
Abstract
NADPH oxidase of the phagocytic cells (Nox2) transfers electrons from cytosolic NADPH to molecular oxygen in the extracellular or intraphagosomal space. The produced superoxide anion (O2 ·−) provides the source for formation of all toxic oxygen derivatives, but continuous O2 ·− generation depends on adequate charge compensation. The vital role of Nox2 in efficient elimination of microorganisms is clearly indicated by human pathology as insufficient activity of the enzyme results in severe, recurrent bacterial infections, the typical symptoms of chronic granulomatous disease. The goals of this contribution are to provide critical review of the Nox2-dependent cellular processes that potentially contribute to bacterial killing and degradation and to indicate possible targets of pharmacological interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neutrophilic granulocytes (or PMN, for polymorphonuclear cells) play a key role in the antimicrobial defense of the body. These cells are able to detect, pursue, engulf, kill, and degrade various microbes. Final elimination of potentially pathogenic microorganisms is a complex process that requires directed migration, phagocytosis, production of toxic oxygen (and nitrogen) metabolites, and release of antibacterial proteins and enzymes stored in the different granule types. Human pathology indicates that proper functioning of all of these processes is vital as a deficiency in integrins or production of toxic oxygen metabolites or secondary granule proteins result in similar symptoms: recurrent, severe, often life-threatening infections [53]. Thus, elimination of microorganisms can not be ascribed to any single function or protein; it should rather be regarded as a joint action of several, equally important mechanisms.
The active NADPH oxidase enzyme of phagocytic cells consists of the transmembrane electron carrier (gp91 or Nox2) and four other subunits (p22, p47, p67 and Rac) that stabilize and activate Nox2. Only the properly assembled complex is able to transfer electrons across the membrane from cytosolic NADPH to molecular oxygen forming thereby superoxide (O2 ·−) either in the extracellular or in the intraphagosomal space. In the present review, we refer to the enzymatically active protein complex as Nox2. The discovery that chronic granulomatous disease (CGD) is the consequence of a genetic alteration in any one of the five essential subunits of Nox2 indicates the vital function of the enzyme. There is, however, controversy as to which “product” of the enzymatic activity has essential function.
The chemical product of Nox2 activity is O2 ·−. There is general agreement that O2 ·− itself is not too toxic but several of its derivatives are [44]. On the other hand, the transmembrane movement of electrons creates an electric potential that serves as driving force for secondary ion movements. It has been proposed that ion movements initiated by the electrogenic nature of Nox2 may contribute to the killing process [40, 120]. A new paradigm, developed in the recent years, suggests that the major—perhaps only—role of Nox2 is to provide driving force for ion (mainly K+ and Cl−) movements in and out of the phagosome creating thereby optimal conditions for the activity of granule enzymes [10, 110, 121, 123].
The purpose of this review is to summarize the experimental data on the role of Nox2 in the elimination of microorganisms. As the basic function of other Nox enzymes is also the transmembrane transport of electrons, most ideas are relevant to the entire Nox/Duox family of proteins.
Toxic effects of oxygen metabolites
The term “reactive oxygen species” (ROS) includes chemically reactive radicals and nonradical derivatives of oxygen. Radicals harbor one or more unpaired electrons, whereas nonradicals do not contain unpaired electrons. All oxygen radicals are ROS, but not all ROS are oxygen radicals [44]. Long before superoxide production by phagocytes has been discovered, the in vitro toxicity of hydrogen peroxide had been already known. Early reports suggested H2O2 to be the bactericidal agent in the antibacterial effect of notatin, a toxin from Penicillium notatum [25, 76]. The antibacterial effect of the xanthine oxidase system has been also shown [42]. But, the physiological importance of superoxide and ROS derived from superoxide was not recognized until phagocytosing polymorphonuclear leukocytes had been shown to produce superoxide anions [6]. Phagocytosing neutrophils undergo an abrupt increase in oxygen uptake, which was first called a “respiratory burst” but is not caused by mitochondrial oxidative metabolism. The phagocyte NADPH oxidase produces exclusively superoxide anions, which are the precursors of more distant ROS (H2O2, HOCl, 1O2, OH.). ROS react with a wide range of biocompounds, including nucleic acids, proteins, lipids, carbohydrates and molecules of other structure.
Superoxide and hydrogen peroxide
Superoxide itself is only weakly reactive and reacts with a few macromolecules, and its direct toxicity is still controversial [118]. Superoxide is unstable, cannot reach very far from the site of its production, and is not able to cross lipid bilayers. Its reactivity, however, can be increased in a hydrophobic environment (e.g., in the proximity of the plasma membrane) or by lowered pH values since its protonated derivative (HO2) is more reactive [72] and has been shown to penetrate into Escherichia coli and inactivate fumarase [79]. A toxic role of superoxide was shown in a few studies where in vitro killing of bacteria was achieved by addition of KO2 or inhibited by superoxide dismutase (SOD; Table 1). However, it is a challenging task to prove that superoxide anions kill directly since superoxide spontaneously dismutates into hydrogen peroxide and two H2O2 molecules are able to produce distal ROS, for example highly reactive OH.. Inhibition of killing by SOD shows only the requirement of superoxide but does not reveal if superoxide kills directly or through distal derivatives. Two of these studies failed to show a clear direct role of superoxide in killing since they also showed complete or partial inhibition of killing by the H2O2 scavenger catalase and the OH. scavenger mannitol [5, 114].
Hydrogen peroxide (H2O2) is formed by the spontaneous or SOD-catalyzed dismutation of superoxide. Two superoxide anions will produce one hydrogen peroxide molecule by incorporating two hydrogen ions and releasing one molecule of oxygen (Fig. 1, reaction no. 1). H2O2 is a well-known oxidizing agent capable of reacting with a wide range of macromolecules. Hydrogen peroxide is relatively stable and membrane permeable, so it can diffuse away from the site of its formation. Early works have shown already that hydrogen peroxide is toxic to a broad spectrum of microorganisms in vitro (Table 2). These studies differed both in the source of hydrogen peroxide and in the target bacterium used. Hydrogen peroxide can be added directly to the assay medium, can be produced directly by the enzymatic reaction of glucose by glucose oxidase (GO), or can be dismutated from superoxide created by xanthine oxidase (XO) and its adequate substrate (purine, hypoxanthine, xanthine, or acetaldehyde).

Reactive oxygen species formed by phagocytes. The figure shows the chemical formulae of radical and nonradical ROS and the stoichiometric equations of chemical reactions leading to their formation
More distal derivatives
Although hydrogen peroxide is reactive, its toxicity can be increased dramatically by forming further derivatives. One of the most abundant neutrophil proteins is myeloperoxidase (MPO), which is expressed only in polymorphonuclear leukocytes, monocytes, and certain types of macrophages in the human body. MPO resides in the primary granules of neutrophils and is released into the phagosome upon engulfment of bacteria. MPO catalyzes the oxidation of different substrates by hydrogen peroxide. The most common substrates are halides: chloride, bromide, fluoride, or iodide; MPO catalyzes their oxidation by hydrogen peroxide into hypohalous acids. The halides by themselves and the peroxidase on its own are not or only slightly antimicrobial, whereas the oxidized products are highly reactive and kill microbes easily [72]. The MPO/H2O2/I− system has been extensively characterized in vitro [70]. E. coli were killed in an MPO-, hydrogen peroxide-, and halide-dependent manner. Iodination or chlorination of bacteria is inversely proportional to their survival meaning that killing must occur—at least in part—by reacting with hypoiodous/hypochlorous acid [20, 70]. Several studies proved the in vitro toxicity of the MPO/H2O2/halide system [51, 71, 90, 111] or HOCl itself [20, 45, 51, 87, 111]. The phagocytic NADPH oxidase is required for the iodination of bacteria since CGD neutrophils fail to do so [73]. Although iodide is the preferred substrate for MPO among the halides [71], due to high concentrations of chloride in the phagosome, hypochlorous acid is formed in the highest amounts. Hypochlorous acid is a strong oxidizing agent, it attacks a broad range of biologically relevant compounds, but the preferred targets are: thiols, thioesters, amines, phenols, unsaturated bonds; it is membrane permeable [45].
Although formation of hydroxyl radical (OH.), a very reactive radical occurs in vitro from hydrogen peroxide catalyzed by iron (Fig. 1 reaction nos. 2–4), its contribution to killing remains doubtful since the iron released into the phagosome by either the bacteria or the neutrophils is bound to lactoferrin [15], and studies using electron paramagnetic resonance failed to detect hydroxyl radical formation in neutrophils following engulfment of iron-rich bacteria [24].
Singlet oxygen is an electronically excited state of molecular oxygen. There are many pathways suggested to form singlet oxygen but the best established mechanism is the one by H2O2 and HOCl [115] (Fig. 1 reaction no. 7). Singlet oxygen is a highly reactive and short-lived radical attacking a wide range of biomolecules. Although singlet oxygen has been suggested to be present in neutrophils’ phagosome [2, 129], the contribution of singlet oxygen to killing in neutrophil phagosome is still difficult to interpret due to imperfect specificity of singlet oxygen scavengers [23].
The production of ozone, a highly reactive ROS, by IgG antibodies opsonizing bacteria has been suggested in phagocytosing neutrophils [7, 137], but doubts have been raised by others on the specificity of the probes used for ozone detection in those studies [68, 69].
Human neutrophils do not produce large amounts of nitric oxide (NO); they only do so when primed with a certain cytokine cocktail [138]. NO can react with superoxide to form peroxynitrite (ONOO−; Fig. 1, reaction no. 8), which can be protonated (peroxynitrous acid) at physiological pH and split into two radicals, hydroxyl radical and nitric dioxide (Fig. 1, reaction no. 9). The reaction between peroxynitrite and carbon dioxide results in nitric dioxide and carbonate, two reactive radicals (Fig. 1, reaction no. 10). It has been shown that the latter species together can destroy molecules of biological origin and kill microbes [3, 98]. Nitrogen oxide and its oxygen-containing derivatives can be classified as both ROS and reactive nitrogen species.
ROS and myeloperoxidase: pro and contra
The efficiency of the MPO/H2O2/Cl− system to kill microbes of different origins has been shown in vitro without any doubt, but concerns have been raised if these experimental settings apply to the neutrophil phagosome as well.
Contra
These reconstituted in vitro systems used much lower levels of MPO than the estimated concentration in the phagosome; they were conducted at acidic pH (instead of the close-to-neutral measured in the phagosome) and did not contain high amounts of granule proteins present under physiological conditions [111]. It has been proposed that the function of Nox2 in the neutrophil phagosome is to liberate and activate bound granule proteins (which would be responsible after all for killing), and ROS produced are only byproducts of the reaction, which need to be eliminated in order to protect the neutrophil itself [121]. High levels of MPO indeed protect Staphylococcus aureus from being killed by the MPO/H2O2/Cl− system in vitro [111]. The activity of MPO at the reported pH in the phagosome (7.0–8.0) is also very low compared to the optimal levels at pH 5.0–5.5 [70, 111]. These findings might raise the possibility that MPO helps to control the robust levels of ROS in the phagosome besides converting hydrogen peroxide into HOCl. Most of the targets of the MPO-mediated chlorination in the phagosome are indeed neutrophil and not bacterial proteins [20, 111].
Pro
On the other hand, many arguments support the importance of ROS and MPO as toxic agents [74]. Two studies have concluded that enough HOCl is produced in the phagosome of neutrophils to kill engulfed bacteria [45, 58]. Chlorination of neutrophil proteins leads to the formation of chloramines, which are still highly reactive and capable of reacting with microbes. It is also questionable if neutrophils do need to be protected against ROS since the cells die anyhow soon after the initiation of phagocytosis. Helicobacter pylori incapable of deoxyribonucleic acid (DNA) damage repair is sensitive to ROS-mediated killing [101]. OxyR-deficient E. coli are killed by neutrophils more efficiently than wild-type bacteria [128]. S. aureus capable of synthesizing antioxidant carotenoids has increased resistance to neutrophil killing [85]. Neutrophil cytoplasts (neutroplasts) do not contain nuclei and lost most of their granules (MPO too) but harbor an intact respiratory burst [78, 113]. Neutroplasts engulf but do not kill S. aureus unless the microbes are coated with MPO, which suggests that under these conditions, the respiratory burst by itself is not sufficient to kill the bacterium, but completed with MPO, it is [100].
The exact mechanism of oxidative killing of microbes in neutrophils and its players (NADPH oxidase, MPO, granule enzymes) is still a subject of debate. From an evolutionary point of view, the ROS-producing phagocytic NADPH oxidase is far too complex and dangerous if its primary purpose is simply to create membrane potential changes across the phagosomal membrane. Since CGD neutrophils can kill most bacteria as efficient as normal neutrophils and the list of pathogens in CGD patients is limited to only a few characteristic bacteria and fungi, most probably both ROS and the oxidase-induced membrane potential changes are very important but only two of the numerous elements of neutrophils’ weaponry.
Nox2: an electrogenic transporter
Transfer of electrons via Nox2 results in net flux of negative charges across the membrane. This electron current can be measured by the patch clamp technique [120], and it leads to the change of the membrane potential. Resting PMNs have a membrane potential of about −60 mV that is quickly raised to approximately +60 mV or around 0 mV upon activation by PMA or the chemotactic agent formyl-methionyl-leucyl-phenylalanine (fMLP), respectively [56]. The electron current was shown to be dependent on the transmembrane potential difference and to cease when the membrane potential reached +160 to +200 mV [31, 104]. Taking into account the rate of O2 ·− generation and the membrane capacitance, it was calculated that the enzymatic activity of Nox2 would shut down in approximately 250 ms in neutrophils and in less than 20 ms in eosinophils [27, 31]. Evidently, the duration of the oxidative burst is significantly longer, whether activation occurs by soluble or particulate agents [30]. Thus, the critical factor in maintaining O2 ·− production is adequate charge compensation.
Theoretically, compensation of the electron flux via Nox2 could occur by moving a cation in the same or an anion in the opposite direction or a combination of both processes (Fig. 2a). In the following paragraphs, we summarize the experimental data on movements of H+, K+, and Cl− as the most probable ions for compensation of electron flux in PMN.

Possibilities of charge compensation for electron transfer via Nox2. a summarizes the theoretical possibilities, whereas b shows the role of H+ movements. Bacterium engulfed in the phagosome and release of protein content of different granules are represented with symbolic figures
Charge compensation by H+ efflux
H+ efflux via electrogenic pathway(s) as charge compensation for O2 ·− production has been proposed more than 20 years ago [49]. Movement of protons parallel to electrons would avoid both strong acidification of the cytoplasm due to liberation of H+ from NADPH and excessive alkalinization of the phagosomal space due to dismutation of O2 ·− ions (Fig. 2b).
Indeed, electrogenic H+ transport has been detected and characterized in PMN both by chemical (following pH changes by fluorescent dyes) and electrophysiological (patch clamp) techniques ([29, 32, 66], reviewed in [27]). Although the molecular identity of the presumed H+ channels has not yet been decided [27, 109, 117], there is general agreement that under resting conditions, H+ conductance is very low. Several factors have been described that increase the opening probability and hence the H+ current (Table 3). All these changes take place upon activation of PMN either by soluble or by particulate stimuli. In accordance with the concerted action of various factors upon the H+ channels, in activated cells, the membrane potential was shown to correspond in a wide range to the equilibrium (Nernst) potential for H+. This finding clearly indicates that in the activated state, H+ conductance largely exceeds the conductance of any other ion [8].
However, there is no perfect synchronization between initiation of electron and H+ flux as most of the factors leading to activation of H+ channels (see Table 3) are consequences of Nox2 turnover. This is evident in changes of the plasma membrane potential: There is a rapid and drastic depolarization upon activation of PMN. After approximately 1 min, the membrane potential stabilizes or begins to reverse depending on the duration of the respiratory burst (see Fig. 3 of [41]). The fact that under identical conditions no change of the membrane potential can be detected in PMN from CGD patients [41] supports that depolarization of normal PMN is due to Nox2 activity. According to our quantitative analysis, there is a highly nonlinear relationship between the rate of O2 ·− production and extent of depolarization [107]: 50% of the maximal depolarization occurs when Nox2 turns only at 2% of its maximal activity. The membrane potential stabilizes when the enzyme activity reaches 10–20% of its maximal activity. Our experimental data correspond very well to the results of model calculations [95].

Effect of valinomycin on the membrane potential of PMN. Membrane potential changes were followed by the potential sensitive fluorescent dye di-O-C5 as described in [107]. Where indicated, valinomycin was applied in a concentration of 3 μg/ml; PMA at 100 nM, and fMLP at 1 μM. An increase in relative fluorescence (RFU) indicates hyperpolarization and a decrease in RFU signifies depolarization of the plasma membrane. Result of one representative experiment out of three similar ones is shown
One study reported two-phase changes also in the intraphagosomal pH: Upon stimulation with PMA, an initial alkalinization was followed by acidification after approximately 2 min [122]. However, these observations could not be reproduced in another study [57].
The above data suggest that charge compensation is insufficient in the initial period or at a very low rate of O2 ·− production, and in this phase, the major compensating ion is probably not H+. Stabilization of the membrane potential is reached when a new steady state is established, where electron transport is fully compensated by the movement of other ions. The fact that manipulation of either the H+ permeability or the driving force for H+ ions results in significant alteration of both the membrane potential and the rate of O2 ·− production (see Table 4) supports that in the steady state, the major compensating ion is H+.
Charge compensation by K+
Recently K+ has been proposed as a cation responsible for partial compensation of the negative charges moved via Nox2 [1, 110]. In the same publications K+ efflux from the cytosol of neutrophils has been interpreted as the key element of the killing process, and blocking of K+ movement was suggested to prevent killing of microorganisms completely [1].
In the following paragraphs we list arguments which support or question the hypothesis on the central role of K + movements in microbial killing.
Data and considerations supporting the role of K+ movements in bacterial killing
Most experiments concluded that the membrane potential of resting neutrophils is around −60 mV (for details, see [27]). This value is fairly close to the equilibrium potential calculated for K+ ions (−89 mV; [96]) indicating that in the resting condition, K+ conductance dominates. Earlier reports estimated the ratio of Na+ to K+ permeability in resting human PMN to be 0.1 [126]. It is thus reasonable to suggest that in the initial phase of O2 ·− generation, K+ efflux could represent a significant component of charge compensation.
Indeed, K+ or 86Rb+ efflux has been measured upon activation of Nox2 [107, 110], and it is significantly enhanced by Zn2+ and Cd2+, known inhibitors of the H+ channels [1]. In our quantitative analysis, we found that a majority of K+ movement takes place at low enzyme activity but gradually declines as depolarization progresses and H+ channel activation increases [107]. This finding is in good agreement with earlier observations showing that H+ conductance is dominant in the fully activated state [8].
In the case of maximal Nox2 activity, approximately 6% of the total charge movement was suggested to be compensated by K+ ions [121]. However, our data indicated that at the lowest measurable activity of Nox2 (2% of the maximal activity), 50% of the transported negative charge could be compensated by K+ ions [106]. Thus, at low enzyme activity, K+ movement may be critical. The major question is whether this K+ flux has any relevance to the killing process.
In our experiments, we observed two phenomena that may indicate a role of K+ movement in the elimination of bacteria. (1) In the presence of 10 μM Zn2+, no increase of bacterial survival was observed in spite of a 30% reduction in O2 ·− generation [106]. Under these conditions, H+ efflux is impaired resulting in decreased activity of Nox2 but increased depolarization and K+ efflux [1, 106]. (2) The relationship between bacterial survival and Nox2 activity was steeper at low enzyme activity, when most of the K+ flux took place, than at a high rate of O2 ·− generation when charge compensation occurred mostly by H+ ions [107].
Both observations provide only indirect support for a role of K+ ions in the elimination of microorganisms, and both refer to conditions in which Nox2 activity is limited. They may, however, model conditions where limited substrate supply—e.g., in hypoxic regions—allows only a low rate of O2 ·− generation.
Data and considerations questioning the central role of K+ movements in bacterial killing
In an earlier publication, it has been claimed that bacterial killing was dependent on the opening or closing of one single type of K+ channel [1]. If K+ movements are so critical in the antimicrobial activity, then one would expect that alterations of the K+ concentration of the environment have drastic effects on killing efficiency. However, in our hands, there was no difference in the killing activity of either human peripheral or murine bone marrow neutrophils whether the killing experiment was carried out in physiological solutions containing 5 mM K+ or in nominally K+-free media or in 145 mM KCl (Hably and Meczner, unpublished observations). These results are in agreement with earlier experiments, where alterations of K+ concentration did not affect O2 ·− generation [50, 97, 130].
Another possibility for manipulation of K+ movements is by application of valinomycin, an ionophore highly selective for K+ and Rb+. Valinomycin is the prototype for mobile carriers, and earlier investigations suggested that valinomycin doubles the K+ permeability of human PMN [126]. The addition of valinomycin to resting neutrophils results in definitive hyperpolarization indicating an increase in K+ permeability (Fig. 3). However, valinomycin has only a moderate effect on the rate or extent of depolarization induced by PMA or fMLP, i.e., when Nox2 is allowed to function at full activity (Fig. 3). Thus, K+ flux carried by valinomycin is able to provide some—but far from full—charge compensation for electron transport via Nox2 (valinomycin is only able to provide significant charge compensation when Nox2 activity is mostly inhibited by diphenyliodonium [DPI] as in Fig. 4 of [41]). The important question is whether an increase of K+ permeability has any effect on the killing activity of PMN. In our experiments, we could not detect any difference in elimination of S. aureus by human PMN in the absence or presence of the ionophore. Valinomycin did not affect the killing efficiency in K+-free or K+-rich medium either. Thus, even if valinomycin allows some charge compensation to occur via K+, this does not seem to have significant impact on killing activity when Nox2 is turning at full capacity. Our findings are at variance with some previous data [110].

Comparison of superoxide production and killing of S. aureus in media with high or low chloride concentration. In a, superoxide production was stimulated at time 0 by 100 nM PMA. pH of the solution at the end of the measurements is shown on the right. In b, killing of S. aureus was followed by the method described in [107]. In Cl−-free solutions, NaCl was replaced by 145 mM Na-HEPES or Na-phosphate pH 7.4, respectively. Mean ± SD of three separate experiments is shown
Our quantitative analysis also argues against an “all-or-none” role of K+ movement in bacterial elimination. When Nox2 turned at 25% of its maximal rate, 70% of the total K+ movement had already occurred, yet 90% of the bacteria survived [107]. An increase in the rate of O2 ·− generation resulted in gradual decrease in bacterial survival although only very little further K+ transport occurred.
A similar condition is known in human pathology in the form of “variant CGD” patients. In these cases, the mutation allows the expression of all the subunits, but the enzymatic activity is impaired so that the PMN of these patients are able to produce O2 ·− only at 10% to 20% of the normal rate [10, 127]. These patients present clinical symptoms, although their Nox2 activity could be sufficient to allow K+ movements comparable to healthy cells.
K+ channels in PMN
It is not clear through which kind of K+ channels the detected K+ flux occurs. Voltage-dependent, inwardly rectifying K+ channels have been described in PMN and eosinophils, but these channels close upon depolarization [80, 130]. Thus, they probably do participate in the generation of the resting membrane potential but do not function under condition of intensive O2 ·− production. Indeed, blocking inwardly rectifying channels does not inhibit superoxide production [130]. Ca2+-activated K+ channels have also been demonstrated [77, 134], but their contribution is questionable, as in activated cells, H+ current was the only detectable outward current [94].
The decisive function of large-conductance voltage- and Ca2+-activated K+(BK or maxi-K+) channels has been proposed recently [1]. However, no trace of the existence or function of BK channels could be demonstrated by several groups (see Table 5).
If K+ movement into the phagosome is important in the establishment of the optimal ionic environment for the function of granule enzymes, then it would be logical to increase K+ conductance selectively in the phagosomal membrane. This goal could be achieved by the activation of some of the two pore domain K+ channels (K2P). Several members (e.g., TALK-1, TALK-2, TASK-1, TASK-2) of this family of K+ channels are pH sensitive, showing increased conductance when the pH on the extracellular side becomes alkaline [33, 62]. In addition, TALK-1 and TALK-2 were shown to be activated by ROS [33]. Conditions prevailing in the phagosome (initial alkalinization, generation of O2 ·− and its metabolites) could be favorable for the opening of TALK-1 or TALK-2. With this idea in mind, we searched for the messenger ribonucleic acid (mRNA) of various K2P channels. We could detect a message for TALK-2 both in peripheral blood PMN and in cultured PLB cells. The amount of TALK-2 mRNA was quantitated in real-time polymerase chain reaction experiments and estimated to be five orders of magnitude lower than that of GADPH. In view of this low level of mRNA, investigations at the protein level by electrophysiology or Western blotting were hitherto not feasible (Enyedi and Lakatos, unpublished observation).
Charge compensation by Cl−
The major mobile anion of both the extracellular and intracellular space is Cl−. In a recent study, it has been proposed that 90% of charge compensation for Nox2 activity occurs via Cl− flux from the extracellular or intraphagosomal space into the cytosol [121]. Further, it has been claimed that both the respiratory burst and microbial killing are abolished in the absence of Cl− ions [121]. There are theoretical as well as experimental problems with Cl− as the major charge-compensating ion. Theoretically, it is hard to envisage the movement of massive amounts of the osmotically active Cl− ions and the consequent volume changes (for details, see [96]). Experimentally, Cl− efflux has been detected during the respiratory burst, i.e., movement of Cl− in the opposite direction than required for charge compensation [18, 89, 97, 125]. Recently, it has been reported that ClC-3-deficient mice have a defect in O2 ·− generation; nevertheless, these mice were fully competent in killing of various microbes [93].
In view of the controversial data, we tested the ability of human peripheral PMN to produce O2 ·− in a Cl−-free environment (Fig. 4). Our results indicate that the ability of O2 ·− generation depends on the species of the anion replacing Cl−: It is significantly decreased in the phosphate-based medium but not altered in a 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-based medium that is widely applied in patch clamp experiments. As high phosphate concentration is known to chelate di- and trivalent cations, resulting in strong acidification, we tested the pH of the solutions. We found a correlation between acidification and inability of O2 ·− generation. Accordingly, in the presence of adequate buffering capacity, a high rate of O2 ·− production could be restored also in the phosphate-based medium.
Thus, Nox2 activity per se does not seem to depend on the possibility of Cl− influx. In accordance with this finding, we did not observe any difference in the killing activity whether the extracellular space contained Cl− ions or not (Fig. 4b).
Alterations of Ca2+ household in CGD cells
The function of the electrogenic NADPH oxidase also affects the movement of another ion, calcium. Ca2+ does not have a crucial role in regulating the membrane potential but does play an important role in many cellular processes and signaling events. When the phagocytic NADPH oxidase is activated, the depolarization prevents calcium ions from entering the cell, and it diminishes cytosolic calcium levels. Early studies could not detect any differences in fMLP-induced calcium signals between normal and CGD neutrophils [84]. However, in this report, the investigators used the calcium-sensitive dye, Quin-2, which has low affinity for calcium, and it could have disturbed the calcium signals in the cells. More recent studies of our laboratory using the ratiometric dye Fura-2 with high affinity for calcium have clearly shown that calcium metabolism is affected in neutrophils and neutrophil-like cells in the absence of a functional NADPH oxidase [41, 108]. In fMLP-triggered CGD neutrophils, the influx through store-operated calcium channels was increased and started earlier than in normal cells [41, 131]. In model cells lacking the NADPH oxidase, the fMLP-induced calcium signal was higher than in the normal cells [108]. The inhibition of the oxidase by DPI in neutrophils increased the cytosolic calcium transient stimulated by fMLP [108]. Extracellular production of superoxide by the xanthine/XO system did not have any effect on the calcium influx [41] showing that superoxide anions are not responsible for the difference. Hyperpolarizing the membrane potential in neutrophils after the addition of fMLP does not change superoxide production but increases cytosolic calcium levels immediately (Fig. 5a) due to the oxidase-induced membrane potential changes that increase the driving force for Ca2+ influx (Fig. 5b,c). Altered calcium signaling in CGD neutrophils might cause disturbed cytokine release, apoptosis, and killing [108].

The phagocytic NADPH oxidase inhibits calcium entry in human neutrophils. Human neutrophils were stimulated by fMLP (1 μM, white arrow), and 2 min later, the potassium ionophore, valinomycin (1 μM, black arrow), was added. Changes in cytosolic calcium concentration (a), superoxide production (a, insert), and membrane potential changes (b) were measured. Valinomycin-induced hyperpolarization did not influence fMLP-stimulated superoxide production but increased [Ca2+]ic. c Model showing the effect of the oxidase-induced depolarization on calcium entry. Result of one representative experiment out of four similar ones is shown
Effect of priming on electrophysiological properties of PMN
The effect of priming upon various PMN functions has been investigated in detail, and it is summarized in another paper of this issue (El Benna et al. 2008). However, there are two less well-known aspects that are related to the contribution of electrophysiological factors to the killing process.
Efficient priming during conventional preparation of PMN
In earlier studies, we observed that the release of secretory vesicles, the most easily mobilizable population of PMN vesicles, could only be detected if the cells were prepared under sterile conditions, in pyrogen-free solutions [91]; otherwise, they were eliminated already during the preparation process. We also found significant difference in fMLP-stimulated O2 ·− generation: Conventionally prepared PMN produced fourfold more O2 ·− than PMN prepared under sterile conditions did [61]. These data suggested that PMN might become primed during the conventional preparation process. Systematic investigation supported this suggestion (Fig. 6). PMN prepared under nonsterile conditions produced 3.5 times more O2 ·− upon stimulation with opsonized S. aureus than cells prepared under sterile conditions did (Fig. 6a). Nonsterile PMNs were also more efficient in killing of bacteria (Fig. 6b). We could partially mimic the effect of preparation under nonsterile conditions by treating sterile PMN with tumor necrosis factor-α (TNFα) or granulocyte macrophage colony-stimulating factor (Rada, unpublished observations). Thus, PMNs isolated by conventional techniques, i.e., not in solutions prepared with pyrogen-free water, are definitely primed, and their properties differ from those of resting cells.

Efficient priming of PMN during conventional preparation. ROS production (a), killing of bacteria (b), and rate of depolarization (c and d) were compared for PMN prepared under sterile or nonsterile conditions. In c and d, a decrease in RFU indicates depolarization. Mean ± SEM of eight (a) or seven (b) separate experiments and the result of one representative experiment out of four (c) or eight (d) similar ones is shown
Electrophysiological changes induced by priming
There was no difference in the resting membrane potential of PMN prepared under sterile or nonsterile conditions. However, the rate of depolarization upon stimulation with PMA was increased in nonsterile cells, whereas the extent of depolarization showed no difference (Fig. 6c). These findings indicate that priming increased the activity of Nox2 but did not induce significant alteration in the activity of the charge compensating mechanisms.
Ionic composition of the intracellular space and thereby ionic movements are basically determined by the Na+, K+-ATPase. Inhibition of the Na+/K+ pump by ouabain results in gradual depolarization due to unopposed Na+ influx. Following ouabain treatment of PMN, we observed significantly faster depolarization in nonsterile than in sterile cells (Fig. 6d), indicating an enhanced activity of the Na+, K+-ATPase in the primed cells. Activation of the Na+, K+-ATPase has been shown earlier to occur during differentiation of HL-60 promyelocytic cells [81] and upon chemotactic stimulation [9]. In renal collecting duct cells, it has been demonstrated that LPS and TNFα are able to induce the recruitment of Na+, K+-ATPase molecules to the plasma membrane [135].
Enhancement of the activity of the Na+, K+-ATPase as part of the priming process could increase the intracellular concentration of K+ ions and their movement to the phagosomal space in compensation of the electron flux via Nox2. In this way, upregulation of the Na+/K+ pump may contribute to the observed improvement of the killing activity of primed PMN.
Medical significance of the mechanism of charge compensation for Nox2 activity
O2 ·− generation is vital in the efficient elimination of microorganisms. However, the production of O2 ·− at inappropriate sites or in an inadequate quantity can be deleterious to the host organism [112]. With the help of Nox2-deficient mice, it has been demonstrated that O2 ·− and its derivatives produced by phagocytes contribute to the pathogenesis of ischemia/reperfusion injury [103, 124], stroke [59, 136], neurodegenerative diseases [139], and alcohol-induced liver disease [77].
Thus, the inhibition of Nox2 could be advantageous in prevention or amelioration of several pathological conditions. At present, no efficient inhibitors are available that would be specific for any Nox or cell type [82] (the widely used inhibitor DPI interacts with almost any heme protein). The inhibition of charge compensation has been shown to limit electron transfer via Nox2 [31, 66]. As ion channels have many isoforms and tissue-specific expression has been widely documented, careful investigation of the ion channels participating in charge compensation for electron flux through Nox2 may reveal new potential targets for pharmacological intervention.
On the other hand, if specific ion movement into the phagosome can—under certain conditions—indeed improve the efficiency of killing, then initiation of such ion movements may substitute or partially compensate for lacking O2 ·− generation. This could provide a basis for new approaches in the therapy of CGD.
Alterations in CGD in addition to impaired phagosomal killing
The failure of CGD neutrophils to kill certain bacteria and fungi has been extensively studied, but numerous recent emerging reports suggest that the malfunction of the phagocytic NADPH oxidase in these patients has other consequences, as well.
It has been shown that CGD patients develop inflammatory granulomas in hollow organs without clinical signs of infections [19]; these granulomas are sterile, free of bacteria. Mice deficient in either gp91phox or p47phox show also exaggerated inflammatory reactions to fungus with enhanced accumulation of mononuclear cells and neutrophils [55, 95, 105]. These data suggest that disregulated inflammatory processes contribute to the symptoms of CGD. Neutrophils leave the blood stream and migrate to the site of infection by following the chemical gradient of inflammatory cytokines and bacterial products. On the site of bacterial invasion, neutrophils undergo apoptosis soon after the initiation of phagocytosis and are phagocytosed by macrophages. Delayed apoptosis and disregulated cytokine production could explain the experienced abnormalities in CGD.
Apoptosis and cytokine production in CGD
Many studies suggested that ROS induce apoptosis in neutrophils [4, 16, 35, 36, 46, 67, 86, 119, 140] and showed delayed onset of apoptosis in CGD neutrophils [26, 35, 39, 102, 116].
Several reports have shown that CGD neutrophils and CGD macrophages have altered production of inflammatory cytokines and mediators to a range of stimuli and also showed increased expression of inflammatory molecules in the lung of X-linked CGD (X-CGD) mice (Table 6). Hydrogen peroxide produced by the phagocytic NADPH oxidase has been presented to inhibit fMLP-triggered interleukin-8 (IL-8) production in human neutrophils in vitro, and this inhibition was absent in PMNs from CGD patients [83]. The altered calcium homeostasis of CGD neutrophils discussed above might also contribute to delayed apoptosis and altered cytokine production since calcium has been shown to be important in both processes in neutrophils.
Neutrophil extracellular traps
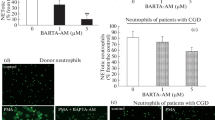
Recently, a very important novel function of ROS has been suggested in the formation of Neutrophil extracellular traps (NETs) [38]. NETs are extracellular fibers made by activated but not by naïve neutrophils. NETs contain smooth stretches and globular structures and are not surrounded by membrane [13]. DNA is the structural backbone of NETs, but it also contains proteins (histones and proteins from primary, secondary, and tertiary granules) [13]. The formation of NETs is an active process and can be initiated by different stimuli (PMA, IL-8, lipopolysaccharide [LPS], bacteria). NETs have been shown to colocalize with and kill bacteria extracellularly through their granular proteases [13]. NETs also capture and kill fungi [132]. As the formation of the NETs, a novel cell death program (“netosis”) has been suggested [38]. During NET formation, stimulated neutrophils change their shape, homogenization of the hetero- and euchromatin occurs, later the membrane of the nucleus and the granules disintegrate, and their content becomes mixed [38]. Subsequently, the plasma membrane breaks, and the extracellular traps are released. The authors claim the mechanism for NET formation being different from both, necrosis and apoptosis, involving ROS formed by the phagocyte NADPH oxidase [38]. CGD neutrophils failed to form NETs when stimulated with stimuli that were effective in normal PMNs (S. aureus, PMA) but elicited NET formation when hydrogen peroxide was provided enzymatically by the glucose/GO system [38]. Type I and II interferons have been suggested to prime neutrophils for subsequent NET formation after stimulation by complement factor 5a [88]. With the discovery of NETs, the long-described antimicrobial effect of the histones and their derivatives also gain physiological importance [14, 52]. Beyond the in vitro studies, NETs have already been shown to be important in vivo in human preeclampsia [43], appendicitis [13], and streptococcal infections [92] resulting in necrotizing fascitis [17] and pneumococcal pneumonia [11]. NET formation revealed a new mechanism by which neutrophils kill microbes but also by which they might contribute to destruction of host tissues or to both at the same time, e.g., in the pathogenesis of sepsis [133].
Our view on the function of neutrophilic granulocytes in the human body has been changing tremendously recently. This cell type, previously thought to be very simple, fulfills a central role in coordinating the inflammatory process between the innate and the adaptive immunity [99] and goes through a second major burst of transcriptional and protein synthetic activities when leaving the blood stream [12]. Besides the novel roles of the best-studied neutrophil enzyme, the NADPH oxidase in bacterial killing (activation of certain granule enzymes and NET formation), a report investigating global gene expression changes in phagocytosing healthy and X-CGD neutrophils showed more than 200 genes whose expression levels are dependent on ROS produced by the NADPH oxidase [75]. CGD neutrophils have increased expression of proinflammatory and lowered expression of anti-inflammatory genes compared with normal neutrophils. These data of NADPH oxidase-dependent transcriptional changes of numerous genes support the observations that neutrophils deficient in gp91phox show prolonged inflammation and delayed resolution of the inflammatory process.
Conclusion
The experimental findings detailed above can be summarized in a very simple message: Nox2 plays multiple important roles in elimination of microorganisms (Fig. 7). Intraphagosomal killing is the result of concerted action of (1) various ROS formed from the primary product of Nox2, (2) different granule enzymes and antimicrobial peptides, and (3) ion transport that allows sustained Nox2 activity and provides optimal microenvironment. In addition to this classical function, ROS participate in induction of apoptosis, influence cytokine synthesis and secretion, and modify gene expression. Last but not least, they also play a role in extracellular killing of various microorganisms. Intensity and relative importance of these processes may differ in the case of different microorganisms or under different conditions (such as hypoxic environment), but apparently, they all contribute to the complex clinical picture that characterizes the immundefficiency in CGD.

Possible roles of the Nox2-based NADPH oxidase in PMN function
References
Ahluwalia J, Tinker A, Clapp LH, Duchen MR, Abramov AY, Pope S et al (2004) The large-conductance Ca2+-activated K+ channel is essential for innate immunity. Nature 427:853–858 doi:10.1038/nature02356
Allen RC, Stjernholm RL, Steele RH (1972) Evidence for the generation of an electronic excitation state(s) in human polymorphonuclear leukocytes and its participation in bactericidal activity. Biochem Biophys Res Commun 47:679–684 doi:10.1016/0006-291X(72)90545-1
Alvarez B, Radi R (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25:295–311 doi:10.1007/s00726-003-0018-8
Arruda MA, Barcellos-de-Souza P, Sampaio AL, Rossi AG, Graca-Souza AV, Barja-Fidalgo C (2006) NADPH oxidase-derived ROS: key modulators of heme-induced mitochondrial stability in human neutrophils. Exp Cell Res 312:3939–3948 doi:10.1016/j.yexcr.2006.08.022
Babior BM, Curnutte JT, Kipnes RS (1975) Biological defense mechanisms. Evidence for the participation of superoxide in bacterial killing by xanthin oxidase. J Lab Clin Med 85:235–244
Babior BM, Kipnes RS, Curnutte JT (1973) Biological defense machanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest 52:741–744 doi:10.1172/JCI107236
Babior BM, Takeuchi C, Ruedi J, Gutierrez A, Wentworth P Jr (2003) Investigating antibody-catalyzed ozone generation by human neutrophils. Proc Natl Acad Sci USA 100:3031–3034 doi:10.1073/pnas.0530251100
Bánfi B, Schrenzel J, Nüsse O, Lew DP, Ligeti E, Krause KH et al (1999) A novel H(+) conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J Exp Med 190:183–194 doi:10.1084/jem.190.2.183
Becker EL, Talley V, Showell HJ, Naccache PH, Sha’afi RI (1978) Activation of the rabbit polymorphonuclear leukocyte membrane “Na+, K+”-ATPase by chemotactic factor. J Cell Biol 77:329–333 doi:10.1083/jcb.77.2.329
Behe P, Segal AW (2007) The function of the NADPH oxidase of phagocytes, and its relationship to other NOXs. Biochem Soc Trans 35:1100–1103 doi:10.1042/BST0351100
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B (2006) An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 16:401–407 doi:10.1016/j.cub.2006.01.056
Borregaard N, Sorensen OE, Theilgaard-Mönch K (2007) Neutrophil granules: a library of innate immunity proteins. Trends Immunol 28:340–345 doi:10.1016/j.it.2007.06.002
Brinkmann V, Reichard U, Goosman C, Fauler B, Uhlemann Y, Weiss DS et al (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535 doi:10.1126/science.1092385
Brinkmann V, Zychlinsky A (2007) Beneficial suicide: why neutrophils die to make NETs. Natl Rev 5:577–582 doi:10.1038/nrmicro1710
Britigan BE, Hassett DJ, Rosen GM, Hammil DR, Cohen MS (1989) Neutrophil degranulation inhibits potential hydroxyl-radical formation. Relative impact on myeloperoxidase and lactoferrin release on hydroxyl-radical production by iron-supplemented neutrophils assessed by spin-trapping techniques. Biochem J 264:447–455
Brown JR, Goldblatt D, Buddle J, Morton L, Thrasher AJ (2003) Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease. J Leukoc Biol 73:591–599 doi:10.1189/jlb.1202599
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M et al (2006) DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 16:396–400 doi:10.1016/j.cub.2005.12.039
Busetto S, Trevisan E, Decleva E, Dri P, Menegazzi R (2007) Chloride movements in human neutrophils during phagocytosis: characterization and relationship to granule release. J Immunol 179:4110–4124
Cale CM, Jones AM, Goldblatt D (2000) Follow up of patients with chronic granulomatous disease diagnosed since 1990. Clin Exp Immunol 120:351–355 doi:10.1046/j.1365-2249.2000.01234.x
Chapman ALP, Hampton MB, Santhilmohan R, Winterbourn CC (2002) Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcu aureus. J Biol Chem 277:9757–9762 doi:10.1074/jbc.M106134200
Cherny VV, Henderson LM, Xu W, Thomas LL, DeCoursey TE (2001) Activation of NADPH oxidase-related proton and electron currents in human eosinophils by arachidonic acid. J Physiol 535:783–794 doi:10.1111/j.1469-7793.2001.00783.x
Cherny VV, Markin VS, DeCoursey TE (1995) The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J Gen Physiol 105:861–896 doi:10.1085/jgp.105.6.861
Clifford DP, Repine JE (1982) Hydrogen peroxide mediated killing of bacteria. Mol Cell Biochem 49:143–149 doi:10.1007/BF00231175
Cohen MS, Britigan BE, Chai YS, Pou S, Roeder TL, Rosen GM (1991) Phagocyte-derived free radicals stimulated by ingestion of iron rich Staphylococcus aureus: a spin trapping study. J Infect Dis 163:819–824
Coulthard CE, Short WF, Michaelis R, Sykes G, Skrimshire GEH, Standfast AFB et al (1942) Notatin: an anti-bacterial glucose-aerodehydrogenase from Penicillium notatum wrestling. Nature 150:148–150 doi:10.1038/150634a0
Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH et al (1996) A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5:653–666 doi:10.1016/S1074-7613(00)80278-2
DeCoursey TE (2004) Voltage-gated proton channels and other proton transfer pathways. Physiol Rev 83:475–579
DeCoursey TE (2007) Electrophysiology of the phagocyte respiratory burst. Focus on “Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity”. Am J Physiol Cell Physiol 293:C30–C32 doi:10.1152/ajpcell.00093.2007
DeCoursey TE, Cherny VV (1993) Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys J 65:1590–1598
DeCoursey TE, Ligeti E (2005) Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci 62:2173–2193 doi:10.1007/s00018-005-5177-1
DeCoursey TE, Morgan D, Cherny VV (2003) The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 422:531–534 doi:10.1038/nature01523
Demaurex N, Grinstein S, Jaconi M, Schlegel W, Lew DP, Krause KH (1993) Proton currents in human granulocytes: regulation by membrane potential and intracellular pH. J Physiol 466:329–344
Duprat F, Girard C, Jarretou G, Lazdunski M (2005) Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J Physiol 562:235–244 doi:10.1113/jphysiol.2004.071266
Essin K, Salanova B, Kettritz R, Sausbier M, Luft FC, Kraus D et al (2007) Large-conductance calcium-activated potassium channel activity is absent in human and mouse neutrophils and is not required for innate immunity. Am J Physiol Cell Physiol 293:C45–C54 doi:10.1152/ajpcell.00450.2006
Fadeel B, Ahlin A, Henter JI, Orrenius S, Hampton MB (1998) Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood 92:4808–4818
Fay AJ, Qian X, Jan YN, Jan LY (2006) SK channels mediate NADPH oxidase-independent reactive oxygen species production and apoptosis in granulocytes. Proc Natl Acad Sci USA 103:17548–17553 doi:10.1073/pnas.0607914103
Femling JK, Cherny VV, Morgan D, Rada B, Davis AP, Czirják G et al (2006) The antibacterial activity of human neutrophils and eosinophils requires proton channels but not BK channels. J Gen Physiol 127:659–672 doi:10.1085/jgp.200609504
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V et al (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241 doi:10.1083/jcb.200606027
Gamberale R, Giordano M, Trevani AS, Andonegui G, Geffner JR (1998) Modulation of human neutrophil apoptosis by immune complexes. J Immunol 161:2666–3674
Geiszt M, Kapus A, Ligeti E (2001) Chronic granulomatous disease: more than the lack of superoxide? J Leukoc Biol 69:191–196
Geiszt M, Kapus A, Német K, Farkas L, Ligeti E (1997) Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J Biol Chem 272:26471–26478 doi:10.1074/jbc.272.42.26471
Green DE, Pauli R (1943) The antibacterial action of the xanthine oxidase system. Proc Soc Exp Biol Med 54:148–150
Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S (2005) Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 66:1146–1154 doi:10.1016/j.humimm.2005.11.003
Halliwell B (2006) Phagocyte-derived reactive species: salvation or suicide? Trends Biochem Sci 31:509–515 doi:10.1016/j.tibs.2006.07.005
Hampton MB, Kettle AJ, Winterbourn CC (1998) Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92:3007–3017
Hampton MB, Vissers MC, Keenan JI, Winterbourn CC (2002) Oxidant-mediated phosphatidylserine exposure and macrophage uptake of activated neutrophils: possible impairment in chronic granulomatous disease. J Leukoc Biol 71:775–781
Hatanaka E, Carvalho BTC, Condino-Neto A, Campa A (2004) Hyperresponsiveness of neutrophils from gp91phox deficient patients to lipopolysaccharide and serum amyloid A. Immunol Lett 94:43–46 doi:10.1016/j.imlet.2004.04.016
Henderson LM, Chappell JB (1992) The NADPH-oxidase-associated H+ channel is opened by arachidonate. Biochem J 283:171–175
Henderson LM, Chappell JB, Jones OT (1987) The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem J 246:325–329
Henderson LM, Chappell JB, Jones OT (1988) Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem J 255:285–290
Hirche TO, Gaut JP, Heinecke JW, Belaaouaj A (2005) Myeloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating neutrophil elastase: effects on host defense. J Immunol 174:1557–1565
Hirsch JG (1958) Bactericidal action of histone. J Exp Med 108:925–944 doi:10.1084/jem.108.6.925
Holland SM, Gallin JI (1999) Disorders of phagocytic cells. In: Gallin JI, Snyderman R (eds) Inflammation. 3rd edn. Lippincott Williams & Wilkins, Philadelphia
Ismail G, Sawyer WD, Wegener WS (1977) Effect of hydrogen peroxide and superoxide radicals on viability of Neisseria gonorrhoeae and related bacteria. Proc Soc Exp Biol Med 155:264–269
Jackson S, Gallin J, Holland S (1995) The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 182:751–758 doi:10.1084/jem.182.3.751
Jankowski A, Grinstein S (1999) A noninvasive fluorimetric procedure for measurement of membrane potential. Quantification of the NADPH oxidase-induced depolarization in activated neutrophils. J Biol Chem 274:26098–26104 doi:10.1074/jbc.274.37.26098
Jankowski A, Scott CC, Grinstein S (2002) Determinants of the phagosomal pH in neutrophils. J Biol Chem 277:6059–6066 doi:10.1074/jbc.M110059200
Jiang Q, Griffin DA, Barofsky DF, Hurst JK (1997) Intraphagosomal chlorination dynamics and yields determined using unique fluorescent bacterial mimics. Chem Res Toxicol 10:1080–1089 doi:10.1021/tx9700984
Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R et al (2007) NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 38:3000–3006 doi:10.1161/STROKEAHA.107.489765
Káldi K, Szászi K, Suszták K, Kapus A, Ligeti E (1994) Lymphocytes possess an electrogenic H(+)-transporting pathway in their plasma membrane. Biochem J 301:329–334
Káldi K, Szeberényi J, Rada BK, Kovács P, Geiszt M, Mócsai A et al (2002) Contribution of phopholipase D and a brefeldin A-sensitive ARF to chemoattractant-induced superoxide production and secretion of human neutrophils. J Leukoc Biol 71:695–700
Kang D, Kim D (2004) Single-channel properties and pH sensitivity of two-pore domain K+ channels of the TALK family. Biochem Biophys Res Commun 315:836–844 doi:10.1016/j.bbrc.2004.01.137
Kapus A, Romanek R, Grinstein S (1994) Arachidonic acid stimulates the plasma membrane H+ conductance of macrophages. J Biol Chem 269:4736–4745
Kapus A, Romanek R, Qu AY, Rotstein OD, Grinstein S (1993) A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J Gen Physiol 102:729–760 doi:10.1085/jgp.102.4.729
Kapus A, Suszták K, Ligeti E (1993) Regulation of the electrogenic H+ channel in the plasma membrane of neutrophils: possible role of phospholipase A2, internal and external protons. Biochem J 292:445–450
Kapus A, Szászi K, Ligeti E (1992) Phorbol 12-myristate 13-acetate activates an electrogenic H(+)-conducting pathway in the membrane of neutrophils. Biochem J 281:697–701
Kasahara Y, Iwai K, Yachie A, Ohta K, Konno A, Seki H et al (1997) Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 89:1748–1753
Kettle AJ, Clark BM, Winterbourn CC (2004) Superoxide converts indigo carmine to isatin sulfonic acid: implications for the hypothesis that neutrophils produce ozone. J Biol Chem 279:18521–18525 doi:10.1074/jbc.M400334200
Kettle AJ, Winterbourn CC (2005) Do neutrophils produce ozone? An appraisal of current evidence. Biofactors 24:41–45
Klebanoff SJ (1967) Iodination of bacteria: a bactericidal mechanism. J Exp Med 126:1063–1076 doi:10.1084/jem.126.6.1063
Klebanoff SJ (1968) Myeloperoxidase–halide–hydrogen peroxide antibacterial system. J Bacteriol 95:2131–2138
Klebanoff SJ (1999) Oxygen metabolites from phagocytes. In: Gallin JI, Synderman R (eds) Inflammation. Basic principles and clinical correlates. 3rd edn. Lippincott Williams & Wilkins, Philadelphia, pp 721–769
Klebanoff SJ, White LR (1969) Iodination defect in the leukocytes of a patient with chronic granulomatous disease of childhood. N Engl J Med 280:460–466
Klebanoff SJ (2005) Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625 doi:10.1189/jlb.1204697
Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL et al (2004) Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol 172:636–643
Kocholaty W (1943) Purification and properties of the second antibacterial substance produced by Penicillium notatum. Science 97:186–187 doi:10.1126/science.97.2512.186
Kono H, Rusyn I, Yin M, Gäbele E, Yamashina S, Dikalova A et al (2000) NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest 106:867–872 doi:10.1172/JCI9020
Korchak HM, Roos D, Giedd KN, Wynkoop EM, Vienne K, Rutherford LE et al (1983) Granulocytes without degranulation: neutrophil function in granule-depleted cytoplasts. Proc Natl Acad Sci USA 80:4968–4972 doi:10.1073/pnas.80.16.4968
Korshunov SS, Imlay JA (2002) A potential role for periplasmic superoxide dismutase in blocking the penetration of external superoxide into the cytosol of Gram-negative bacteria. Mol Microbiol 43:95–106 doi:10.1046/j.1365-2958.2002.02719.x
Krause KH, Welsh MJ (1990) Voltage-dependent and Ca2(+)-activated ion channels in human neutrophils. J Clin Invest 85:491–498 doi:10.1172/JCI114464
Ladoux A, Abita JP, Geny B (1986) Retinoic-acid-induced differentiation of HL-60 cells is associated with biphasic activation of the Na+–K+ pump. Differentiation 33:142–147 doi:10.1111/j.1432-0436.1986.tb00419.x
Lambeth D, Krause KH, Clark RA (2008) NOX enzymes as novel targets of drug development. Semin Immunopathol (in press), (in this volume)
Lekstrom-Himes JA, Kuhns DB, Alvord WG, Gallin JI (2005) Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J Immunol 174:411–417
Lew PD, Wollheim C, Seger RA, Pozzan T (1984) Cytosolic free calcium changes induced by chemotactic peptide in neutrophils from patients with chronic granulomatous disease. Blood 63:231–233
Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF et al (2005) Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202:209–215 doi:10.1084/jem.20050846
Lundqvist-Gustafsson H, Bengtsson T (1999) Activation of the granule pool of the NADPH oxidase accelerates apoptosis in human neutrophils. J Leukoc Biol 65:196–204
Marcinkiewicz J, Biedroń R, Białecka A, Kasprowicz A, Mak M, Targosz M (2006) Susceptibility of Propionibacterium acnes and Staphylococcus epidermidis to killing by MPO–halide system products. Implication for taurine bromamine as a new candidate for topical therapy in treating acne vulgaris. Arch Immunol Ther Exp (Warsz) 54:61–68 doi:10.1007/s00005-006-0007-1
Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF et al (2004) Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 279:44123–44132 doi:10.1074/jbc.M405883200
Menegazzi R, Busetto S, Dri P, Cramer R, Patriarca P (1996) Chloride ion efflux regulates adherence, spreading, and respiratory burst of neutrophils stimulated by tumor necrosis factor-alpha (TNF) on biologic surfaces. J Cell Biol 135:511–522 doi:10.1083/jcb.135.2.511
Miyasaki KT, Wilson ME, Genco RJ (1986) Killing of Actinobacillus actinomycetemcomitans by the human neutrophil myeloperoxidase–hydrogen peroxide–chloride system. Infect Immun 53:161–165
Mócsai A, Jakus Z, Vántus T, Berton G, Lowell CA, Ligeti E (2000) Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol 164:4321–4331
Molloy S (2006) Bacterial pathogenesis: escaping the net. Nat Rev Microbiol 4:242–243 doi:10.1038/nrmicro1409
Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS (2006) Anion channels, including ClC-3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J Biol Chem 281:12277–12288 doi:10.1074/jbc.M511030200
Morgan D, Cherny VV, Murphy R, Xu W, Thomas LL, DeCoursey TE (2003) Temperature dependence of NADPH oxidase in human eosinophils. J Physiol 550:447–558 doi:10.1113/jphysiol.2003.041525
Morgenstern DE, Gifford MAC, Li LL, Doerschuk CM, Dinauer MC (1997) Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med 185:207–218 doi:10.1084/jem.185.2.207
Murphy R, DeCoursey TE (2006) Charge compensation during the phagocyte respiratory burst. Biochim Biophys Acta 1757:996–1011 doi:10.1016/j.bbabio.2006.01.005
Myers JB, Cantiello HF, Schwartz JH, Tauber AI (1990) Phorbol ester-stimulated human neutrophil membrane depolarization is dependent on Ca2(+)-regulated Cl− efflux. Am J Physiol 259:C531–C540
Nathan C, Shiloh MU (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA 97:8841–8848 doi:10.1073/pnas.97.16.8841
Nathan C (2006) Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6:173–182 doi:10.1038/nri1785
Odell EW, Segal AW (1988) The bactericidal effects of the respiratory burst and the myeloperoxidase system isolated in neutrophil cytoplasts. Biochim Biophys Acta 971:266–274
O’Rourke EJ, Chevalier C, Pinto AV, Thiberge JM, Ielpi L, Labigne A et al (2003) Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc Natl Acad Sci USA 100:2789–2794 doi:10.1073/pnas.0337641100
Ottonello L, Frumento G, Arduino N, Dapino P, Tortolina G, Dallegri F (2001) Immune complex stimulation of neutrophil apoptosis: investigating the involvement of oxidative and nonoxidative pathways. Free Radic Biol Med 30:161–169 doi:10.1016/S0891-5849(00)00453-6
Ozaki M, Deshpande SS, Angkeow P, Bellan J, Lowenstein CJ, Dinauer MC et al (2000) Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J 14:418–429
Petheö GL, Demaurex N (2005) Voltage- and NADPH dependence of electron currents generated by the phagocytic NADPH. Biochem J 388:485–491 doi:10.1042/BJ20041889
Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J et al (1995) Mouse model of X-linked chronic granulomatous disease: an inherited defect in phagocyte superoxide production. Nat Genet 9:202–209 doi:10.1038/ng0295-202
Rada BK, Geiszt M, Hably C, Ligeti E (2005) Consequences of the electrogenic function of the phagocytic NADPH oxidase. Philos Trans R Soc Lond B Biol Sci 360:2293–2300 doi:10.1098/rstb.2005.1768
Rada BK, Geiszt M, Káldi K, Timár C, Ligeti E (2004) Dual role of phagocytic NADPH oxidase in bacterial killing. Blood 104:2947–2953 doi:10.1182/blood-2004-03-1005
Rada BK, Geiszt M, Van Bruggen R, Nemet K, Roos D, Ligeti E (2003) Calcium signalling is altered in myeloid cells with a deficiency in NADPH oxidase activity. Clin Exp Immunol 132:53–60 doi:10.1046/j.1365-2249.2003.02138.x
Ramsey IS, Moran MM, Chong JA, Clapham DE (2006) A voltage-gated proton-selective channel lacking the pore domain. Nature 440:1213–1216 doi:10.1038/nature04700
Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G et al (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291–297 doi:10.1038/416291a
Reeves EP, Nagl M, Godovac-Zimmermann J, Segal AW (2003) Reassessment of the microbicidal activity of reactive oxygen species and hypochlorous acid with reference to the phagocytic vacuole of the neutrophil granulocyte. J Med Microbiol 52:643–651 doi:10.1099/jmm.0.05181-0
Ricevuti G (1997) Host tissue damage by phagocytes. Ann NY Acad Sci 832:426–448 doi:10.1111/j.1749-6632.1997.tb46269.x
Roos D, Voetman AA, Meerhof LJ (1983) Functional activity of enucleated human polymorphonuclear leukocytes. J Cell Biol 97:368–377 doi:10.1083/jcb.97.2.368
Rosen H, Klebanoff SJ (1979) Bactericidal activity of a superoxide-generating system. A model for the polymorphonuclear system. J Exp Med 149:27–39 doi:10.1084/jem.149.1.27
Rosen H, Klebanoff SJ (1977) Formation of singlet oxygen by the myeloperoxidase-mediated antimicrobial system. J Biol Chem 252:4803–4810
Sanford AN, Suriano AR, Herche D, Dietzmann K, Sullivan KE (2006) Abnormal apoptosis in chronic granulomatous disease and autoantibody production characteristic of lupus. Rheumatology 45:178–181 doi:10.1093/rheumatology/kei135
Sasaki M, Takagi M, Okamura Y (2006) A voltage sensor-domain protein is a voltage-gated proton channel. Science 312:589–592 doi:10.1126/science.1122352
Sawyer DT, Valentine JS (1981) How super is superoxide? Acc Chem Res 14:393–400 doi:10.1021/ar00072a005
Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK et al (2004) Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood 104:2557–2564 doi:10.1182/blood-2004-01-0191
Schrenzel J, Serrander L, Bánfi B, Nüsse O, Fouyouzi R, Lew DP et al (1998) Electron currents generated by the human phagocyte NADPH oxidase. Nature 392:734–737 doi:10.1038/33725
Segal AW (2005) How neutrophils kill microbes. Annu Rev Immunol 23:197–223 doi:10.1146/annurev.immunol.23.021704.115653
Segal AW, Geisow M, Garcia R, Harper A, Miller R (1981) The respiratory burst of phagocytic cells is associated with a rise in vacuolar pH. Nature 290:406–409 doi:10.1038/290406a0
Segal AW (2008) The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int J Biochem Cell Biol 40:604–618
Seligmann C, Bock A, Leitsch T, Schimmer M, Simsek Y, Schultheiss HP (2001) Polymorphonuclear granulocytes induce myocardial dysfunction during ischemia and in later reperfusion of hearts exposed to low-flow ischemia. J Leukoc Biol 69:727–731
Shimizu Y, Daniels RH, Elmore MA, Finnen MJ, Hill ME, Lackie JM (1993) Agonist-stimulated Cl− efflux from human neutrophils. A common phenomenon during neutrophil activation. Biochem Pharmacol 45:1743–1751 doi:10.1016/0006-2952(93)90429-Z
Simchowitz L, Spilberg I, De Weer P (1982) Sodium and potassium fluxes and membrane potential of human neutrophils: evidence for an electrogenic sodium pump. J Gen Physiol 79:453–479 doi:10.1085/jgp.79.3.453
Stasia MJ, Li XJ (2008) Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol (in press), (in this volume)
Staudinger BJ, Oberdoerster MA, Lewis PJ, Rosen H (2002) mRNA expression profiles for Escherichia coli ingested by normal and phagocyte oxidase-deficient human neutrophils. J Clin Invest 110:1151–1163
Steinbeck MJ, Khan AU, Karnovsky MJ (1992) Intracellular singlet oxygen generation by phagocytosing neutrophils in response to particles coated with a chemical trap. J Biol Chem 267:13425–13433
Tare M, Prestwich SA, Gordienko DV, Parveen S, Carver JE, Robinson C et al (1998) Inwardly rectifying whole cell potassium current in human blood eosinophils. J Physiol 506:303–318 doi:10.1111/j.1469-7793.1998.303bw.x
Tintinger GR, Theron AJ, Steel HC, Andersen R (2001) Accelerated calcium influx and hyperactivation of neutrophils in chronic granulomatous disease. Clin Exp Immunol 123:254–263 doi:10.1046/j.1365-2249.2001.01447.x
Urban CF, Reichard U, Brinkmann V, Zychlinksy A (2006) Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 8:668–676 doi:10.1111/j.1462-5822.2005.00659.x
Urban C, Zychlinsky A (2007) Netting bacteria in sepsis. Nat Med 13:403–404 doi:10.1038/nm0407-403
Várnai P, Demaurex N, Jaconi M, Schlegel W, Lew DP, Krause KH (1993) Highly co-operative Ca2+ activation of intermediate-conductance K+ channels in granulocytes from a human cell line.. J Physiol 472:373–390
Vinciguerra M, Hasler U, Mordasini D, Roussel M, Capovilla M, Ogier-Denis E et al (2005) Cytokines and sodium induce protein kinase A-dependent cell-surface Na,K-ATPase recruitment via dissociation of NF-kappaB/IkappaB/protein kinase A catalytic subunit complex in collecting duct principal cells. J Am Soc Nephrol 16:2576–2585 doi:10.1681/ASN.2005040448
Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC et al (1997) Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke 28:2252–2258
Wentworth P Jr, McDunn JE, Wentworth AD, Takeuchi C, Nieva J, Jones T et al (2002) Evidence for antibody-catalyzed ozone formation in bacterial killing and inflammation. Science 298:2195–2199 doi:10.1126/science.1077642
Wheeler MA, Smith SD, García-Cardeña G, Nathan CF, Weiss RM, Sessa WC (1997) Bacterial infection induces nitric oxide synthase in human neutrophils. J Clin Invest 99:110–116 doi:10.1172/JCI119121
Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H et al (2003) NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. Proc Natl Acad Sci USA 100:6145–6150 doi:10.1073/pnas.0937239100
Zhang B, Hirahashi J, Cullere X, Mayadas TN (2003) Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis: cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J Biol Chem 278:28443–28454 doi:10.1074/jbc.M210727200
Acknowledgements
The authors are indebted to Professor Thomas E. DeCoursey for valueable suggestions and critical reading of the manuscript. Experimental work carried out in the laboratory of L.E. has been supported by the Hungarian Research Fund (grants no. 62221 and 49851) and from the Hungarian Ministry of Health (grant no. 181/2006).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rada, B., Hably, C., Meczner, A. et al. Role of Nox2 in elimination of microorganisms. Semin Immunopathol 30, 237–253 (2008). https://doi.org/10.1007/s00281-008-0126-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-008-0126-3