol 11:688–699
Piper RC, Katzmann DJ (2007) Biogenesis and function of multivesicular bodies. Ann Rev Cell Dev Biol 23:519–547
Doring T, Gotthardt K, Stieler J, Prange R (2010) γ2-Adaptin is functioning in the late endosomal sorting pathway and interacts with ESCRT-I and -III subunits. Biochim Biophys Acta 1803:1252–1264
DeGassart A, Geminard C, Hoekstra D, Vidal M (2004) Exosome secretion: the art of reutilizing non-recycled proteins? Traffic 5:896–903
Buschow SI, Liefhebber JM, Wubbolts R, Stoorvogel W (2005) Exosomes contain ubiquitinated proteins. Blood Cells Mol Dis 35:398–403
Qu JL, Qu XJ, Zhao MF, Teng YE, Zhang Y, Hou KZ, Jiang YH, Yang XH, Liu YP (2009) The role of cbl family of ubiquitin ligases in gastric cancer exosome-induced apoptosis of Jurkat T cells. Acta Oncol 15:1–8
Wollert T, Hurley JH (2010) Molecular mechanisms of multivesicular body biogenesis by ESCRT complexes. Nature 464:864–869
Katzmann DJ, Stefan CJ, Babst M, Emr SD (2003) Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 162:413–423
Skinner AM, O’Neill SL, Kurre P (2009) Cellular microvesicle pathways can be targeted to transfer genetic information between non-immune cells. PLoS One 4:e6219
Min CK, Bang SY, Cho BA, Choi YH, Yang JS, Lee SH, Seong SY, Kim KW, Kim S, Jung JU, Choi MS, Kim IS, Cho NH (2008) Role of amphipathic helix of a herpes viral protein in membrane deformation and T cell receptor downregulation. PLoS Pathog 4:e1000209
Chen J, Wang J, Meyers KR, Enns CA (2009) Transferrin-directed internalization and cycling of transferring receptor 2. Traffic 10:1488–1501
Simpson RJ, Jensen SS, Lim JW (2008) Proteomic profiling of exosomes: current perspectives. Proteomics 8:4083–4099
Mathivanan S, Simpson RJ (2009) ExoCarta: a compendium of exosomal proteins and RNA. Proteomics 9:4997–5000
Muntasell A, Berger AC, Roche PA (2007) T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J 26:4263–4272
Janiszewski M, DoCarmo AO, Pedro MA, Silva E, Knobel E, Laurindo FR (2004) Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: a novel vascular redox pathway. Crit Care Med 32:818–825
Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, Flament C, Pouzieux S, Faure F, Tursz T, Angevin E, Amigorena ZL (2001) Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med 7:297–303
Pfeffer SR (2010) Two Rabs for exosome release. Nat Cell Biol 12:3–4
McCready J, Sims JD, Chan D, Jay DG (2010) Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10:294
Lee HM, Choi EJ, Kim JH, Kim TD, Kim YK, Kang C, Gho YS (2010) A membranous for of ICAM-1 on exosomes efficiently blocks leukocyte adhesion to activated endothelial cells. Biochem Biophys Res Commun 397:251–256
Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ (2010) Exosome release of β-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol 190:1079–1091
Zöller M (2009) Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer 9:40–55
Stinton LM, Eystathioy T, Selak S, Chan EK, Fritzler MJ (2004) Autoantibodies to protein transport and messenger RNA pathways: endosomes, lysosomes, Golgi complex, proteasomes, assemblyosomes, exosomes, and GW bodies. Clin Immunol 110:30–44
Cheruvanky A, Zhou H, Pisitkun T, Kopp JB, Knepper MA, Yuen PS, Star RA (2007) Rapid isolation of urinary exosomal biomarkers using a nanomembrane ultrafiltration concentrator. Am J Physiol Renal Physiol 292:F1657–F1661
Viaud S, Théry C, Ploix S, Tursz T, Lapierre V, Lantz O, Zitvogel L, Chaput N (2010) Dendritic cell-derived exosomes for cancer immuntherapy: what's next? Cancer Res 70:1281–1285
Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, Pomel C, Lhomme C, Escudier B, Le Chevalier T, Tursz T, Amigorena S, Raposo G, Angevin E, Zitvogel L (2002) Malignant effusions and immunogenic tumour-derived exosomes. Lancet 360:295–305
Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, Coscia C, Iessi E, Logozzi M, Molinari A, Colone M, Tatti M, Sargiacomo M, Fais S (2009) Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem 284:34211–34222
Simons M, Raposo G (2009) Exosomes: vesicular carriers for intercellular communication. Curr Opin Cell Biol 21:575–581
Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W, Lim SK, Sze SK (2010) Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteomics 9:1085–1099
Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, Zoeller M (2008) CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med 8:784–804
Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, Cheng Z, Shah SV, Wang GJ, Zhang L, Grizzle WE, Mobley J, Zhang HG (2009) Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer 124:2621–2633
Hong BS, Cho JH, Kim H, Choi EJ, Rho S, Kim J, Kim JH, Choi DS, Kim YK, Hwang D, Gho YS (2009) Colorectal cancer cell-derived microvesicles are enriched in cell cycle-related mRNAs that promote proliferation of endothelial cells. BMC Genomics 10:556
Nazarenko I, Rana S, Baumann A, McAlear J, Hellwig A, Trendelenburg M, Lochnit G, Preissner KT, Zöller M (2010) Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res 70:1668–1678
Keller S, König AK, Marmé F, Runz S, Wolterink S, Koensgen D, Mustea A, Sehouli J, Altevogt P (2009) Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Lett 278:73–81
Dolo V, D'Ascenzo S, Violini S, Pompucci L, Festuccia C, Ginestra A, Vittorelli ML, Canevari S, Pavan A (1999) Matrix-degrading proteinases are shed in membrane vesicles by ovarian cancer cells in vivo and in vitro. Clin Exp Metastasis 17:131–140
Dolo V, Ginestra A, Cassara D, Ghersi G, Nagase H, Vittorelli ML (1999) Shed membrane vesicles and selective localization of gelatinases and MMP-9/TIMP-1 complexes. Ann N Y Acad Sci 878:497–499
Graves LE, Ariztia EV, Navari JR, Matzel HJ, Stack MS, Fishman DA (2004) Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res 64:7045–7049
Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res 66:605–612
McLellan AD (2009) Exosome release by primary B cells. Crit Rev Immunol 29:203–217
Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ (2006) Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 20:1487–1495
Ichim TE, Zhong Z, Kaushal S, Zheng X, Ren X, Hao X, Joyce JA, Hanley HH, Riordan NH, Koropatnick J, Bogin V, Minev BR, Min WP, Tullis RH (2008) Exosomes as a tumor immune escape mechanism: possible therapeutic implications. J Transl Med 6:37
Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y, Krogan NJ, Plemenitas A, Peterlin BM (2010) HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic 11:110–122
Izquierdo-Useros N, Naranjo-Gómez M, Archer J, Hatch SC, Erkizia I, Blanco J, Borràs FE, Puertas MC, Connor JH, Fernández-Figueras MT, Moore L, Clotet B, Gummuluru S, Martinez-Picado J (2009) Capture and transfer if HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood 113:2732–2741
Izquierdo-Useros N, Naranjo-Gómez M, Erkizia I, Puertas MC, Borràs FE, Blanco J, Martinez-Picado J (2010) HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? PLoS Pathog 6:e1000740
Al-Nedawi K, Meehan B, Rak J (2009) Microvesicles: messengers and mediators of tumor progression. Cell Cycle 8:2014–2018
Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J (2008) Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 10:619–624
Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J (2009) Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA 106:3794–3799
Viswanathan M, Sangiliyandi G, Vinod SS, Mohanprasad BK, Shanmugam G (2003) Genomic instability and tumor-specific alterations in oral squamous cell carcinomas assessed by inter-(simple sequence repeat) PCR. Clin Cancer Res 9:1057–1062
Camussi G, Deregibus MC, Bruno S, Cantaluppi V, Biancone L (2010) Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int 78:838–848
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10:1470–1476
Janowska-Wieczorek A, Wysoczynski M, Kijowski J, Marquez-Curtis L, Machalinski B, Ratajczak J, Ratajczak MZ (2005) Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 113:752–760
Koh W, Sheng CT, Tan B, Lee QY, Kuznetsov V, Kiang LS, Tanavde V (2010) Analysis of deep sequencing microRNA expression profile from human embryonic stem cells derived mesenchymal stem cells reveals possible role of let-7 microRNA family in downstream targeting of hepatic nuclear factor 4 alpha. BMC Genomics 11(Suppl 1):S6
Yuan XL, Chen L, Li MX, Dong P, Xue J, Wang J, Zhang TT, Wang XA, Zhang FM, Ge HL, Shen LS, Xu D (2010) Elevated expression of Foxp3 in tumor-infiltrating Treg cells suppresses T-cell proliferation and contributes to gastric cancer progression in a COX-2-dependent manner. Clin Immunol 134:277–288
Whiteside TL (2005) Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system? Br J Cancer 92:209–211
Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L (2007) Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res 67:2912–2915
Cocucci E, Racchetti G, Meldolesi J (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51
Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, Zaccheddu A, Colone M, Arancia G, Gentile M, Seregni E, Valenti R, Ballabio G, Belli F, Leo E, Parmiani G, Rivoltini L (2005) Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterol 128:1796–1804
Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL (2009) Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol 183:3720–3730
Huber V, Filipazzi P, Iero M, Fais S, Rivoltini L (2008) More insights into the immunosuppressive potential of tumor exosomes. J Transl Med 6:63
Taylor DD, Gercel-Taylor C, Lyons KS, Stanson J, Whiteside TL (2003) T-cell apoptosis and suppression of T-cell receptor/CD3-zeta by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin Cancer Res 9:5113–5119
Taylor DD, Gercel-Taylor C, Weese JL (1989) Expression and shedding of mdr-1 antigen by variants of the murine B16 melanoma. Surg Forum 40:406–408
Taylor DD, Gercel-Taylor C, Gall SA (1996) Expression and shedding of CD44 isoforms by gynecologic cancer patients. J Soc Gynecol Invest 3:289–294
Taylor DD, Lyons KS, Gercel-Taylor C (2002) Shed membrane fragment-associated markers for endometrial and ovarian cancers. Gynecol Oncol 84:443–448
Schorey JS, Bhatnagar S (2008) Exosome function: from tumor immunology to pathogen biology. Traffic 9:871–881
Zindl CL, Chaplin DD (2010) Immunology: tumor immune evasion. Science 328:697–698
Moserle L, Amadori A, Indraccolo S (2009) The angiogenic switch: implications in the regulation of tumor dormancy. Curr Mol Med 9:935–941
Gao D, Nolan D, McDonnell K, Vahdat L, Benezra R, Altorki N, Mittal V (2009) Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim Biophys Acta 1796:33–40
Zwirner NW, Croci DO, Domaica CI, Rabinovich GA (2010) Overcoming the hurdles of tumor immunity by targeting regulatory pathways in innate and adaptive immune cells. Curr Pharm Des 16:255–267
Soloski MJ (2001) Recognition of tumor cells by the innate immune system. Curr Opin Immunol 13:154–162
Ashiru O, Boutet P, Fernández-Messina L, Agüera-González S, Skepper JN, Valés-Gómez M, Reyburn HT (2010) Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res 70:481–489
Groh V, Wu J, Yee C, Spies T (2002) Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 419:734–738
Frey AB (2006) Myeloid suppressor cells regulate the adaptive immune response to cancer. J Clin Invest 116:2587–2590
Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB (2006) CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol 176:2085–2094
Yu S, Liu C, Su K, Wang J, Liu Y, Zhang L, Li C, Cong Y, Kimberly R, Grizzle WE, Falkson C, Zhang HG (2007) Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J Immunol 178:6867–6875
Soderberg A, Barral AM, Soderstrom M, Sander B, Rosen A (2007) Redox-signaling transmitted in trans to neighboring cells by melanoma-derived TNF-containing exosomes. Free Radic Biol Med 43:90–99
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rébé C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120:457–471
Liu Y, Xiang X, Zhuang X, Zhang S, Liu C, Cheng Z, Michalek S, Grizzle W, Zhang HG (2010) Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol 176:2490–2499
Iero M, Valenti R, Huber V, Filipazzi P, Parmiani G, Fais S, Rivoltini L (2008) Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ 15:80–88
Wieckowski E, Whiteside TL (2006) Human tumor-derived vs dendritic cell derived exosomes have distinct biologic roles and molecular profiles. Immunol Res 36:247–254
Tomihari M, Chung JS, Akiyoshi H, Cruz PD Jr, Ariizumi K (2010) DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res 70:5778–5787
Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL (2005) Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res 11:1010–1020
Laulagnier K, Grand D, Dujardin A, Hamdi S, Vincent-Schneider H, Lankar D, Salles JP, Bonnerot C, Perret B, Record M (2004) PLD2 is enriched on exosomes and its activity is correlated to the release of exosomes. FEBS Lett 572:11–14
Morelli AE (2006) The immune regulatory effect of apoptotic cells and exosomes on dendritic cells: its impact on transplantation. Am J Transplant 6:254–261
Abusamra AJ, Zhong Z, Zheng X, Li M, Ichim TE, Chin JL, Min WP (2005) Tumor exosomes expressing Fas ligand mediate CD8+ T cell apoptosis. Blood Cells Mol Dis 35:169–173
Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR (2004) Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest 114:560–568
Clayton A, Tabi Z (2005) Exosomes and the MICA-NKG2D system in cancer. Blood Cells Mol Dis 34:206–213
Liu C, Yu S, Zinn K, Wang J, Zhang L, Jia Y, Kappes JC, Barnes S, Kimberly RP, Grizzle WT, Zhang HG (2006) Murine mammary carcinoma exosomes promote tumor growth by suppression of NK cell function. J Immunol 176:1375–1385
Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z (2008) Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol 180:7249–7258
Perone MJ, Larregina AT, Shufesky WJ, Papworth GD, Sullivan ML, Zahorchak AF, Stolz DB, Baum LG, Watkins SC, Thomson AW, Morelli AE (2006) Transgenic galectin-1 induces maturation of dendritic cells that elicit contrasting responses in naïve and activated T cells. J Immunol 176:7207–7220
Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, Raposo G, Hivroz C (2002) TCR activation of human T cells induces the production of exosomes bearing TCR/CD3/zeta complex. J Immunol 168:3235–3241
Bamias A, Tsiatas ML, Kafantari E, Liakou C, Rodolakis A, Voulgaris Z, Vlahos G, Papageorgiou T, Tsitsilonis O, Bamia C, Papatheodoridis G, Politi E, Archimandritis A, Antsaklis A, Dimopoulos MA (2007) Significant differences of lymphocytes isolated from ascites of patients with ovarian cancer compared to blood and tumor lymphocytes. Association of CD3+ CD56+ cells with platinum resistance. Gynecol Oncol 106:75–81
Kalinski P, Okada H (2010) Polarized dendritic cells as cancer vaccines: directing effector-type T cells to tumors. Semin Immunol 22(3):173–182
Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, Camus M, Gillard M, Bruneval P, Fridman WH, Pagès F, Trajanoski Z, Galon J (2010) Biomolecular network reconstruction identifies T cell homing factors associated with colorectal cancer. Gastroenterology 138(4):1429–1440
Lee CH, Chiang YH, Chang SE, Chong CL, Cheng BM, Roffler SR (2009) Tumor-localized ligation of CD3 and CD28 with systemic regulatory T-cell depletion induces potent innate and adaptive antitumor responses. Clin Cancer Res 15:2756–2766
Maccalli C, Pisarra P, Vegetti C, Sensi M, Parmiani G, Anichini A (1999) Differential loss of T cell signaling molecules in metastatic melanoma patients' T lymphocyte subsets expressing distinct TCR variable regions. J Immunol 163:6912–6923
Nieuwland R, van der Post JA, Gemma CA, Kenter G, Sturk A (2010) Microparticles and exosomes in gynecologic neoplasias. Semin Thromb Hemost 36:925–929
Ristorcelli E, Beraud E, Verrando P, Villard C, Lafitte D, Sbarra V, Lombardo D, Verine A (2008) Human tumor nanoparticles induce apoptosis of pancreatic cancer cells. FASEB J 22:3358–3369
Khazaie K, von Boehmer H (2006) The impact of CD4+ CD25+ Treg on tumor specific CD8+ T cell cytotoxicity and cancer. Semin Cancer Biol 16:124–136
Shen LS, Wang J, Shen DF, Yuan XL, Dong P, Li MX, Xue J, Zhang FM, Ge HL, Xu D (2009) CD4+CD25+CD127low/- regulatory T cells express Foxp3 and suppress effector T cell proliferation and contribute to gastric cancers progression. Clin Immunol 131:109–118
Wada J, Onishi H, Suzuki H, Yamasaki A, Nagai S, Morisaki T, Katano M (2010) Surface-bound TGF-beta1 on effusion-derived exosomes participates in maintenance of number and suppressive function of regulatory T cells in malignant effusions. Anticancer Res 30:3747–3757
Gardella S, Gardella S, Andrei C, Lotti LV, Poggi A, Torrisi MR, Zocchi MR, Rubartelli A (2001) CD8+ T lymphocytes induce polarized exocytosis of secretory lysosomes by dendritic cells with release of interleukin-1B and cathepsin D. Blood 98:2152–2159
Xie Y, Bai O, Yuan J, Chibbar R, Slattery K, Wei Y, Deng Y, Xiang J (2009) Tumor apoptotic bodies inhibit CTL responses and antitumor immunity via membrane-bound transforming growth factor-beta-1 inducing CD8+ T-cell anergy and CD4+ Tr1 cell responses. Cancer Res 69:7756–7766
Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL (2010) Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One 5:e11469
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Anderson HC, Mulhall D, Garimella R (2010) Role of extracellular membrane vesicles in the pathogenesis of various diseases, including cancer, renal diseases, atherosclerosis, and arthritis. Lab Invest 90:1549–1557
Temme S, Eis-Hubinger AM, McLellan AD, Koch N (2010) The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J Immunol 184:236–243
Miguet L, Béchade G, Fornecker L, Zink E, Felden C, Gervais C, Herbrecht R, Van Dorsselaer A, Mauvieux L, Sanglier-Cianferani S (2009) Proteomic analysis of malignant B-cell derived microparticles reveals CD148 as a potentially useful antigenic biomarker for mantle cell lymphoma diagnosis. J Proteome Res 8:3346–3354
Taylor DD, Gercel-Taylor C, Jiang CG, Black PH (1988) Characterization of plasma membrane shedding from murine melanoma cells. Int J Cancer 41:629–635
Taylor DD, Black PH (1985) Inhibition of macrophage Ia antigen expression by shed plasma membrane vesicles from metastatic murine melanoma lines. J Natl Cancer Inst 74:859–867
Poutsiaka DD, Schroder EW, Taylor DD, Levy EM, Black PH (1985) Membrane vesicles shed by murine melanoma cells selectively inhibit the expression of Ia antigen by macrophages. J Immunol 134:138–144
Pelton JJ, Taylor DD, Fowler WC, Gercel-Taylor C, Carp NZ, Weese JL (1991) Lymphokine-activated killer cell suppressor factor in malignant effusions. Arch Surg 126:476–480
Albanese J, Meterissian S, Kontogiannea M, Dubreuil C, Hand A, Sorba S, Dainiak N (1998) Biologically acive Fas antigen and its cognate ligand are expressed on plasma membrane-derived extracellular vesicles. Blood 91:3862–3874
Admyre C, Johansson SM, Qazi KR, Filén JJ, Lahesmaa R, Norman M, Neve EP, Scheynius A, Gabrielsson S (2007) Exosomes with immune modulatory features are present in human breast milk. J Immunol 179:1969–1978
Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD (2007) MHC class II + exosomes in plasma suppress inflammation in an antigen-specific and Fas ligand/Fas-dependent manner. J Immunol 179(4):2235–2241
Martinez FO, Sica A, Mantovani A, Locati M (2008) Macrophage activation and polarization. Front Biosci 13:453–461
Ehrt S, Schnappinger D, Bekiranov S, Drenkow J, Shi S, Gingeras TR, Gaasterland T, Schoolnik G, Nathan C (2001) Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med 194:1123–1140
Martinez FO, Gordon S, Locati M, Mantovani A (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177:7303–7311
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35
Dinarello CA (1996) Biologic basis for interleukin-1 in disease. Blood 87:2095–2147
Scotton CJ, Martinez FO, Smelt MJ, Sironi M, Locati M, Mantovani A, Sozzani S (2005) Transcriptional profiling reveals complex regulation of the monocyte IL-1 beta system by IL-13. J Immunol 174:834–845
Roman J, Ritzenthaler JD, Fenton MJ, Roser S, Schuyler W (2000) Transcriptional regulation of the human interleukin 1beta gene by fibronectin: role of protein kinase C and activator protein 1 (AP-1). Cytokine 12:1581–1589
Roman J, McDonald JA (1997) Fibronectins and fibronectin receptors in lung development, injury and repair. In: Crystal RG, West JB, Barnes P, Weibel ER (eds) The lung: scientific foundations, 2nd edn. Raven, New York, pp 737–755
Graves K, Roman J (1996) Fibronectin modulates the expression of interleukin-1β and its receptor antagonist in human mononuclear cells. Am J Physiol 271:L61–L69
Pacifici R, Roman J, Kimble R, Civitelli R, Brownfield CM, Bizzarri C (1994) Ligand binding to monocyte alpha 5 beta 1 integrin activates the alpha 2 beta 1 receptor via the alpha 5 subunit cytoplasmic domain and protein kinase C. J Immunol 153:2222–2233
Takizawa T, Nishinarita S, Kitamura N, Hayakawa J, Kang H, Tomita Y, Mitamura K, Yamagami K, Horie T (1995) Interaction of the cell-binding domain of fibronectin with VLA-5 integrin induces monokine production in cultured human monocytes. Clin Exp Immunol 101:376–382
Ruoslahti E (1996) RGD and other recognition sequences for integrins. Ann Rev Cell Dev Biol 12:697–715
Ardoin SP, Shanahan JC, Pisetsky DS (2007) The role of microparticles in inflammation and thrombosis. Scan J Immunol 66:159–165
Distler JH, Distler O (2010) Inflammation: microparticles and their roles in inflammatory arthritides. Nat Rev Rheumatol 6(7):385–386
Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA (2008) Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol 180:817–824
Gasser O, Schifferli JA (2004) Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 104:2543–2548
Mack M, Kleinschmidt A, Brühl H, Klier C, Nelson PJ, Cihak J, Plachý J, Stangassinger M, Erfle V, Schlöndorff D (2000) Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med 6:769–775
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Small Vesicles as Immune Modulators.
Rights and permissions
About this article
Cite this article
Taylor, D.D., Gercel-Taylor, C. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Semin Immunopathol 33, 441–454 (2011). https://doi.org/10.1007/s00281-010-0234-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-010-0234-8