
Abstract
Reactive oxygen species (ROS) have long been studied in the context of their direct toxic effects on cells. As a result, ROS have conventionally been thought of as a necessary nuisance to aerobic living. However, in recent years, much work has been done to examine the contribution of ROS to the field of immunity. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases were identified as one of the key sources of ROS in immune cells. The NOX2 NADPH oxidase in particular has been assigned multiple roles, functioning as a source of antimicrobial ROS, an activator of many signaling pathways, a participant in chemotaxis, an immune modulator, and a critical player in the initiation of antigen cross-presentation. Furthermore, recent studies have revealed a novel role for the NOX2 NADPH oxidase in the activation of autophagy, a cellular degradative pathway. Here, we examine these functions of NOX2 NADPH oxidase in immunity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactive oxygen species (ROS) are a group of highly reactive free radical and non-radical molecules [1]. ROS have a number of important functions in immunity that have only been recently realized. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) are major sources of ROS in immune cells. The critical role that NOX2 NADPH oxidase-derived ROS play in host immunity was clearly demonstrated by genetic studies done in the late 1980s. These studies showed that patients with mutations in NOX2 NADPH oxidase component genes develop chronic granulomatous disease (CGD) (reviewed by [2]). CGD is characterized by recurrent and severe infections by a particular spectrum of bacterial and fungal infections as a result of the host's inability to mount an effective innate immune response [3]. Since that time, much work has been done to elucidate the contribution of ROS to innate immunity and the defense against invading pathogens. The current opinion on the specific roles that ROS perform has evolved greatly beyond ROS simply as microbicidal compounds, developing into a vastly more complex model of ROS function. The purpose of this review is to discuss the current understanding of all aspects of NOX2 NADPH oxidase-derived ROS contribution to immunity.
The assembly and function of NOX2 NADPH oxidase
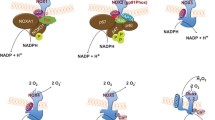
There are a number of cellular sources that generate ROS including NOXs, xanthine oxidase, the mitochondrial electron transport chain, peroxisomes, and the endoplasmic reticulum (ER) [4–8]. The NOX family comprises seven members (NOX1-5 and DUOX1-2) with NOX2 NADPH oxidase being the predominant source of ROS production in humans [9]. The main producers of ROS are phagocytic cells—neutrophils and macrophages. NOX2 NADPH oxidase is composed of functional transmembrane heterodimers, gp91phox and p22phox (also known collectively as the cytochrome b558), and four regulatory cytosolic subunits—p40phox, p47phox, p67phox, and the small GTPase, Rac2 (Fig. 1). In the dormant state, cytochrome b558 resides in intracellular vesicles [10], while cytosolic Rac2 remains inactive in the guanosine diphosphate (GDP) bound state via interaction with RhoGDI [11, 12]. Upon the initiation of phagocytosis, GDP-Rac2 is converted to GTP-Rac2 through the activity of a Rac guanine nucleotide exchange factor. This allows for Rac2 translocation to the plasma or phagosomal membrane, thereby allowing the subsequent transit of cytochrome b558 from the vesicle to the membrane [13]. Concurrently, p47phox is phosphorylated and undergoes a conformational change that now exposes two SRC-homology 3 regions to interact with the proline rich motif on p22phox [14]. Furthermore, Phox homology domains on p47phox allow for binding to phosphatidylinositol 3-phosphate (PI(3)P) and PI(3,4)P2, transient phosphoinositides that are generated only at the plasma membrane upon phagocytosis, thus further stabilizing p47phox localization to cytochrome b558 [9].

A schematic of NOX2 NADPH oxidase assembly and activation. a In the resting stage, cytochrome b558 (gp91phox and p22phox) resides in vesicles. Rac2 in the inactive GDP bound form remains in the cytosol. The regulatory subunits, p47phox, p67phox, and p40phox, are trimerized in the cytosol. b Upon receiving signals for activation, cytochrome b558 and the trimeric regulatory subunits are recruited to the membrane. RhoGDI inhibition of Rac2 is now released to allow GTP binding. c The assembled complex functions at the membrane
Since p47phox, p67phox, and p40phox are trimerized in the cytosol, the translocation of p47phox brings the other two regulatory subunits to the membrane as well [15, 16]. However, there are specific regulatory mechanisms in place that control the activation state of both p67phox and p40phox, which are also required for the proper functioning of the NOX2 NADPH oxidase complex. Phosphorylated p67phox interacts with Rac2 and cytochrome b558, inducing a conformational change in the functional subunit that is necessary for ROS production [17]. The role that p40phox plays is less clear, but studies have indicated that phosphorylated p40phox, via its interaction with PI(3)P, is also critical for NOX2 NADPH oxidase activation [18–20]. Recent work suggests that p40phox binds to p67phox via its PB1 domain [21] and assists in p67phox regulation of NOX2 NADPH oxidase activity [22].
There have been many reports about the identity of the kinases responsible for p47phox, p67phox, and p40phox activation, which include several protein kinase C (PKC) isoforms (PKCα [23, 24], PKCδ [24, 25], PKCβ [24, 26], PKCγ [27], and PKCζ [24, 28]), protein kinase A [29], p21 activated kinase [30], ERK1/2 [31, 32], AKT [33], PI3K [34, 35], and possibly others. The complexity of NOX2 NADPH oxidase regulation by such a large number of kinases not only suggests that perhaps a high threshold of activating signals may be required for NOX2 NADPH oxidase activity but also that there may be differing amounts of ROS produced by each individual NOX2 NADPH oxidase complex, depending on the type of local activating signal it receives. In fact, a recent study indicated that NOX2 NADPH oxidase assembly and activity is highly heterogeneous—where only 50% of phagosomes formed upon Fc-γ receptor (FcγR)-mediated phagocytosis have proper p40phox localization and NOX2 NADPH oxidase function [36]. After assembly and activation, NOX2 NADPH oxidase then produces ROS in a reaction on the cytoplasmic region of the gp91phox subunit that converts NADPH to NADP+, resulting in the liberation of two electrons and one H+. The two electrons are transported through cytochrome b558 to the lumen of the phagosome where they react with two oxygen molecules to form two superoxide ions.
While NOX2 NADPH oxidase is the main producer of ROS upon phagocytosis, it must be noted that other NOX family members also generate ROS in macrophages given specific stimulations. In the pathobiology of atherosclerosis, one of the key steps in plaque formation is macrophage to foam cell conversion, mediated by chronic macrophage intake of oxidized low-density lipoproteins (oxLDL) [37]. It was recently shown that lipopolysaccharide (LPS), a toll-like receptor (TLR)-4 agonist, can lead to increased NOX1 NADPH oxidase activity, thereby expediting macrophage conversion to foam cells by increasing the levels of oxLDL [38, 39]. Other groups have found that NOX4 NADPH oxidase also plays a critical role in the formation of oxLDL, leading to macrophage death [40]. Additionally, NOX3 NADPH oxidase expression, albeit low, has been reported in RAW macrophages as well [41]. Thus, while NOX2 NADPH oxidase is the predominant form of NADPH oxidase expressed in macrophages and other phagocytes, the expression and contributions of other NOX members during infection warrants further investigation.
Functions of ROS
Microbicidal activity
Direct inhibitory mechanisms
Upon pathogen infection, neutrophils, macrophages, and dendritic cells (DCs) have a number of defensive strategies that are employed to contain and eliminate pathogens. Such strategies include phagocytosis-mediated lysosomal degradation as well as the production of antimicrobial peptides, defensins, lactoferrins (and other metal chelators), proteases, cathepsins, reactive nitrogen species (RNS), and ROS (reviewed in [42]). Of these strategies, NOX2 NADPH oxidase activity is among the earliest and most robust defenses that phagocytes have against microbes. The importance of NOX2 NADPH oxidase is clearly seen in CGD patients with deficient NOX2 NADPH oxidase activity in which they develop severe innate immune deficiency. Therefore, it is not surprising to find that a number of microbial pathogens have evolved mechanisms to modulate ROS production by NOX2 NADPH oxidase (Table 1).
Despite the predominant production of superoxide by NOX2 NADPH oxidase [9], it remains controversial whether superoxide is the main antimicrobial compound. Some researchers have noted that superoxide is relatively unreactive when compared with the rest of the ROS family and thus may not be a sufficient defensive strategy on its own [43]. Conversely, others have argued that, in the low pH environment of the phagosome, the majority of superoxide is protonated (HO2•), becoming a much more reactive compound [44].
While the direct antimicrobial effect of superoxides remains contentious, the indirect antimicrobial effect of superoxides, via its reactive products, is well established (Fig. 2). Due to its highly unstable nature, superoxide readily forms a number of other compounds (reviewed in [45, 46]). Superoxide reacts with nitric oxide to produce peroxynitrite, an even stronger oxidizing agent [47]. The combination of two molecules of superoxide ions, catalyzed by the activity of the enzyme superoxide dismutase (SOD), results in the production of hydrogen peroxide (H2O2). H2O2 acts mainly upon thiol groups in cysteine residues, leading to either oxidation [48, 49] or disulfide bond formation [50, 51]. H2O2, in turn, produces hypochloric acid (HOCl) when combined with Cl− in a reaction catalyzed by myeloperoxidase. Both H2O2 and HOCl have been shown to be present in sufficient concentrations in the phagosome to kill microbes [52, 53]. H2O2 also interacts with transition metal ions, such as ferrous and ferric ions, to produce hydroxyl radicals (OH•), or with superoxides to generate singlet oxygen (1O2). OH•, in particular, while short lived, is the most highly oxidizing member of the ROS family, reacting rapidly and non-discriminatorily with DNA, lipids, and proteins.

The generation of common ROS products in phagocytes by NOX2 NADPH oxidase
Indirect inhibitory mechanisms
In addition to its direct microbicidal effects on pathogens, ROS also arrests pathogen survival and growth either via the inactivation of critical bacterial products or the modulation of the phagosomal or extracellular environment.
Leukotoxin, a key virulence factor of Actinobacillus actinomycetemcomitans, has been shown to induce apoptosis in human macrophages and neutrophils [54, 55]. Myeloperoxidase, through the production of HOCl, has been implicated in the oxidation and subsequent inactivation of leukotoxin [56–58]. Pneumolysin, a vital virulence factor secreted by Streptococcus pneumophilus that is required for survival in human neutrophils, is likewise oxidized by HOCl. Pneumolysin is a pore-forming toxin (PFT) whose function is regulated by the ability to form disulfide bonds, without which proper oligomerization for pore formation cannot occur [59, 60]. Myeloperoxidase was found to be effective in inhibiting pneumolysin activity, presumably via the prevention of disulfide bond formation [61]. Another PFT, listeriolysin O (LLO), has been suggested to be inhibited via a similar mechanism [62]. LLO is one of the key virulence factors produced by Listeria monocytogenes that mediates bacterial survival and growth in both human and murine macrophages [63, 64]. It has long been known that LLO requires a reducing environment for its activity [65, 66]. Recent work has identified the host protein GILT (γ-interferon inducible lysosomal thiolreductase) to be responsible for the reduction of LLO to mediate disulfide bond formation [67]. It is therefore likely that, similar to pneumolysin, LLO is also inhibited by ROS via the prevention of disulfide bond formation. However, virulence factors are not the only reported microbial target of ROS as quorum sensing by Staphylococcus aureus is also affected both in vitro and in vivo [68]. Quorum sensing is the process by which bacteria modulate their behavior once they “sense” that their population has expanded above a certain threshold number. Rothfork and colleagues demonstrated that HOCl and peroxynitrite inhibit the autoinducer peptide activity, a critical player in quorum sensing, leading to an inability to upregulate virulence factor expression once the bacteria has reached sufficient numbers.
In addition to effects on bacterial products, ROS also play a role in the mobilization of other host microbicidal factors. NOX2 NADPH oxidase is in essence an electron transporter, potentially generating a negatively charged environment in the lumen of the phagosome. This buildup requires the concurrent compensation of positive charges to maintain membrane neutrality. While there has been much speculation on which ions participate in this charge compensation, it has been shown that K+ contributes, at least in part, to this process [69, 70]. The influx of K+ increases ionic strength in the phagosome, thereby allowing the release of proteases, such as cathepsin G and elastases, from the negatively charged proteoglycan matrix. This allows the activation and targeting of these proteases to the pathogen for degradation [71].
Another host antimicrobial process activated by ROS is neutrophil extracellular traps (NETs) formation. NETs are fibrous meshworks comprising DNA and protein projected into the extracellular space by mature stimulated neutrophils for the purpose of entrapping and killing pathogens [72, 73]. While the mechanism of NETs formation remains unclear, a recent study suggests that NETs may form as a result of dying neutrophils undergoing a series of cellular reorganization events (i.e., granule disintegration and dissolution of the nuclear envelope) such that, upon the loss of plasma membrane integrity, the toxic intracellular contents are released extracellularly [74]. It has been reported that a number of classical pro-inflammatory signals—interleukin (IL)-8, phorbol 12-myristate 13-acetate (PMA), LPS—serve as activating signals for NET formation [72]. ROS have also been shown to act as signals for induced cell death or “netosis” with the explicit purpose of creating antimicrobial NETs. Interestingly, CGD patients cannot form NETs, further implicating NOX2 NADPH oxidase-derived ROS in this antimicrobial response [72].
Immune modulatory effects of ROS
In addition to its antimicrobial activity, ROS have also been implicated in the modulation of the immune system to help create an environment that allows for an efficient and effective immune response. As such, there has been a wealth of research focused on the interaction between NOX2 NADPH oxidase-derived ROS and various key members of both the innate and adaptive immune systems. The results of these efforts demonstrate a diverse array of ROS functions as they have been reported to participate in both pro- and anti-inflammatory signaling.
Pro-inflammatory effects
The majority of the work linking ROS to the activation of the immune system has focused on the interaction between ROS and DCs. Considered to be the “bridge” between the innate and adaptive immune systems, DCs are professional antigen presenting cells that mediate the activation of different types of adaptive immune responses. Work done by Rutault and colleagues indicate that treatment with H2O2 promotes human DC activation and subsequent T-cell engagement and proliferation as a result of enhanced expression of major histocompatibility complex (MHC) II and the co-stimulatory molecules, CD40 and CD86 [75]. Similarly, superoxide produced by xanthine oxidase has also been found to induce dendritic cell maturation [76]. Further examination of the dendritic cell cytokine profile revealed the predominance of two pro-inflammatory cytokines—IL-8 and tumor necrosis factor (TNF)-α—in particular [77]. Thus, while the role of NOX2 NADPH oxidase-derived ROS in DC development has yet to be examined, ROS derived from alternate sources can efficiently induce DC maturation.
Anti-inflammatory effects
While it may seem counterintuitive for ROS to participate in an anti-inflammatory role, a number of studies on the subject suggest that ROS can play a critical role in the prevention of autoimmunity and in the regulation of immune activation. A study of the differences in gene expression between human neutrophils in healthy and CGD patients revealed that a large number of inflammatory genes were upregulated upon phagocytosis in the CGD neutrophils when compared with healthy controls [78]. Further in vitro investigations confirmed that neutrophils from CGD patients produced a more prolonged inflammatory profile accompanied with a delay in apoptosis and clearance. Interestingly, the chronic granulomas in CGD patients are often found to be sterile, suggesting that the hyperinflammation seen in the absence of ROS production may result in aberrant immune responses to the host itself [79]. An examination of the CGD patient population reveals a high prevalence of concomitant autoimmune diseases such as inflammatory bowel disease (IBD), lupus erythematosus, and chorioretinitis [3, 80]. Experimentally, evidence for the link between ROS and the control of autoimmunity is seen in p47phox-deficient mice and rats as they develop a more severe arthritis when challenged with collagen-specific T cells [81]. Transgenic mice that received normal p47phox-expressing macrophages via adoptive transfer reduced the number of arthritic animals to the level of wild type (WT) in the T-cell-dependent collagen-induced model but not the T-cell-independent anti-collagen antibody-induced model. Further in vitro investigations revealed that two measures of T-cell activation—proliferation and IL-2 production—are inhibited by macrophage ROS production. p47phox knockout mice also display an increased autoimmune phenotype with more severe arthritis that is reduced with pharmacological activators of NOX2 NADPH oxidase [82]. Taken together, these observations suggest that macrophage-derived ROS are sufficient in inhibiting autoreactive T-cell responses.
Beyond autoimmunity, ROS deficiency has been shown to be a detriment to the host's ability to generate an appropriate immune response against pathogens. Hyperinflammation has also been observed in the context of Aspergillus fumigatus extract challenge in murine neutrophils [83], Helicobacter pylori mouse infection in vivo [84, 85], and influenza infection in mouse lungs [86].
Cellular signaling effects
ROS, in particular H2O2, have a wide range of reported signaling effects that will only be briefly discussed (this topic is extensively reviewed by [87, 88]).
One mechanism by which ROS plays a signaling role is through the modification of cysteine residues. Oxidation of the sulfur molecule to form sulfenic (Cys-SOH), sulfinic (Cys-SO2H), or sulfonic (Cys-SO3H) acid can occur [89]. Additionally, oxidation of cysteines can result in reactive thiols, leading to the formation of disulfide bonds [50, 51]. Changes in cysteine oxidation may alter cellular signaling via the inhibition of tyrosine phosphatases, G proteins, and certain ion channels [48, 49], as well as the activation of kinases, such as mitogen-activated protein kinases [90]. Another major effect of ROS on signaling is the modulation of Ca2+ signaling. Oxidation of the ryanodine receptor, which contains ROS-sensitive cysteine residues [91], results in the release of intracellular Ca2+ stores [92–94]. ROS also activates the inositol triphosphate (IP3) receptor family Ca2+ release channels [95, 96]. Increasing the intracellular concentration of Ca2+ in immune cells results in differentiation, proliferation, and/or activation, depending on the cell type [97]. An additional consequence of ROS signaling is the activation of transcription factors. Stimulation of TLR results in the increase of ROS signaling, leading to enhanced expression of nuclear factor-kappa B [98]. ROS signaling also results in the expression of a number of genes involved in antioxidative and tumor suppressive action, such as Nrf2 [99], forkhead box containing transcription factor O (FoxO) [100], and p53 [101].
Roles for ROS in chemotaxis
In addition to the intracellular effects of ROS, recent studies have revealed a critical extracellular role that ROS play in ensuring the proper recruitment of immune cells to the site of infection. Through a small-molecule screen for drugs capable of inhibiting neutrophil chemotaxis, Hattori and colleagues found that the NOX2 NADPH oxidase inhibitor, diphenyleneiodonium chloride (DPI), most effectively impaired neutrophil directionality during migration [102]. The DPI effect was found to be ROS specific as the induction of ROS production resulted in an increase in the directed mobility of neutrophils, and this phenotype was reversed when gp91phox or p22phox was silenced by siRNA. Furthermore, neutrophils from CGD patients were found to be slow and disorderly in movement when compared with healthy patients. Finally, a comparison of gp91phox−/− and WT murine neutrophils in an adoptive transfer system revealed that gp91phox−/− but not WT neutrophils were severely stunted in their ability to be properly recruited to the peritoneum in the thioglycolate-induced peritonitis model.
The involvement of ROS in chemotaxis is also seen in other settings. Herpes virus entry mediator binds to host tumor necrosis factor family ligand, LIGHT, leading to the enhanced killing potential of both human macrophages and neutrophils [103]. The mechanism of this increased activation is due to the ROS-mediated increase in migration and expression of chemokine receptors, CCR1 and CCR2 [104]. The 30-kDa antigen produced by Mycobacterium tuberculosis elevates both mRNA and protein levels of the chemokine receptors, CXCL8 and CCL2, in primary monocytes via the induction of NOX2 NADPH oxidase-derived ROS [105]. More recently, LPS induction of matrix metalloproteinase (MMP) production and migration of both peritoneal and RAW 264.7 macrophages were found to require ROS [106]. It was reported that the presence of the antioxidant, N-acetyl-cysteine (NAC), was sufficient to inhibit both MMP production and migration. Furthermore, it is only the specific silencing of NOX2 NADPH oxidase, and not NOX1 NADPH oxidase, that results in a decrease in MMPs. Interestingly, NOX1 NADPH oxidase has recently been implicated in a very specific step in chemotaxis—the degradation of extracellular matrix (ECM) [107]. Invadopodia, cellular protrusions found in many metastatic cancers, are thought to proteolytically degrade the ECM. It was shown that NOX1 NADPH oxidase-derived ROS are responsible for this local degradative ability in the invadopodia of human colon cancer cells. In preeclampsia, neutrophil migration and activation at the placental endothelium have been shown to be key pathogenesis events. Recent studies linked neutrophil production of H2O2 to the mediation of neutrophil adhesion to the endothelia [108, 109]. It is known that elevated homocysteine levels in the blood correlate with the increased risk of cardiovascular diseases. The mechanism of this relationship appears to hinge upon ROS. It was discovered that homocysteine is a signal for ROS production in macrophages, which results in the production of monocyte chemoattractant protein-1 (MCP-1) [110]. In the model of airway irritation in rats, nicotine induced mRNA expression of the chemokine, macrophage inflammatory protein-1 alpha, an observation that is reversed in the presence of NAC [111]. Together, these studies suggest that, regardless of the model system, ROS appear to be a critical player in phagocyte chemoattraction and migration.
ROS in antigen cross-presentation
Recent studies have demonstrated a pivotal role for ROS in the context of antigen cross-presentation in DCs. Antigen cross-presentation is the process by which antigens taken up via phagocytosis are presented on MHCI molecules, in addition to the conventional MHCII presentation, and vice versa [112]. Upon uptake, antigens are partially degraded in the phagolysosomal pathway, transported into the cytosol, further degraded by the proteasome, and finally transited into the lumen of the ER for loading onto nascent MHCI molecules [113]. However, for proper MHCI or MHCII loading, peptides must, in general, be eight to nine amino acids long. Thus, a degree of control must be exerted over antigen degradation in order for the “right” amount of degradation to occur if antigen cross-presentation is to take place. There are a number of strategies that phagocytes use to degrade engulfed antigens, one of which is the NOX2 NADPH oxidase production of superoxides. The consequence of superoxides in a low pH environment, such as that present in a phagosome, is that the superoxides will readily react with the available H+ to make H2O2 and other ROS. This results in a rapid increase in pH. In neutrophils, the production of ROS is regulated to provide an “oxidative burst,” aimed at killing phagocytosed microbes with a high but temporary dose of ROS, thereby leaving the pH largely unchanged. However, DCs express a much lower level of proteolytic enzymes, and engulfed antigen persists longer than in macrophages [114]. A close examination of NOX2 NADPH oxidase activity in human DCs upon phagocytosis revealed that, while there is a tenfold lower level of NOX2 NADPH oxidase activity, it is more prolonged when compared with macrophages [115]. The consequence of the longer-lasting ROS production is a sustained alkalinization of the DC phagosome, as opposed to the rapid acidification of macrophage phagosomes. The increase in pH therefore leads to a consistently lower degree of proteolytic activity, thus attenuating the efficiency of antigen cross-presentation of phagocytosed particles. The evidence for this stems from the comparison of NOX2−/− DCs to WT DCs [116] and gp91phox deficient CGD patient DCs to those of the healthy controls [115]. NOX2−/− DCs of either mouse or human origin showed enhanced phagosomal acidification and antigen degradation, leading to a drastic decrease in antigen cross-presentation. Thus, ROS function as a mode of pH regulation to allow for antigen cross-presentation in DCs, making them important players in innate immunity and in the initiation of adaptive immunity.
ROS and the induction of autophagy
Autophagy is the controlled process of cellular self-digestion where cellular contents are delivered to the lysosome for degradation such that cellular homeostasis may be maintained [117]. Autophagy is characterized by the presence of autophagosomes, double membranous lamellar vesicles bearing the autophagy marker, microtubule-associated protein light chain 3 (LC3) (reviewed in [118]). LC3, or Atg8 in yeast, is a member of a family of over 30 autophagy-related (Atg) proteins that act sequentially to mediate the formation of autophagosomes. While the initial steps of autophagosome formation are currently under much debate, what is known is that the autophagic proteins recruit a crescent-shaped isolation membrane (the source of which is currently unclear) to elongate around and capture cytoplasmic cargo, be it damaged organelles, cytosol, macromolecules, or pathogens. Autophagy is subdivided into three types—chaperone-mediated autophagy, microautophagy, and macroautophagy (reviewed in [119]).
With respect to immunity, there has been a wealth of research demonstrating that macroautophagy, hereafter referred to as autophagy, is a key component of the innate immune defense against many pathogenic microorganisms by removing them from the cytosol, limiting their escape from phagosomes and promoting phagosome maturation (reviewed in [118, 120]). The survival and growth of a number of pathogens have been shown to be restricted by autophagy. Detection of the presence of streptolysin O, a virulence factor produced by Streptococcus pyogens, activates autophagy, resulting in rapid lysosomal degradation and clearance of the bacteria [121]. Salmonella enterica serovar Typhimurium resides in vacuoles inside infected host cells. These bacteria can, through the vacuole damaging ability of its type III secretion system, gain access to the cytoplasm. In the presence of the damaged vacuoles, autophagy is activated to limit intracellular spread and replication of the bacteria [122, 123]. Autophagy targeting and clearance of bacteria have likewise been shown to restrict the survival and growth of L. monocytogenes [124, 125], Francisella tulerensis [126], M. tuberculosis [127], Coxiella burnetii [128], and others. Furthermore, autophagy has been implicated in the clearance of viruses, such as parvovirus B19 [129], and parasites, such as Toxoplasma gondii [130].
A closer examination of the activation and role of autophagy and NOX2 NADPH oxidase in immunity reveals a striking similarity in many regards. Both NOX2 NADPH oxidase and autophagy are early antimicrobial events that occur upon pathogen phagocytosis [131, 132]. There are a number of common activating signals reported for them, including TLR activation [133–135], TNF-α [136, 137], and others. The function of autophagy and NOX2 NADPH oxidase is sensitive to class III PI3K inhibitors [18, 19, 138], and they both can lead to apoptosis [7, 139]. Furthermore, both autophagy and NOX2 NADPH oxidase genes have been implicated in the development of IBD and Crohn's Disease [140–142]. There is also evidence to link ROS to the induction of autophagy. It is known that mounting oxidative stress results in damaged organelles, proteins, and DNA, the clearance of which is one of the main functions of autophagy in a number of cell types [143–145] (reviewed in [146]). If proper clearance cannot take place, increasing ROS levels may then lead to autophagic cell death [147], as seen in the context of TNF-α-induced autophagic cell death in Ewing sarcoma cells [136] or LPS-induced autophagic macrophage cell death [148]. Thus, given the overlap of function and the relationship between ROS and autophagy, it is therefore logical to explore the role that ROS plays in the induction of autophagy in immunity.
The first direct link between ROS and autophagy was provided in the context of starvation-induced autophagy and mitochondrial-derived ROS [149–151]. Starvation-induced autophagy is the induction of autophagy to allow for the autodigestion of the cytoplasm in support of critical cellular processes in times of nutrient deprivation [119]. In response to starvation, it was found that a number of cell lines—HeLa, HEK293, CHO, and mouse embryonic fibroblasts (MEFs)—greatly induced both superoxide and H2O2 production in a class III PI3K-dependent manner when compared with controls. Furthermore, in MEFs with and without Atg5, an essential autophagy component, no difference in the ROS levels was observed [151]. These observations combined indicate that PI3K is somehow upstream of ROS production, leading to autophagy. ROS production was proposed to inhibit the redox-sensitive cysteine protease, Atg4—specifically the cysteine residue residing in the active site of the enzyme. Atg4 is one of the autophagy proteins involved in the regulation of LC3 phosphatidylethanolamine (PE) lipidation and delipidation [152], a post-translational event essential for autophagy to occur. When Atg4 encounters H2O2, Cys81 becomes oxidized in a reversible reaction, rendering the enzymatic activity null [151]. The lack of Atg4 activity is thought to allow LC3 to remain lipidated and localized to the autophagosomal membrane, leading to autophagy.
The link between mitochondrial ROS and starvation-induced autophagy was further confirmed by Chen and colleagues [153]. However, unlike the study done by Scherz-Shouval and colleagues, superoxides but not H2O2 were found to be the critical ROS member for the induction of autophagy in HEK293, human glioma cell line U87, and HeLa cells. By pharmacologically inhibiting or siRNA silencing SOD, which converts superoxide to H2O2, this study found that there was an increase in autophagy when compared with WT controls. Conversely, the overexpression of SOD resulted in a decrease in superoxide levels and an increase in H2O2, leading to decreased autophagosome formation.
More recently, in the context of muscular atrophy, it was found that transgenic mice with SOD deletion exhibit increased numbers of autophagosomes [154]. Since the superoxides are quickly converted to H2O2 and other reactive products, there are limitations in ROS assays that make it hard to distinguish one particular type from another. Furthermore, ROS products are linked together in a web of reactions (Fig. 2); thus, it becomes difficult to take away any one enzyme without affecting any other products in a compensatory manner. For example, by knocking out SOD, not only is the level of H2O2 affected but also the levels of HOCl, peroxynitrite, and OH•. The limitation of detection techniques available, coupled with the transient nature of ROS members, makes it challenging to pinpoint specific triggers of autophagy. Regardless of the type of ROS, however, it is clear that mitochondrial ROS are indeed involved in the activation of starvation-induced autophagy.
Further evidence has surfaced relating ROS to starvation-induced autophagy. One of the mechanisms of glycolysis downregulation is mediated through TP53-induced glycolysis and apoptosis regulator (TIGAR). An indirect consequence of TIGAR activity on glucose metabolism is the decrease in global intracellular ROS levels [155]. TIGAR activity was found to block autophagosome formation in the human osteosarcoma U2OS cells, even under starvation conditions [156]. This inhibition of autophagy was correlated with lowered ROS levels. Adding TIGAR exogenously results in an even greater decrease in ROS and autophagy. Thus, TIGAR is potentially expressed only under nutrient-rich conditions to inhibit autophagy activation via the inhibition of ROS.
NOX2 NADPH oxidase-derived ROS induction of antimicrobial autophagy
Unlike starvation-induced autophagy, the mechanism by which antimicrobial autophagy is activated is less clear. The first detection of invading pathogens occurs via the binding of pattern recognition receptors, such as TLRs, to their pathogen-associated molecular patterns, resulting in the activation of autophagy in both murine RAW 264.7 macrophage cell line and primary bone marrow-derived macrophages [135, 157, 158] (reviewed by [159]). Nucleotide-binding oligomerization domain (NOD) and NOD-like receptor signaling has also been shown to regulate autophagy [160, 161]. Upon the activation of TLR, FcγR, complement receptors, or others, phagocytosis of microbes is initiated, and autophagy is triggered [135, 162]. Seminal work done by Sanjuan and colleagues showed that the activation of TLR upon phagocytosis results in the recruitment of LC3 to the phagosome [135], forming a single membrane LC3+ phagosome. The induction of autophagy leads to phagosomal maturation and subsequent degradation of its contents.
Given that TLR activation leads to both NOX2 NADPH oxidase activation and autophagy, we hypothesized that ROS might also play a role in the activation of antimicrobial autophagy. To this end, we first examined if TLR- or FcγR-activated autophagy is mediated through ROS production. Using bone marrow-derived neutrophils (BMDN), we challenged cells with IgG-coated latex beads or dead yeast cells (zymosan particles), and found that LC3 was recruited to the phagosome [163]. This recruitment was further examined using either pharmacological agents to block/neutralize ROS or comparing WT with NOX2−/− BMDN with respect to LC3 recruitment upon stimulation. We found that, in the absence of NOX2 NADPH oxidase activity, LC3 recruitment to the phagosome was impaired (Fig. 3c, d). It is hypothesized that ROS recruits the assembly of the Atg5–Atg12–Atg16L1 complex, a core event in the autophagic pathway to allow for LC3II (the lipidated form of LC3) localization on the phagosomal membrane. An analysis of the purified phagosomes revealed enhanced localization of Atg5–Atg12 conjugate and LC3-II on IgG-coated latex bead containing phagosomes, and this recruitment was inhibited by the addition of DPI. Recently, another independent study confirmed some of our observations in human neutrophils, suggesting that NOX2 NADPH oxidase-derived ROS induction of autophagy is not a murine-only phenomenon [164].

ROS induction of autophagy. a Phagocytosis of microbes results in the recruitment of NOX2 NADPH oxidase complex to the phagosome. The presence of ROS signals the induction of autophagy through an unknown mechanism, resulting in the recruitment of autophagy protein, LC3, to the phagosome, where LC3 is then conjugated to PE. Subsequent fusion with the lysosome allows for microbial degradation in the autophagolysosome. b Upon nutrient starvation, ROS are generated in the mitochondria as a result of electron transport chain activity. Loss of mitochondrial integrity results in the detection of ROS, triggering autophagy. The damaged mitochondria and partial cytoplasm are then contained in an autophagosome that is likewise marked for lysosomal fusion and degradation. c Epifluorescence images of WT or d NOX2 NADPH oxidase deficient bone marrow-derived neutrophils (BMDN) fed with IgG-coated latex beads for 1 h in presence or absence of DPI. Cells were transfected with LC3-GFP and stained with LAMP-1 in red
We further established that other cell types (epithelial cells) also employ other member(s) of the NOX family for antimicrobial autophagy, suggesting that the NOX family may be a general mechanism of antimicrobial defense in a range of cell types. Using p22phox siRNA silencing in the human embryonic intestinal epithelial cells, Henle-407, we observed a reduction in autophagy targeting of Salmonella Typhimurium. The specific NOX family member(s) that mediate antimicrobial autophagy in non-phagocytic cells remain undetermined. Thus, ROS plays an important role in the induction of both starvation-induced (Fig. 3b) and antimicrobial autophagy (Fig. 3a).
Conclusion
The current understanding of NOX2 NADPH oxidase-derived ROS has evolved tremendously over the past few years to give a more holistic and complex picture of just how important ROS are to host immunity. Researchers now have a greater appreciation of ROS function beyond simply that of the antimicrobial agent but also as an integral player in immunity—participating in immune modulation, adaptive immune activation, intracellular signaling, chemoattraction, and the induction of autophagy (Fig. 4). Given the plethora of immunological roles that ROS are both directly and indirectly involved in, perhaps a new paradigm of thinking about immunological diseases and microbial pathogenesis in general is in order. Certainly, an analysis of the CGD population reveals that a number of immunological diseases are reported to be comorbidities in CGD patients; chiefly among them is IBD [165]. In a recent genome-wide association study examining the Crohn's disease patient population, Ncf4, the human gene encoding p40phox, was identified as a significant susceptibility gene [142]. Together, these observations suggest that NOX2 NADPH oxidase deficiency can be thought of as a syndrome of related diseases that are characterized by a distinct panel of immunological disorders resulting from the loss of NOX2 NADPH oxidase function. Therefore, further studies into the role of NOX2 NADPH oxidase in immunity would contribute greatly to the understanding of not only CGD but also a number of other diseases as well.

Summary of the known roles that ROS play in immunity
References
Rada B, Hably C, Meczner A, Timar C, Lakatos G, Enyedi P, Ligeti E (2008) Role of Nox2 in elimination of microorganisms. Semin Immunopathol 30:237–253
Segal AW (1996) The NADPH oxidase and chronic granulomatous disease. Mol Med Today 2:129–135
Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H (2000) Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79:155–169
Antonenkov VD, Grunau S, Ohlmeier S, Hiltunen JK (2010) Peroxisomes are oxidative organelles. Antioxid Redox Signal 13:525–537
Harrison R (2002) Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med 33:774–797
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Nauseef WM (2008) Biological roles for the NOX family NADPH oxidases. J Biol Chem 283:16961–16965
Santos CX, Tanaka LY, Wosniak J, Laurindo FR (2009) Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid Redox Signal 11:2409–2427
Nauseef WM (2004) Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122:277–291
DeLeo FR, Allen LA, Apicella M, Nauseef WM (1999) NADPH oxidase activation and assembly during phagocytosis. J Immunol 163:6732–6740
Ando S, Kaibuchi K, Sasaki T, Hiraoka K, Nishiyama T, Mizuno T, Asada M, Nunoi H, Matsuda I, Matsuura Y et al (1992) Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J Biol Chem 267:25709–25713
Mizuno T, Kaibuchi K, Ando S, Musha T, Hiraoka K, Takaishi K, Asada M, Nunoi H, Matsuda I, Takai Y (1992) Regulation of the superoxide-generating NADPH oxidase by a small GTP-binding protein and its stimulatory and inhibitory GDP/GTP exchange proteins. J Biol Chem 267:10215–10218
Diebold BA, Bokoch GM (2001) Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nat Immunol 2:211–215
Dusi S, Della Bianca V, Grzeskowiak M, Rossi F (1993) Relationship between phosphorylation and translocation to the plasma membrane of p47phox and p67phox and activation of the NADPH oxidase in normal and Ca(2+)-depleted human neutrophils. Biochem J 290(Pt 1):173–178
Lapouge K, Smith SJ, Groemping Y, Rittinger K (2002) Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem 277:10121–10128
Park JW, Benna JE, Scott KE, Christensen BL, Chanock SJ, Babior BM (1994) Isolation of a complex of respiratory burst oxidase components from resting neutrophil cytosol. Biochemistry 33:2907–2911
Zhao T, Benard V, Bohl BP, Bokoch GM (2003) The molecular basis for adhesion-mediated suppression of reactive oxygen species generation by human neutrophils. J Clin Invest 112:1732–1740
Ellson CD, Anderson KE, Morgan G, Chilvers ER, Lipp P, Stephens LR, Hawkins PT (2001) Phosphatidylinositol 3-phosphate is generated in phagosomal membranes. Curr Biol 11:1631–1635
Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, Thuring JW, Cooper MA, Lim ZY, Holmes AB, Gaffney PR, Coadwell J, Chilvers ER, Hawkins PT, Stephens LR (2001) PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40(phox). Nat Cell Biol 3:679–682
Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3:675–678
Honbou K, Minakami R, Yuzawa S, Takeya R, Suzuki NN, Kamakura S, Sumimoto H, Inagaki F (2007) Full-length p40phox structure suggests a basis for regulation mechanism of its membrane binding. EMBO J 26:1176–1186
Bissonnette SA, Glazier CM, Stewart MQ, Brown GE, Ellson CD, Yaffe MB (2008) Phosphatidylinositol 3-phosphate-dependent and -independent functions of p40phox in activation of the neutrophil NADPH oxidase. J Biol Chem 283:2108–2119
Bengis-Garber C, Gruener N (1996) Protein kinase A downregulates the phosphorylation of p47 phox in human neutrophils: a possible pathway for inhibition of the respiratory burst. Cell Signal 8:291–296
Fontayne A, Dang PM, Gougerot-Pocidalo MA, El-Benna J (2002) Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 41:7743–7750
Dang PM, Cross AR, Babior BM (2001) Assembly of the neutrophil respiratory burst oxidase: a direct interaction between p67PHOX and cytochrome b558. Proc Natl Acad Sci USA 98:3001–3005
Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J, Segal AW (2000) Protein kinase C-beta contributes to NADPH oxidase activation in neutrophils. Biochem J 347(Pt 1):285–289
Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, Subbulakshmi V, Feldman GM, Wientjes FB, Cathcart MK (2004) Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol 173:5730–5738
Dang PM, Fontayne A, Hakim J, El Benna J, Perianin A (2001) Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol 166:1206–1213
Kramer IM, van der Bend RL, Verhoeven AJ, Roos D (1988) The 47-kDa protein involved in the NADPH:O2 oxidoreductase activity of human neutrophils is phosphorylated by cyclic AMP-dependent protein kinase without induction of a respiratory burst. Biochim Biophys Acta 971:189–196
Martyn KD, Kim MJ, Quinn MT, Dinauer MC, Knaus UG (2005) p21-activated kinase (Pak) regulates NADPH oxidase activation in human neutrophils. Blood 106:3962–3969
Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J (2000) The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol 165:5238–5244
Dang PM, Morel F, Gougerot-Pocidalo MA, El Benna J (2003) Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochemistry 42:4520–4526
Chen Q, Powell DW, Rane MJ, Singh S, Butt W, Klein JB, McLeish KR (2003) Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J Immunol 170:5302–5308
Lehmann K, Muller JP, Schlott B, Skroblin P, Barz D, Norgauer J, Wetzker R (2009) PI3Kgamma controls oxidative bursts in neutrophils via interactions with PKCalpha and p47phox. Biochem J 419:603–610
Yamamori T, Inanami O, Nagahata H, Kuwabara M (2004) Phosphoinositide 3-kinase regulates the phosphorylation of NADPH oxidase component p47(phox) by controlling cPKC/PKCdelta but not Akt. Biochem Biophys Res Commun 316:720–730
Tian W, Li XJ, Stull ND, Ming W, Suh CI, Bissonnette SA, Yaffe MB, Grinstein S, Atkinson SJ, Dinauer MC (2008) Fc{gamma}R-stimulated activation of the NADPH oxidase: phosphoinositide-binding protein p40phox regulates NADPH oxidase activity after enzyme assembly on the phagosome. Blood 112:3867–3877
Carnevale R, Pignatelli P, Lenti L, Buchetti B, Sanguigni V, Di Santo S, Violi F (2007) LDL are oxidatively modified by platelets via GP91(phox) and accumulate in human monocytes. FASEB J 21:927–934
Lee SH, Park DW, Park SC, Park YK, Hong SY, Kim JR, Lee CH, Baek SH (2009) Calcium-independent phospholipase A2beta-Akt signaling is involved in lipopolysaccharide-induced NADPH oxidase 1 expression and foam cell formation. J Immunol 183:7497–7504
Park DW, Baek K, Kim JR, Lee JJ, Ryu SH, Chin BR, Baek SH (2009) Resveratrol inhibits foam cell formation via NADPH oxidase 1-mediated reactive oxygen species and monocyte chemotactic protein-1. Exp Mol Med 41:171–179
Lee CF, Qiao M, Schroder K, Zhao Q, Asmis R (2010) Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ Res 106:1489–1497
Sasaki H, Yamamoto H, Tominaga K, Masuda K, Kawai T, Teshima-Kondo S, Rokutan K (2009) NADPH oxidase-derived reactive oxygen species are essential for differentiation of a mouse macrophage cell line (RAW264.7) into osteoclasts. J Med Investig 56:33–41
Flannagan RS, Cosio G, Grinstein S (2009) Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7:355–366
Reeves EP, Nagl M, Godovac-Zimmermann J, Segal AW (2003) Reassessment of the microbicidal activity of reactive oxygen species and hypochlorous acid with reference to the phagocytic vacuole of the neutrophil granulocyte. J Med Microbiol 52:643–651
Klebanoff SJ (2005) Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625
Bartosz G (2009) Reactive oxygen species: destroyers or messengers? Biochem Pharmacol 77:1303–1315
Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4:181–189
Ramos CL, Pou S, Rosen GM (1995) Effect of anti-inflammatory drugs on myeloperoxidase-dependent hydroxyl radical generation by human neutrophils. Biochem Pharmacol 49:1079–1084
Cho SH, Lee CH, Ahn Y, Kim H, Kim H, Ahn CY, Yang KS, Lee SR (2004) Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett 560:7–13
Rhee SG, Bae YS, Lee SR, Kwon J (2000) Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci STKE 2000:pe1
Biswas S, Chida AS, Rahman I (2006) Redox modifications of protein-thiols: emerging roles in cell signaling. Biochem Pharmacol 71:551–564
Georgiou G (2002) How to flip the (redox) switch. Cell 111:607–610
Jiang Q, Griffin DA, Barofsky DF, Hurst JK (1997) Intraphagosomal chlorination dynamics and yields determined using unique fluorescent bacterial mimics. Chem Res Toxicol 10:1080–1089
Nauseef WM (2001) Contributions of myeloperoxidase to proinflammatory events: more than an antimicrobial system. Int J Hematol 74:125–133
Simpson DL, Berthold P, Taichman NS (1988) Killing of human myelomonocytic leukemia and lymphocytic cell lines by Actinobacillus actinomycetemcomitans leukotoxin. Infect Immun 56:1162–1166
Tsai CC, Taichman NS (1986) Dynamics of infection by leukotoxic strains of Actinobacillus actinomycetemcomitans in juvenile periodontitis. J Clin Periodontol 13:330–331
Korostoff J, Wang JF, Kieba I, Miller M, Shenker BJ, Lally ET (1998) Actinobacillus actinomycetemcomitans leukotoxin induces apoptosis in HL-60 cells. Infect Immun 66:4474–4483
Yamaguchi N, Kieba IR, Korostoff J, Howard PS, Shenker BJ, Lally ET (2001) Maintenance of oxidative phosphorylation protects cells from Actinobacillus actinomycetemcomitans leukotoxin-induced apoptosis. Cell Microbiol 3:811–823
Clark RA, Leidal KG, Taichman NS (1986) Oxidative inactivation of Actinobacillus actinomycetemcomitans leukotoxin by the neutrophil myeloperoxidase system. Infect Immun 53:252–256
Geoffroy C, Gilles AM, Alouf JE (1981) The sulfhydryl groups of the thiol-dependent cytolytic toxin from Bacillus alvei evidence for one essential sulfhydryl group. Biochem Biophys Res Commun 99:781–788
Hotze EM, Wilson-Kubalek EM, Rossjohn J, Parker MW, Johnson AE, Tweten RK (2001) Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane beta-sheet from a prepore intermediate. J Biol Chem 276:8261–8268
Clark RA (1986) Oxidative inactivation of pneumolysin by the myeloperoxidase system and stimulated human neutrophils. J Immunol 136:4617–4622
Lam GY, Brumell JH (2008) Cell biology: a Listeria escape trick. Nature 455:1186–1187
Portnoy DA, Jacks PS, Hinrichs DJ (1988) Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med 167:1459–1471
Schnupf P, Portnoy DA (2007) Listeriolysin O: a phagosome-specific lysin. Microbes Infect 9:1176–1187
Geoffroy C, Gaillard JL, Alouf JE, Berche P (1987) Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect Immun 55:1641–1646
Portnoy DA, Chakraborty T, Goebel W, Cossart P (1992) Molecular determinants of Listeria monocytogenes pathogenesis. Infect Immun 60:1263–1267
Singh R, Jamieson A, Cresswell P (2008) GILT is a critical host factor for Listeria monocytogenes infection. Nature 455:1244–1247
Rothfork JM, Timmins GS, Harris MN, Chen X, Lusis AJ, Otto M, Cheung AL, Gresham HD (2004) Inactivation of a bacterial virulence pheromone by phagocyte-derived oxidants: new role for the NADPH oxidase in host defense. Proc Natl Acad Sci USA 101:13867–13872
Ahluwalia J, Tinker A, Clapp LH, Duchen MR, Abramov AY, Pope S, Nobles M, Segal AW (2004) The large-conductance Ca2+-activated K+ channel is essential for innate immunity. Nature 427:853–858
Segal AW (2005) How neutrophils kill microbes. Annu Rev Immunol 23:197–223
Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291–297
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Urban CF, Reichard U, Brinkmann V, Zychlinsky A (2006) Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8:668–676
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241
Rutault K, Alderman C, Chain BM, Katz DR (1999) Reactive oxygen species activate human peripheral blood dendritic cells. Free Radic Biol Med 26:232–238
Kantengwa S, Jornot L, Devenoges C, Nicod LP (2003) Superoxide anions induce the maturation of human dendritic cells. Am J Respir Crit Care Med 167:431–437
Verhasselt V, Goldman M, Willems F (1998) Oxidative stress up-regulates IL-8 and TNF-alpha synthesis by human dendritic cells. Eur J Immunol 28:3886–3890
Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, DeLeo FR (2004) Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol 172:636–643
Cale CM, Jones AM, Goldblatt D (2000) Follow up of patients with chronic granulomatous disease diagnosed since 1990. Clin Exp Immunol 120:351–355
Foster CB, Lehrnbecher T, Mol F, Steinberg SM, Venzon DJ, Walsh TJ, Noack D, Rae J, Winkelstein JA, Curnutte JT, Chanock SJ (1998) Host defense molecule polymorphisms influence the risk for immune-mediated complications in chronic granulomatous disease. J Clin Invest 102:2146–2155
Gelderman KA, Hultqvist M, Pizzolla A, Zhao M, Nandakumar KS, Mattsson R, Holmdahl R (2007) Macrophages suppress T cell responses and arthritis development in mice by producing reactive oxygen species. J Clin Invest 117:3020–3028
Olofsson P, Holmberg J, Tordsson J, Lu S, Akerstrom B, Holmdahl R (2003) Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet 33:25–32
Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, Dinauer MC (1997) Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med 185:207–218
Blanchard TG, Yu F, Hsieh CL, Redline RW (2003) Severe inflammation and reduced bacteria load in murine Helicobacter infection caused by lack of phagocyte oxidase activity. J Infect Dis 187:1609–1615
Keenan JI, Peterson RA 2nd, Hampton MB (2005) NADPH oxidase involvement in the pathology of Helicobacter pylori infection. Free Radic Biol Med 38:1188–1196
Snelgrove RJ, Edwards L, Rae AJ, Hussell T (2006) An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol 36:1364–1373
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Oakley FD, Abbott D, Li Q, Engelhardt JF (2009) Signaling components of redox active endosomes: the redoxosomes. Antioxid Redox Signal 11:1313–1333
Bindoli A, Fukuto JM, Forman HJ (2008) Thiol chemistry in peroxidase catalysis and redox signaling. Antioxid Redox Signal 10:1549–1564
Djordjevic T, Pogrebniak A, BelAiba RS, Bonello S, Wotzlaw C, Acker H, Hess J, Gorlach A (2005) The expression of the NADPH oxidase subunit p22phox is regulated by a redox-sensitive pathway in endothelial cells. Free Radic Biol Med 38:616–630
Liu G, Pessah IN (1994) Molecular interaction between ryanodine receptor and glycoprotein triadin involves redox cycling of functionally important hyperreactive sulfhydryls. J Biol Chem 269:33028–33034
Favero TG, Zable AC, Abramson JJ (1995) Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J Biol Chem 270:25557–25563
Kawakami M, Okabe E (1998) Superoxide anion radical-triggered Ca2+ release from cardiac sarcoplasmic reticulum through ryanodine receptor Ca2+ channel. Mol Pharmacol 53:497–503
Suzuki YJ, Cleemann L, Abernethy DR, Morad M (1998) Glutathione is a cofactor for H2O2-mediated stimulation of Ca2+ induced Ca2+ release in cardiac myocytes. Free Radic Biol Med 24:318–325
Germano G, Sanguigni V, Pignatelli P, Caccese D, Lenti L, Ragazzo M, Lauro R, Violi F (2004) Enhanced platelet release of superoxide anion in systemic hypertension: role of AT1 receptors. J Hypertens 22:1151–1156
Hu Q, Yu ZX, Ferrans VJ, Takeda K, Irani K, Ziegelstein RC (2002) Critical role of NADPH oxidase-derived reactive oxygen species in generating Ca2+ oscillations in human aortic endothelial cells stimulated by histamine. J Biol Chem 277:32546–32551
Grinstein S, Klip A (1989) Calcium homeostasis and the activation of calcium channels in cells of the immune system. Bull NY Acad Med 65:69–79
Bubici C, Papa S, Dean K, Franzoso G (2006) Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene 25:6731–6748
Copple IM, Goldring CE, Jenkins RE, Chia AJ, Randle LE, Hayes JD, Kitteringham NR, Park BK (2008) The hepatotoxic metabolite of acetaminophen directly activates the Keap1-Nrf2 cell defense system. Hepatology 48:1292–1301
Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128:325–339
Liu B, Chen Y, St Clair DK (2008) ROS and p53: a versatile partnership. Free Radic Biol Med 44:1529–1535
Hattori H, Subramanian KK, Sakai J, Jia Y, Li Y, Porter TF, Loison F, Sarraj B, Kasorn A, Jo H, Blanchard C, Zirkle D, McDonald D, Pai SY, Serhan CN, Luo HR (2010) Small-molecule screen identifies reactive oxygen species as key regulators of neutrophil chemotaxis. Proc Natl Acad Sci U S A 107:3546–3551
Heo SK, Ju SA, Lee SC, Park SM, Choe SY, Kwon B, Kwon BS, Kim BS (2006) LIGHT enhances the bactericidal activity of human monocytes and neutrophils via HVEM. J Leukoc Biol 79:330–338
Heo SK, Yun HJ, Park WH, Park SD (2008) NADPH oxidase activation is required for migration by LIGHT in human monocytes. Biochem Biophys Res Commun 371:834–840
Lee HM, Shin DM, Kim KK, Lee JS, Paik TH, Jo EK (2009) Roles of reactive oxygen species in CXCL8 and CCL2 expression in response to the 30-kDa antigen of Mycobacterium tuberculosis. J Clin Immunol 29:46–56
Kim SY, Lee JG, Cho WS, Cho KH, Sakong J, Kim JR, Chin BR, Baek SH (2010) Role of NADPH oxidase-2 in lipopolysaccharide-induced matrix metalloproteinase expression and cell migration. Immunol Cell Biol 88:197–204
Gianni D, Diaz B, Taulet N, Fowler B, Courtneidge SA, Bokoch GM (2009) Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Sci Signal 2:ra54
Lee VM, Quinn PA, Jennings SC, Ng LL (2003) NADPH oxidase activity in preeclampsia with immortalized lymphoblasts used as models. Hypertension 41:925–931
Tsukimori K, Komatsu H, Fukushima K, Kaku T, Nakano H, Wake N (2008) Inhibition of nitric oxide synthetase at mid-gestation in rats is associated with increases in arterial pressure, serum tumor necrosis factor-alpha, and placental apoptosis. Am J Hypertens 21:477–481
Zeng X, Dai J, Remick DG, Wang X (2003) Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ Res 93:311–320
Chong IW, Lin SR, Hwang JJ, Huang MS, Wang TH, Hung JY, Paulauskis JD (2002) Expression and regulation of the macrophage inflammatory protein-1 alpha gene by nicotine in rat alveolar macrophages. Eur Cytokine Netw 13:242–249
Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S (2003) ER-phagosome fusion defines an MHC class I cross-presentation compartment in dendritic cells. Nature 425:397–402
Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA (2005) Mechanisms of MHC class I-restricted antigen processing and cross-presentation. Immunol Rev 207:145–157
Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES (2005) Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307:1630–1634
Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, Amigorena S (2008) NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 112:4712–4722
Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126:205–218
Levine B, Deretic V (2007) Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 7:767–777
Hussey S, Travassos LH, Jones NL (2009) Autophagy as an emerging dimension to adaptive and innate immunity. Semin Immunol 21:233–241
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Deretic V, Levine B (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549
Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T (2004) Autophagy defends cells against invading group A Streptococcus. Science 306:1037–1040
Birmingham CL, Brumell JH (2006) Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2:156–158
Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH (2006) Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem 281:11374–11383
Birmingham CL, Canadien V, Gouin E, Troy EB, Yoshimori T, Cossart P, Higgins DE, Brumell JH (2007) Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3:442–451
Py BF, Lipinski MM, Yuan J (2007) Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3:117–125
Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J (2006) Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci USA 103:14578–14583
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V (2004) Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766
Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI (2007) The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 9:891–909
Nakashima A, Tanaka N, Tamai K, Kyuuma M, Ishikawa Y, Sato H, Yoshimori T, Saito S, Sugamura K (2006) Survival of parvovirus B19-infected cells by cellular autophagy. Virology 349:254–263
Wang Y, Weiss LM, Orlofsky A (2009) Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J Biol Chem 284:1694–1701
Minakami R, Sumimotoa H (2006) Phagocytosis-coupled activation of the superoxide-producing phagocyte oxidase, a member of the NADPH oxidase (nox) family. Int J Hematol 84:193–198
Sanjuan MA, Green DR (2008) Eating for good health: linking autophagy and phagocytosis in host defense. Autophagy 4:607–611
Quinn MT, Gauss KA (2004) Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukoc Biol 76:760–781
Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C (2005) Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of gram-negative bacteria. J Immunol 175:5596–5600
Sanjuan MA, Milasta S, Green DR (2009) Toll-like receptor signaling in the lysosomal pathways. Immunol Rev 227:203–220
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P (2006) NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 281:30373–30382
Yazdanpanah B, Wiegmann K, Tchikov V, Krut O, Pongratz C, Schramm M, Kleinridders A, Wunderlich T, Kashkar H, Utermohlen O, Bruning JC, Schutze S, Kronke M (2009) Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460:1159–1163
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ (1997) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243:240–246
Kroemer G, Jaattela M (2005) Lysosomes and autophagy in cell death control. Nat Rev Cancer 5:886–897
Cheng JF, Ning YJ, Zhang W, Lu ZH, Lin L (2010) T300A polymorphism of ATG16L1 and susceptibility to inflammatory bowel diseases: a meta-analysis. World J Gastroenterol 16:1258–1266
Hausmann M, Spottl T, Andus T, Rothe G, Falk W, Scholmerich J, Herfarth H, Rogler G (2001) Subtractive screening reveals up-regulation of NADPH oxidase expression in Crohn's disease intestinal macrophages. Clin Exp Immunol 125:48–55
Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR (2008) Confirmation of association of IRGM and NCF4 with ileal Crohn's disease in a population-based cohort. Genes Immun 9:561–565
Kaushik S, Cuervo AM (2006) Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med 27:444–454
Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8:3–5
Xiong Y, Contento AL, Nguyen PQ, Bassham DC (2007) Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol 143:291–299
Kiffin R, Bandyopadhyay U, Cuervo AM (2006) Oxidative stress and autophagy. Antioxid Redox Signal 8:152–162
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB (2008) Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 15:171–182
Xu Y, Kim SO, Li Y, Han J (2006) Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem 281:19179–19187
Scherz-Shouval R, Elazar Z (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17:422–427
Scherz-Shouval R, Shvets E, Elazar Z (2007) Oxidation as a post-translational modification that regulates autophagy. Autophagy 3:371–373
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26:1749–1760
Nakatogawa H, Ichimura Y, Ohsumi Y (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130:165–178
Chen Y, Azad MB, Gibson SB (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 16:1040–1052
Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, Rosenthal N, Molinaro M, Protasi F, Fano G, Sandri M, Musaro A (2008) Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 8:425–436
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126:107–120
Bensaad K, Cheung EC, Vousden KH (2009) Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28:3015–3026
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V (2008) Toll-like receptors control autophagy. EMBO J 27:1110–1121
Xu Y, Liu XD, Gong X, Eissa NT (2008) Signaling pathway of autophagy associated with innate immunity. Autophagy 4:110–112
Delgado MA, Deretic V (2009) Toll-like receptors in control of immunological autophagy. Cell Death Differ 16:976–983
Suzuki T, Nunez G (2008) A role for Nod-like receptors in autophagy induced by Shigella infection. Autophagy 4:73–75
Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11:55–62
Park JB (2003) Phagocytosis induces superoxide formation and apoptosis in macrophages. Exp Mol Med 35:325–335
Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH (2009) Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 106:6226–6231
Mitroulis I, Kourtzelis I, Kambas K, Rafail S, Chrysanthopoulou A, Speletas M, Ritis K (2010) Regulation of the autophagic machinery in human neutrophils. Eur J Immunol 40:1461–1472
Holland SM (2010) Chronic granulomatous disease. Clin Rev Allergy Immunol 38:3–10
Garcia-Garcia JC, Rennoll-Bankert KE, Pelly S, Milstone AM, Dumler JS (2009) Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect Immun 77:2385–2391
Keith KE, Hynes DW, Sholdice JE, Valvano MA (2009) Delayed association of the NADPH oxidase complex with macrophage vacuoles containing the opportunistic pathogen Burkholderia cenocepacia. Microbiology 155:1004–1015
Boncompain G, Schneider B, Delevoye C, Kellermann O, Dautry-Varsat A, Subtil A (2010) Production of reactive oxygen species is turned on and rapidly shut down in epithelial cells infected with Chlamydia trachomatis. Infect Immun 78:80–87
Siemsen DW, Kirpotina LN, Jutila MA, Quinn MT (2009) Inhibition of the human neutrophil NADPH oxidase by Coxiella burnetii. Microbes Infect 11:671–679
McCaffrey RL, Allen LA (2006) Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol 80:1224–1230
Schulert GS, McCaffrey RL, Buchan BW, Lindemann SR, Hollenback C, Jones BD, Allen LA (2009) Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect Immun 77:1324–1336
Buchan BW, McCaffrey RL, Lindemann SR, Allen LA, Jones BD (2009) Identification of migR, a regulatory element of the Francisella tularensis live vaccine strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect Immun 77:2517–2529
Mohapatra NP, Soni S, Rajaram MV, Dang PM, Reilly TJ, El-Benna J, Clay CD, Schlesinger LS, Gunn JS (2010) Francisella acid phosphatases inactivate the NADPH oxidase in human phagocytes. J Immunol 184:5141–5150
Allen LA, McCaffrey RL (2007) To activate or not to activate: distinct strategies used by Helicobacter pylori and Francisella tularensis to modulate the NADPH oxidase and survive in human neutrophils. Immunol Rev 219:103–117
Harada T, Miyake M, Imai Y (2007) Evasion of Legionella pneumophila from the bactericidal system by reactive oxygen species (ROS) in macrophages. Microbiol Immunol 51:1161–1170
Lodge R, Diallo TO, Descoteaux A (2006) Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cell Microbiol 8:1922–1931
Descoteaux A, Matlashewski G, Turco SJ (1992) Inhibition of macrophage protein kinase C-mediated protein phosphorylation by Leishmania donovani lipophosphoglycan. J Immunol 149:3008–3015
Vazquez-Torres A, Fang FC (2001) Salmonella evasion of the NADPH phagocyte oxidase. Microbes Infect 3:1313–1320
Vazquez-Torres A, Fantuzzi G, Edwards CK 3rd, Dinarello CA, Fang FC (2001) Defective localization of the NADPH phagocyte oxidase to Salmonella-containing phagosomes in tumor necrosis factor p55 receptor-deficient macrophages. Proc Natl Acad Sci USA 98:2561–2565
Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, Fang FC (2000) Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science 287:1655–1658
Chung KJ, Cho EJ, Kim MK, Kim YR, Kim SH, Yang HY, Chung KC, Lee SE, Rhee JH, Choy HE, Lee TH (2010) RtxA1-induced expression of the small GTPase Rac2 plays a key role in the pathogenicity of Vibrio vulnificus. J Infect Dis 201:97–105
Hartland EL, Green SP, Phillips WA, Robins-Browne RM (1994) Essential role of YopD in inhibition of the respiratory burst of macrophages by Yersinia enterocolitica. Infect Immun 62:4445–4453
Aguirre-Garcia MM, Okhuysen PC (2007) Cryptosporidium parvum: identification and characterization of an acid phosphatase. Parasitol Res 101:85–89
Gruhne B, Sompallae R, Marescotti D, Kamranvar SA, Gastaldello S, Masucci MG (2009) The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc Natl Acad Sci USA 106:2313–2318
Bureau C, Bernad J, Chaouche N, Orfila C, Beraud M, Gonindard C, Alric L, Vinel JP, Pipy B (2001) Nonstructural 3 protein of hepatitis C virus triggers an oxidative burst in human monocytes via activation of NADPH oxidase. J Biol Chem 276:23077–23083
Thoren F, Romero A, Lindh M, Dahlgren C, Hellstrand K (2004) A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J Leukoc Biol 76:1180–1186
Salmen S, Colmenares M, Peterson DL, Reyes E, Rosales JD, Berrueta L (2010) HIV-1 Nef associates with p22-phox, a component of the NADPH oxidase protein complex. Cell Immunol 263:166–171
Vilhardt F, Plastre O, Sawada M, Suzuki K, Wiznerowicz M, Kiyokawa E, Trono D, Krause KH (2002) The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J Biol Chem 277:42136–42143
Kaul P, Biagioli MC, Singh I, Turner RB (2000) Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J Infect Dis 181:1885–1890
Acknowledgements
The authors would like to thank Michelle Ang and Michal Bohdanowicz for their technical assistance in the generation of the figures. The authors would also like to thank Veronica Canadien for the use of her images in Figure 3c-d. John H. Brumell, PhD, holds an Investigators in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. Infrastructure for the Brumell Laboratory was provided by a New Opportunities Fund from the Canadian Foundation for Innovation and the Ontario Innovation Trust. G.Y.L. is supported by a M.D/Ph.D Studentship and Canadian Graduate Scholarship Doctoral Research Award from the Canadian Institutes of Health Research. J.H. holds a Canadian Association of Gastroenterology/Canadian Institutes of Health Research/Crohn's and Colitis Foundation of Canada postdoctoral fellowship administered by the Canadian Association of Gastroenterolgy.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Autophagy.
Rights and permissions
About this article
Cite this article
Lam, G.Y., Huang, J. & Brumell, J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol 32, 415–430 (2010). https://doi.org/10.1007/s00281-010-0221-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-010-0221-0