
Abstract
Obstructive sleep apnea (OSA) is a highly prevalent sleep disorder leading to cardiovascular and metabolic complications. OSA is also a multicomponent disorder, with intermittent hypoxia (IH) as the main trigger for the associated cardiovascular and metabolic alterations. Indeed, recurrent pharyngeal collapses during sleep lead to repetitive sequences of hypoxia–reoxygenation. This IH induces several consequences such as hemodynamic, hormonometabolic, oxidative, and immuno-inflammatory alterations that may interact and aggravate each other, resulting in artery changes, from adaptive to degenerative atherosclerotic remodeling. Atherosclerosis has been found in OSA patients free of other cardiovascular risk factors and is related to the severity of nocturnal hypoxia. Early stages of artery alteration, including functional and structural changes, have been evidenced in both OSA patients and rodents experimentally exposed to IH. Impaired vasoreactivity with endothelial dysfunction and/or increased vasoconstrictive responses due to sympathetic, endothelin, and renin–angiotensin systems have been reported and also contribute to vascular remodeling and inflammation. Oxidative stress, inflammation, and vascular remodeling can be directly triggered by IH, further aggravated by the OSA-associated hormonometabolic alterations, such as insulin resistance, dyslipidemia, and adipokine imbalance. As shown in OSA patients and in the animal model, genetic susceptibility, comorbidities (obesity), and life habits (high fat diet) may aggravate atherosclerosis development or progression. The intimate molecular mechanisms are still largely unknown, and their understanding may contribute to delineate new targets for prevention strategies and/or development of new treatment of OSA-related atherosclerosis, especially in patients at risk for cardiovascular disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Definition
Obstructive sleep apnea (OSA) is characterized by recurrent pharyngeal collapses occurring during sleep [1]. The upper airway closure could be complete, leading to obstructive apneas (defined by airflow cessation of at least 10 s with persistent thoracic and abdominal movements), or partial, leading to hypopneas (i.e., reduction in ventilation of more than 50% from baseline, or less than 50% with oxygen desaturation or microarousal). The diagnosis of OSA is based on polysomnography. Patients are considered to suffer from OSA when they present more than five respiratory events per hour of sleep (apnea–hypopnea index (AHI) > 5), and symptoms, such as excessive daytime sleepiness (EDS), disruptive snoring, witnessed apnea or gasping, obesity, and/or enlarged neck size. Other signs and symptoms include male gender, increased blood pressure (BP), morning headache, and sexual dysfunction [2]. OSA pathophysiology is complex resulting from reduction in pharyngeal dimensions owing to anatomical and functional alterations, such as obesity or maxillofacial structural changes [3, 4], increased pharyngeal collapsibility due to reduced neuromuscular compensation, and altered pharyngeal protective reflex during sleep [5]. Repeated obstructive events during sleep lead to intermittent hypoxia (IH), sleep fragmentation [1], and increased respiratory efforts (Fig. 1). These stimuli are presumably responsible for numerous consequences, including cardiovascular and metabolic complications, and the underlying mechanisms are currently under investigation [6].

Polysomnography. Obstructive sleep apnea (OSA) is diagnosed using polysomnography which consists in several recordings, such as nasal pressure, thoraco-abdominal movements and efforts (pulse transit time), oxygen saturation (SaO2), and sleep stages. The polysomnographic tracing shows complete cessation of flow (nasal pressure) associated with persistent respiratory efforts and oxygen desaturation at each apneic episode. The SaO2 nadir occurs with a delay compared to the end of the apneic episode due to circulation time and the measurement technique and ceases due to microarousal-related reopening of the upper airway. The sum of thorax and abdominal movements during apneas equals zero due to opposite phase movements of thorax and abdomen
OSA is a pathology as frequent as asthma, affecting 5% to 20% of the general population, increasing linearly up to 60 years old and becoming more variable above this threshold value [7, 8]. Regarding OSA morbidity, there is substantial evidence for a causal relationship between OSA and excessive daytime sleepiness leading to increased risk of traffic accidents [9, 10]. In addition, OSA is linked to cardiovascular morbidity and mortality [11, 12], with a higher risk of fatal and nonfatal cardiovascular events [11], and OSA represents an independent risk factor for death from any cause [13].
OSA, cardiovascular risk, and atherosclerosis
OSA is now recognized as a risk factor for cardiovascular morbidity, leading to hypertension, coronary heart disease, arrhythmias, and stroke [14]. The 18-year follow-up of the Wisconsin sleep cohort has showed that untreated severe OSA patients are five times more likely to die from cardiovascular causes [15]. OSA leads to early atherosclerosis, as evidenced by increased intima–media thickness (IMT) and atherosclerotic plaque occurrence in carotid arteries, in the absence of significant comorbidity [16–18]. The severity of nocturnal oxygen desaturation and blood pressure levels seem to be the best predictors for carotid wall hypertrophy in OSA patients, whereas plaques occurrence may be preferentially related to oxygen desaturation [16]. Very recently, coronary atherosclerotic volume has been found increased in OSA patients, with a positive correlation between obstructive AHI and atherosclerotic plaque volume [19]. This strong association between OSA and cardiovascular disease has been recently highlighted by both the American Heart Association and the American College of Cardiology [2, 14] that also acknowledged that the pathophysiological mechanisms are still poorly understood.
Current available treatment
The main treatments of OSA include weight loss, positional treatment, mandibular advancement, and application of a positive pressure through a nasal or facial mask during sleep (CPAP) [20]. CPAP is the recommended treatment in moderate to severe OSA since its effectiveness on sleepiness [21] and daytime function [22] as well as on the cardiovascular morbidity have been firmly established on randomized control trials [12, 23] and large observational studies [11]. In addition, there is good evidence that CPAP ameliorates several mechanisms involved in the genesis of atherosclerosis, including sympathetic activity, endothelial and vascular dysfunction, systemic inflammation, and oxidative stress [6, 24]. A low effective long-term compliance is classically reported [25]. However, CPAP compliance is amazingly higher than most of chronic treatments adherence, particularly in Europe [25–27]. This is essentially related to the perceived clinical benefit, such as improvement in daytime functioning (e.g., reduction in EDS) and life quality. It should be kept in mind, however, that CPAP does not completely reverse all OSA-related consequences. The reduction in BP during CPAP is of limited magnitude (i.e., mean 24-h BP diminished by less than 3 mmHg) which suggests either persistent lesions at the vascular level or additional factors or comorbidities that are not affected by treating sleep-disordered breathing. Obese subjects, for instance, seem to have limited benefits from CPAP treatment regarding metabolic changes [28–30].
Experimental models of OSA
In order to improve the understanding of the pathophysiological link between OSA and cardiovascular disease, several experimental models have been developed for the last 15 years. We have extensively described these models in a recent review [31]. Experimental models have permitted to evaluate each component of the human disease, without unknown and/or confounding factors (disease duration, chronology of events, variable contribution of the different OSA components, genetic, and environmental factors). Some animal models have attempted to reproduce repeated upper airway obstructions [32–34]. However, based on the hypothesis that IH is likely the most important component of OSA with respect to the cardiovascular system, most experimental research in this field has used the animal models of IH where animals intermittently breathe air with low oxygen concentration [35–37]. Cellular models were also developed to study IH-related signaling alterations through different cell lines exposed to IH [38–40]. More specifically, different cell types involved in atherosclerosis were studied (i.e., endothelial cells, monocytes, lymphocytes). The major technical difficulty in the IH model, especially the in vitro IH model, is to reproduce the rapid cyclic changes in oxygen concentration, as occurring in OSA patients. Notwithstanding this technical limitation, animal and cellular models have largely contributed to characterize the role of IH on OSA-related cardiovascular consequences, therefore contributing to the development of alternative or complementary therapeutic strategies to CPAP, especially in patients at risk to develop cardiovascular complications. In this review, using both clinical and experimental findings, we will dissect the mechanisms potentially involved in OSA-related atherosclerosis.
Chronology of atherosclerosis development
Atherosclerosis is characterized by arterial lesions containing cholesterol, immune infiltrates, and fibrosis [41, 42]. Furthermore, development of atherosclerotic lesions results from a dynamic interplay between native cells of the vasculature and leukocytes issued from the general circulation.
Atherosclerosis is known to be initiated principally at the sites of endothelial dysfunction by retention, accumulation, and oxidation of lipoproteins in the arterial wall. Local endothelial cell activation results in leukocytes and activated platelets adhesion to the endothelium, associated with the entry of small-dense low-density lipoprotein (LDL) cholesterol into the vascular intima. The subsequent oxidation of these LDL cholesterol particles leads to the expression and release from endothelial cells of proinflammatory factors, such as cytokines, chemokines, and adhesion molecules. Circulating monocytes adhere to the endothelium and migrate into the subendothelial space where they uptake cholesterol and become cholesterol-laden foam cells to form fatty streak within the intima. These cells exhibit a proinflammatory phenotype and contribute to the evolution of the lesion. Immune cells are recruited in the vascular wall and continuously release proinflammatory cytokines, chemokines, and growth factors that cause proliferation and migration of smooth muscle cells from the media to the intima, as well as recruitment of new immuno-inflammatory cells from the blood circulation. Smooth muscle cells secrete extracellular matrix components within the intima. This leads to collagen and proteoglycans accumulation, initiating fibrous cap formation, a major element in plaque maturation and stabilization. Over time, immuno-inflammatory cells secrete and activate local matrix metalloproteinases, leading to thinner fibrous cap and increased vulnerability of the plaque. The atherosclerotic plaque can be therefore considered as a final stage of artery remodeling, leading to ultimate clinical manifestations such as chronic tissue hypoxia and acute ischemic events, i.e., myocardial infarction and strokes (for review, see [41]).
Thus, atherosclerosis involves a complex pathophysiological process in which coexistence of several risk factors, such as hypertension and dyslipidemia, may act synergistically at the arterial wall level to enhance atherosclerosis [43]. As mentioned above, clinical complications of atherosclerosis are preceded by biological events that may be initiated or aggravated by the hypoxic stimulus of OSA. Therefore, investigating these biological early events is critical to understand the specific role of hypoxia in OSA-related atherogenesis and possibly to prevent and treat atherosclerosis in OSA patients.
Mechanisms of OSA-related atherosclerosis
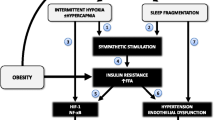
The mechanisms leading to OSA-related atherosclerosis include hemodynamic, metabolic, oxidative, and inflammatory alterations that interact in a complex manner (Fig. 2). More precisely, hypoxia may have either direct or indirect effects on the vasculature, through local and systemic alterations triggered by IH.

Intermittent hypoxia-induced atherosclerosis. Intermittent hypoxia triggers hemodynamic, metabolic, and inflammatory alterations interacting with each other and leading to vascular changes resulting in atherosclerosis. Preatherosclerotic modifications include increased intima (1)–media (2) thickness with smooth muscle cell hypertrophy (5), elastic fiber alterations (6), mucoid degeneration (7), and leukocyte infiltration (8) in the adventitia (3) and peri-adventitia (4) tunica
OSA-related hemodynamic alterations
Independently of any comorbidity, about 50% of OSA patients develop systemic hypertension which depends upon the severity of the pathology [44]. Several pathophysiological mechanisms have been proposed. Using a dog model, Brooks et al. have shown that IH resulting from repeated occlusions seems to be the main determining factor for BP elevation [45]. Although the exact mechanisms of this causal relationship are not fully understood, a growing body of evidence suggests that chronic IH leads to several hormonal changes, including alterations of the renin–angiotensin and endothelin systems [36], as well as abnormal stimulation of the peripheral chemoreflex leading to sympathetic activation [46, 47]. In addition to sustained BP elevation, further BP surges occur at each apnea episode, associated with increased bursts of the sympathetic nerve activity [47]. All these blood pressure changes have been replicated in the animal model of IH [48] and are well known to induce functional and structural alterations of the vascular endothelium, partly through vascular shear stress impairment which is a critical determinant of vascular homeostasis, regulating remodeling and atherogenesis [49].
Functional alterations
Clinical investigations in OSA patients have shown vasoreactivity dysfunction, with increased vasoconstrictive responses and/or decreased endothelial relaxation [50–53]. Several experimental studies have replicated this vasoreactivity dysfunction in animal models of IH, thus suggesting a major role for the hypoxic component of the disease in the vascular complications [48, 54–56]. Furthermore, although vascular alterations may contribute to BP elevation (see above), the concomitant assessment of BP and vasoresponses in mice exposed to IH has showed that BP changes may represent the triggering mechanism [48].
Endothelial dysfunction is associated with a reduced bioavailability of nitric oxide that has also been observed in OSA patients [57–64] and could represent an early marker of atherosclerosis [65]. Beside sympathoadrenal and renin–angiotensin systems, the endothelin system is also possibly involved as a major vasoconstrictive factor. We have studied in rats genetically prone to develop hypertension (spontaneously hypertensive rat, SHR) and their normotensive controls [66] exposed to IH the activation of the endothelin system and particularly the isoform 1 (ET-1) of endothelin which is upregulated by hypoxia inducible factor-1 (HIF-1). Chronic IH resulted in greater hypertension development and infarct size following myocardial ischemia in SHR compared to control rats. This was accompanied by an increase in myocardial big-ET-1 and ET-1 levels, and ET-A receptor expression and by an enhanced coronary vasoconstriction to ET-1 in SHR only. HIF-1 was linked to the promoter of the myocardial ET-1 gene following chronic IH only. Moreover, administration of bosentan, an ET-A and ET-B endothelin receptor antagonist, during chronic IH prevented the increase in both BP and infarct size. Therefore, activation of the endothelin system in SHR appears to be mediated by HIF-1 activity and to be responsible for the enhanced susceptibility to chronic IH and for its associated cardiovascular consequences leading to hypertension and ischemic injuries [66].
Structural alterations
Hemodynamic alterations of hypertension are known to result in eutrophic and/or hypertrophic remodeling of the vascular wall [67]. In OSA patients without the usual risk factors for atherosclerosis, we and others have found an increased carotid IMT which is an early sign of atherosclerosis, the carotid IMT being positively correlated with the severity of nocturnal oxygen desaturation [16–18]. It has also been shown that this increased IMT was reversible when treating OSA, at least in some individuals [68]. Very recently, we have confirmed these data in aorta from mice exposed to IH [48, 69] (see below). Taken together, these data suggest a major role for IH in OSA-associated vascular remodeling. In mice, the enlarged aorta IMT was observed after 2 weeks of IH and involved the media, with a magnitude similar to what has been reported in OSA patients, without aortic dilation. In addition to the hemodynamic stress, hypoxia may contribute to these structural adaptive and degenerative alterations through the release of several factors, including endothelin. For instance, we have recently demonstrated in rodents the role of the endothelin system in the hypoxia-induced vascular consequences, since an endothelin antagonist prevented BP elevation [70], as well as increased IMT, as shown by our preliminary results [71]. These histological findings are consistent with vasoreactivity studies that showed an increased contractile response to endothelin-1 in carotid [56] and mesenteric arteries [72] in rats exposed to IH. We have also studied the role of leukotrienes in early atherosclerosis associated with OSA [73]. More specifically, we looked at leukotriene B4 (LTB4) which is a lipid mediator involved in inflammation and atherogenesis. We found in OSA patients that LTB4 levels were correlated with the mean and minimal arterial oxygen saturation and that CPAP treatment significantly reduced LTB4 production by 50%. LTB4 production correlated with luminal carotid artery diameter in patients with moderate to severe nocturnal hypoxia, but not with carotid IMT. Therefore, LTB4 could contribute to early vascular remodeling in moderate-to-severe hypoxic obstructive sleep apnea patients [73]. These early artery morphological changes could also contribute to impaired shear stress, vascular inflammation, with subsequent aggravation of the degenerative atherosclerotic process through vicious circles (Fig. 2).
Metabolic alterations
As previously reported, metabolic dysregulation represents an important risk factor for atherosclerosis [74]. While there is a strong association between OSA and obesity [75], the number of characteristic features of the metabolic syndrome increases with OSA severity, independently of body mass index [76, 77]. This supports a relationship between OSA and metabolic dysfunction independently of obesity, although the latter one represents an aggravating factor for metabolic dysregulation.
Insulin resistance, visceral fat, and adipokines
Insulin resistance is a key element leading to atherogenic phenotype, promoting hyperglycemia, dyslipidemia, and hypertension [74]. In clinical populations, OSA was found to be associated with an increased frequency of impaired glucose tolerance and type-2 diabetes [78], and this increased prevalence of glucose dysregulation was independent of obesity and age [78]. A number of reports have found increased insulin resistance and impaired glucose tolerance in OSA patients that were independent of body weight [79–82], while worsening with OSA severity, as shown by insulin resistance with increasing AHI [83]. However, other studies failed to demonstrate an independent effect of AHI, owing to the major impact of obesity [84, 85]. EDS may also be of importance, as underlined by the recent findings that OSA patients with EDS had higher glucose and insulin levels, suggesting that EDS in OSA could be a clinical marker for OSA patients susceptible to develop metabolic complications [86]. Lastly, in a recent evaluation of obese individuals prior to bariatric surgery [87], oxygen desaturation greater than 4.6% was associated with a 1.5-fold increase in insulin resistance. Moreover, liver histopathology suggested that severe IH might predispose to hepatic lesions, as OSA patients with more severe nocturnal hypoxia had more hepatic lesions, including inflammation, hepatocyte ballooning, and fibrosis. In contrast, there was no relationship between AHI and insulin resistance or liver histopathology. Together, these results suggest that the hypoxic stress may be implicated in the development of insulin resistance and steatohepatitis in severely obese OSA patients [87]. It is well known that high levels of plasma insulin, as occurring in insulin resistance, are detrimental for the vasculature, leading to impaired vasoreactivity and further artery remodeling, affecting both native cells of the vascular wall and inflammatory cells (for review, see [88, 89]).
In several studies, the murine model of IH has been used to study the mechanisms of glucose dysregulation associated with OSA without any confounding factor. By studying both lean C57BL6 and obese–leptin-deficient mice, it was shown that long term-exposure to IH led to a time-dependent increase in fasting serum insulin levels and worsening of glucose tolerance in obese–leptin-deficient mice only. This suggests that IH-induced glucose dysregulation was dependent on the leptin pathways [90]. Leptin is mainly an adipocyte-derived cytokine that regulates satiety and food intake via central mechanisms and has also a large range of peripheral effects, including regulation of glucose homeostasis (i.e., suppression of insulin secretion and improvement in insulin sensitivity) [91]. Furthermore, through several mechanisms, leptin may contribute to the pathogenesis of atherosclerosis [92, 93]. It has been shown that leptin induces endothelial dysfunction, stimulates inflammatory cytokines production by immune cells (dendritic cells, macrophages, T cells), and promotes platelet aggregation and oxidative stress, as well as migration, hypertrophy, and proliferation of vascular smooth muscle cells [92, 93].
In addition to its relationship with adiposity, serum leptin levels seem to be also modulated by hypoxia, as shown by increased levels of plasma leptin in mice exposed to IH [90], or in normal subjects exposed to altitude [94]. This may be related to a direct effect of hypoxia that may induce HIF-1-alpha-mediated leptin transcription [95].
In OSA subjects, leptin has been found elevated [82], and in some cases, leptin levels seem to be more related to obesity than to OSA [96]. However, most studies found a significant impact of OSA treatment on leptin levels [97]. Therefore, the higher circulating leptin levels observed in OSA could contribute to OSA-related atherogenesis [98–101].
Other adipokines produced by the adipose tissue could be involved in OSA-associated atherosclerosis. Adiponectin is the most abundant cytokine-like hormone secreted by adipose tissue [102] and emerging evidence suggest that adiponectin exerts antiatherogenic effects leading to endothelial function improvement, as well as anti-inflammatory properties [103, 104]. In OSA patients, in spite of few conflicting studies reporting unchanged [105, 106] or even augmented [107] adiponectin levels, most published data support a significant reduction in adiponectin which seems independent of the obesity status [108–114]. Indeed, recent studies have demonstrated that hypoxia per se reduced adiponectin, independently of obesity. Nakagawa et al have reported a reduction in adiponectin in OSA, agreeing with previous studies [115–118], and in mice or adipocytes exposed to sustained hypoxia [112]. Similarly, a recent study has shown that IH suppressed adiponectin secretion in adipocytes [119].
Finally, the adipokine resistin is also produced by adipose tissue but mostly derives from macrophages, and its role is still uncertain in humans. It is known to impair insulin sensitivity at least in rodents and to be associated with increased cardiovascular risk, possibly owing to regulation of proinflammatory and proatherogenic markers and through endothelial dysfunction [103]. A recent study has revealed higher serum resistin levels in untreated OSA patients compared to control subjects [120]. Resistin levels were only related to the severity of OSA, independently of any confounding factor including obesity. Furthermore, resistin levels were reduced by CPAP treatment, and AHI before treatment was predictive of the reduction rate [120].
Taken together, these data suggest a significant activation of adipose tissue in OSA, presumably in response to direct and indirect effects of IH and potentially contributing to the development of atherosclerosis. Indeed, several studies have evidenced a major role of perivascular and visceral adipose tissue in atherosclerosis progression [121–127]. The mechanisms of adipose tissue activation in the apneic pathology should be further studied, since inflammation of the adipose tissue may be a key factor in the OSA-associated cardiovascular and metabolic morbidities. This could represent a potential pharmacological target in the OSA treatment since, as already mentioned, some of these consequences may not be fully reversed by CPAP treatment [6].
Lipid metabolism
OSA patients exhibit multiple anomalies of lipid metabolism. Dyslipidemia, characterized by increase in total serum cholesterol and triglycerides and decrease in high-density lipoprotein (HDL), has been observed in many studies [97, 99, 100, 128, 129]. Perturbation of lipid metabolism results in part from oxidative stress which is often present in OSA patients [130]. This could contribute to HDL dysfunction, i.e., loss of HDL antioxidant capacity [131], and to increase in lipid peroxidation [132]. As previously mentioned, other lipid mediators, such as LTB4, could also contribute to early vascular remodeling in moderate-to-severe hypoxic OSA patients [73]. However, a causal link between OSA and the development of dyslipidemia is difficult to establish due to the many confounding factors, particularly visceral obesity.
Thus, the group of Polotsky has largely investigated lipid metabolism in mice exposed to IH. First, they demonstrated that IH induced hyperlipidemia in lean mice, but obesity with baseline hypercholesterolemia could mask the effects of IH on lipid metabolism [133]. In addition to hyperlipidemia and in accordance with data in OSA [132], they have evidenced lipid peroxidation in IH mice that correlated to the severity of the hypoxic stimulus [134]. Using a human cell model, Lattimore et al. further demonstrated that IH increased in vitro foam cell formation in primary human macrophages, which is consistent with a major proatherogenic role of hypoxia in OSA patients [135].
However, although these data suggest a major contributory role of IH to atherogenesis, only two studies have demonstrated a direct link between experimental IH and the formation of atherosclerotic plaques in animals non susceptible to spontaneously develop atherosclerosis, such as C57BL6 mice [133, 136, 137]. Long-term exposure to IH was required (10–12 weeks), as well as concomitant high-cholesterol diet, suggesting that IH alone is not sufficient for promoting atherogenesis, but genetic susceptibility as well as comorbidities might also be determinant.
Inflammation
Systemic inflammation
Similarly to atherosclerosis, OSA is now considered as a chronic low grade inflammatory disease [37, 138, 139]. Although it has been initially suggested that C-reactive protein (CRP) could be increased in OSA [140–142], this has not been confirmed in the general population [143], and the increase in CRP is probably more related to associated comorbidities, such as obesity [144]. This is further supported by results regarding CPAP treatment which allows suppression of all the OSA components, including IH. In a recently published controlled study, CRP levels were highly variable at baseline between patients and active CPAP had no effect on CRP levels [145]. However, beside CRP, OSA is also associated with elevated levels of prothrombotic and proinflammatory factors. Carpagnano et al. have showed an increased interleukin (IL)-6 and 8-isoprostane in breath condensate of OSA patients [146], and further studies have showed elevated plasmatic levels of inflammatory cytokines, chemokines, and adhesion molecules, such as IL-6, IL-8, intercellular adhesion molecule-1 (ICAM-1), monocyte chemoattractant protein-1/C–C chemokine ligand 2 (MCP-1/CCL2), and tumor necrosis factor-alpha (TNF-α) [141, 147, 148]. These augmented levels of circulating adhesion molecules and chemokines (ICAM-1, vascular cell adhesion molecule-1 (VCAM-1), MCP-1/CCL2, respectively) were correlated with both oxygen desaturation [148] and increased carotid IMT [16, 17]. Through the increased levels of circulating proinflammatory mediators, OSA patients also exhibit activated endothelial cells that in turn express adhesion molecules, promoting interaction between leukocytes and vascular endothelium [39]. In parallel to endothelial activation, neutrophils, monocytes, and T cells are also activated in OSA patients. Activated neutrophils show enhanced production of reactive oxygen species (ROS) [149] and selectins [150] which are involved in cell–cell interactions. This results in enhanced neutrophil rolling which is an early step of leukocyte recruitment. In addition, cytokines, leukotrienes, and ROS are further released by neutrophils [73, 150] which, in addition to delayed apoptosis that will further prolong neutrophils–endothelium interactions [150], may contribute to endothelial injury in OSA patients. Monocytes are also activated in OSA patients, leading to increased adhesion molecules expression and adhesion on endothelial cells and ROS production [39]. Finally, the group of Lavie has also shown a specific activation of lymphocyte subpopulation, with enhanced CD8+ T cell cytotoxicity [151] and increased cytokines production by both CD4+ and CD8+ T cell subtypes [152]. All these inflammatory alterations depend on OSA severity and could largely contribute to the development of atherosclerosis in OSA patients.
Similarly, preliminary results from our group confirmed IH-induced systemic inflammation in mice. We observed a significant increase in chemokine expression within splenic tissue, as well as increased migratory and proliferative capacities of spleen-derived T cells after IH exposure [69]. Furthermore, this systemic inflammation appeared to be an early event, starting from the fifth day of IH exposure, and was further associated with vascular inflammation of small and large arteries (see below).
Vascular inflammatory preatherosclerotic alterations
As mentioned above, increased carotid IMT, which is an early sign of atherosclerosis, has been reported in OSA patients free of significant cardiovascular risk factors [16, 18]. An enlarged aortic IMT has been also found in C57BL6 mice [48, 69, 71] and in Wistar rats [70] after only 14 days of IH. Preliminary data demonstrate that the increased IMT affected the media and was characterized by several similar features to humans, including disorganized elastic fiber network, increased distance between fibers with evidence of smooth-muscle cell hypertrophy, and mucoid material depositions. However, as it was likely to be early in the time course, neither aortic dilation nor lipidic depositions occurred yet [69]. These structural changes could represent mechanical adaptations to IH-induced hemodynamic strains, characterized by sustained systemic BP elevation and additional BP surges during IH episodes [48]. Consistent with this hypothesis and at the same time point of IH exposure, we found a reduced endothelial platelet endothelial cell adhesion molecule-1 (PECAM-1) expression at the dorsal wall of the descending thoracic aorta and in the left ventricle myocardium [48]. Indeed, PECAM-1 is a cell–cell adhesion molecule involved in leukocyte adhesion and transmigration through the endothelium [153] but recent evidence has also suggested a mechanoreceptive role. Alteration in PECAM-1 expression could therefore represent a mechanism that modulates endothelial cells sensitivity to mechanical stimuli [154]. Collectively, IH-induced hemodynamic stress could largely contribute to these structural adaptive and degenerative alterations, as BP oscillations occur from the first day of IH exposure [48]. The underlying mechanisms are likely complex, involving oxidative stress and vascular inflammation that could be potentiated by systemic inflammation and hormonometabolic alterations (Fig. 2). However, there is also evidence for direct effects of IH on vessels. It has been reported that IH can activate the proinflammatory transcription factor nuclear factor-kappa B (NFkB) in mice cardiovascular tissue [69, 155] and in cell culture [40]. This NFkB activation could mediate the increased proinflammatory NFkB-dependent genes, such as the adhesion molecules ICAM-1 and VCAM-1 which have been described in OSA patients [147, 148, 156]. The inflammatory alterations could occur very early in the process, as only 3 h of induced obstructive apneas in rats could be sufficient to increase leukocyte rolling and P-selectin expression in colonic venules [34]. Similarly, in a preliminary study, we observed enhanced leukocyte rolling and ICAM-1 expression in mesenteric resistance vessels from mice exposed to 14 or 35 days of IH [69]. In the aorta and concomitantly to structural changes associated with the enlarged IMT described above, we found an overexpression of the chemotactic chemokine regulated upon activation, normal T cell expressed and secreted/C–C chemokine ligand 5 and the adhesion molecule ICAM-1, with NFkB activation and T lymphocytes infiltration in the aortic wall [69]. Both IH-induced systemic and tissular inflammatory alterations should be considered as a major cause of OSA-related atherosclerosis (see below).
Atherosclerosis
As mentioned above, two recent studies from Polotsky’s group have confirmed a direct link between experimental IH and the development of atherosclerotic plaques in C57BL6 mice [136, 137]. However, using this mouse strain that is nonsusceptible to atherogenesis, long-term exposure to IH and concomitant high-fat high-cholesterol diet were required to develop early lesions (i.e., fatty streak) [157, 158]. The dyslipidemia observed in high-cholesterol fed mice was accentuated with IH, which could have contributed to accelerate atherosclerosis development.
To facilitate the study of IH effects on advanced stages of atherosclerosis, we and Polotsky’s group have used a more susceptible animal model, the apolipoprotein E deficient mice (ApoE−/−) that spontaneously develop atherosclerotic plaques [159]. Both preliminary studies have simultaneously demonstrated the accelerating role of IH on atherosclerosis. In one study, the proatherogenic effect of IH was associated with an increase in plasma levels of LDL cholesterol and diastolic BP and oxidative stress [160]. In our study, the proatherogenic role of IH was rather associated with an increase in systemic and vascular inflammation, without significant BP elevation [161]. Notwithstanding some differences between the two studies, they both confirmed in rodents the accelerating role of IH in the atherosclerosis progression.
The studies with high-cholesterol fed C57BL6 mice and with apolipoprotein E-deficient mice are of clinical interest, as they reproduce the heterogeneous susceptibility and associated comorbidities seen in OSA patients. They also provide clues for potential therapeutic targets in an attempt to reverse or prevent hypoxic consequences in OSA patients.
Conclusion
There is now growing evidence that OSA is a risk factor for cardiovascular complications that are mainly mediated by direct and indirect effects of the hypoxic component of OSA. OSA is indeed associated with cardiovascular complications, including atherosclerosis. Atherosclerosis in OSA seems to be related to the severity of nocturnal hypoxia and occurs in patients free of other significant cardiovascular or metabolic risk factor, although synergistic effects with obesity and dyslipidemia are likely to occur. The mechanisms leading to atherosclerosis in OSA are multifactorial, including hemodynamic, hormonometabolic, oxidative, and immuno-inflammatory alterations that may interact and aggravate each other. These mechanisms are currently under investigation in both patients and animal models, i.e., mainly rodents exposed to IH. OSA is now considered as a chronic low grade inflammatory condition at both systemic and tissue levels including vessels, with evidence for an early implication in the course of the disease. More specifically, white adipose tissue and periadventitial fat are likely to play a role in the systemic and vascular inflammation. The intimate molecular mechanisms are still largely unknown, and their understanding may provide new targets for prevention strategies and new treatments of OSA-related atherosclerosis, especially in patients at risk for cardiovascular diseases.
Abbreviations
- AHI:
-
Apnea–hypopnea index
- BP:
-
Blood pressure
- CPAP:
-
Continuous positive airway pressure
- CRP:
-
C-reactive protein
- EDS:
-
Excessive daytime sleepiness
- ET-1:
-
Endothelin-1
- HIF-1:
-
Hypoxia inducible factor-1
- ICAM-1:
-
Intercellular adhesion molecule-1
- IH:
-
Intermittent hypoxia
- IL:
-
Interleukin
- IMT:
-
Intima–media thickness
- LTB4:
-
Leukotriene B4
- MCP-1/CCL2:
-
Monocyte chemoattractant protein-1/C–C chemokine ligand 2
- OSA:
-
Obstructive sleep apnea
- NFkB:
-
Nuclear factor kappa B
- PECAM-1:
-
Platelet endothelial cell adhesion molecule-1
- RANTES/CCL5:
-
Regulated upon activation, normal T cell expressed and secreted/C–C chemokine ligand 5
- TNF-α:
-
Tumor necrosis factor-alpha
- VCAM-1:
-
Vascular cell adhesion molecule-1
References
Malhotra A, White DP (2002) Obstructive sleep apnoea. Lancet 360(9328):237–245. doi:10.1016/S0140-6736(02) 09464-3
Somers VK et al (2008) Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation 118(10):1080–1111. doi:10.1161/CIRCULATIONAHA.107.189420
Mayer P et al (1996) Relationship between body mass index, age and upper airway measurements in snorers and sleep apnoea patients. Eur Respir J 9(9):1801–1809. doi:10.1183/09031936.96.09091801
White DP (2005) Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med 172(11):1363–1370. doi:10.1164/rccm.200412-1631SO
Horner RL (2007) Contributions of passive mechanical loads and active neuromuscular compensation to upper airway collapsibility during sleep. J Appl Physiol 102(2):510–512. doi:10.1152/japplphysiol.01213.2006
Levy P et al (2008) Intermittent hypoxia and sleep-disordered breathing: current concepts and perspectives. Eur Respir J 32(4):1082–1095. doi:10.1183/09031936.00013308
Young T et al (1993) The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med 328(17):1230–1235. doi:10.1056/NEJM199304293281704
Young T, Peppard PE, Gottlieb DJ (2002) Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 165(9):1217–1239. doi:10.1164/rccm.2109080
Teran-Santos J, Jimenez-Gomez A, Cordero-Guevara J (1999) The association between sleep apnea and the risk of traffic accidents. Cooperative Group Burgos-Santander. N Engl J Med 340(11):847–851. doi:10.1056/NEJM199903183401104
Mazza S et al (2002) Analysis of error profiles occurring during the OSLER test: a sensitive mean of detecting fluctuations in vigilance in patients with obstructive sleep apnea syndrome. Am J Respir Crit Care Med 166(4):474–478. doi:10.1164/rccm.2107065
Marin JM et al (2005) Long-term cardiovascular outcomes in men with obstructive sleep apnoea–hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 365(9464):1046–1053
Pepperell JC et al (2002) Ambulatory blood pressure after therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised parallel trial. Lancet 359(9302):204–210. doi:10.1016/S0140-6736(02)07445-7
Yaggi HK et al (2005) Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 353(19):2034–2041. doi:10.1056/NEJMoa043104
Somers VK et al (2008) Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. J Am Coll Cardiol 52(8):686–717. doi:10.1016/j.jacc.2008.05.002
Young T et al (2008) Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 31(8):1071–1078
Baguet JP et al (2005) The severity of oxygen desaturation is predictive of carotid wall thickening and plaque occurrence. Chest 128(5):3407–3412. doi:10.1378/chest.128.5.3407
Minoguchi K et al (2005) Increased carotid intima–media thickness and serum inflammatory markers in obstructive sleep apnea. Am J Respir Crit Care Med 172(5):625–630. doi:10.1164/rccm.200412-1652OC
Drager LF et al (2005) Early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 172(5):613–618. doi:10.1164/rccm.200503-340OC
Turmel J et al (2009) Relationship between atherosclerosis and the sleep apnea syndrome: an intravascular ultrasound study. Int J Cardiol 132(2):203–209. doi:10.1016/j.ijcard.2007.11.063
Basner RC (2007) Continuous positive airway pressure for obstructive sleep apnea. N Engl J Med 356(17):1751–1758. doi:10.1056/NEJMct066953
Jenkinson C et al (1999) Comparison of therapeutic and subtherapeutic nasal continuous positive airway pressure for obstructive sleep apnoea: a randomised prospective parallel trial. Lancet 353(9170):2100–2105. doi:10.1016/S0140-6736(98)10532-9
Montserrat JM et al (2001) Effectiveness of CPAP treatment in daytime function in sleep apnea syndrome. A randomized controlled study with an optimized placebo. Am J Respir Crit Care Med 164(4):608–613
Haentjens P et al (2007) The impact of continuous positive airway pressure on blood pressure in patients with obstructive sleep apnea syndrome: evidence from a meta-analysis of placebo-controlled randomized trials. Arch Intern Med 167(8):757–764. doi:10.1001/archinte.167.8.757
Lévy P et al (2009) Obstructive sleep apnea and atherosclerosis. Prog Cardiovasc Dis 51(5):400–410. doi:10.1016/j.pcad.2008.03.001
Gay P et al (2006) Evaluation of positive airway pressure treatment for sleep related breathing disorders in adults. Sleep 29(3):381–401
Pepin JL et al (1999) Effective compliance during the first 3 months of continuous positive airway pressure. A European prospective study of 121 patients. Am J Respir Crit Care Med 160(4):1124–1129
Weaver TE et al (2007) Relationship between hours of CPAP use and achieving normal levels of sleepiness and daily functioning. Sleep 30(6):711–719
Harsch IA et al (2004) Continuous positive airway pressure treatment rapidly improves insulin sensitivity in patients with obstructive sleep apnea syndrome. Am J Respir Crit Care Med 169(2):156–162. doi:10.1164/rccm.200302-206OC
Coughlin SR et al (2007) Cardiovascular and metabolic effects of CPAP in obese males with OSA. Eur Respir J 29(4):720–727. doi:10.1183/09031936.00043306
West SD et al (2007) Effect of CPAP on insulin resistance and HbA1c in men with obstructive sleep apnoea and type 2 diabetes. Thorax 62(11):969–974. doi:10.1136/thx.2006.074351
Dematteis M et al. (2009) Cardiovascular consequences of sleep disordered breathing: contribution of animal models to understanding the human disease. ILAR J 50(3)
Kimoff RJ et al (1994) Canine model of obstructive sleep apnea: model description and preliminary application. J Appl Physiol 76(4):1810–1817
Brooks D et al (1997) Obstructive sleep apnea as a cause of systemic hypertension. Evidence from a canine model. J Clin Invest 99(1):106–109. doi:10.1172/JCI119120
Nacher M et al (2007) Recurrent obstructive apneas trigger early systemic inflammation in a rat model of sleep apnea. Respir Physiol Neurobiol 155(1):93–96. doi:10.1016/j.resp. 2006.06.004
Neubauer JA (2001) Invited review: physiological and pathophysiological responses to intermittent hypoxia. J Appl Physiol 90(4):1593–1599
Fletcher EC (2001) Invited review: physiological consequences of intermittent hypoxia: systemic blood pressure. J Appl Physiol 90(4):1600–1605
Morgan BJ (2007) Vascular consequences of intermittent hypoxia. Adv Exp Med Biol 618:69–84. doi:10.1007/978-0-387-75434-5_6
Prabhakar NR (2001) Oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J Appl Physiol 90(5):1986–1994
Dyugovskaya L, Lavie P, Lavie L (2002) Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med 165(7):934–939
Ryan S, Taylor CT, McNicholas WT (2005) Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 112(17):2660–2667. doi:10.1161/CIRCULATIONAHA.105.556746
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352(16):1685–1695. doi:10.1056/NEJMra043430
Libby P (2002) Inflammation in atherosclerosis. Nature 420(6917):868–874. doi:10.1038/nature01323
Lusis AJ (2000) Atherosclerosis. Nature 407(6801):233–241. doi:10.1038/35025203
Peppard PE et al (2000) Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 342(19):1378–1384. doi:10.1056/NEJM200005113421901
Brooks D et al (1997) Effect of obstructive sleep apnea versus sleep fragmentation on responses to airway occlusion. Am J Respir Crit Care Med 155(5):1609–1617
Smith ML, Pacchia CF (2007) Sleep apnoea and hypertension: role of chemoreflexes in humans. Exp Physiol 92(1):45–50. doi:10.1113/expphysiol.2006.033753
Somers VK et al (1995) Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest 96(4):1897–1904. doi:10.1172/JCI118235
Dematteis M et al (2008) Intermittent hypoxia induces early functional cardiovascular remodeling in mice. Am J Respir Crit Care Med 177(2):227–235. doi:10.1164/rccm.200702-238OC
Chatzizisis YS et al (2007) Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol 49(25):2379–2393. doi:10.1016/j.jacc.2007.02.059
Kato M et al (2000) Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation 102(21):2607–2610
Kraiczi H et al (2001) Impairment of vascular endothelial function and left ventricular filling: association with the severity of apnea-induced hypoxemia during sleep. Chest 119(4):1085–1091. doi:10.1378/chest.119.4.1085
Imadojemu VA et al (2002) Impaired vasodilator responses in obstructive sleep apnea are improved with continuous positive airway pressure therapy. Am J Respir Crit Care Med 165(7):950–953
Imadojemu VA et al (2002) Obstructive apnea during sleep is associated with peripheral vasoconstriction. Am J Respir Crit Care Med 165(1):61–66
Tahawi Z et al (2001) Altered vascular reactivity in arterioles of chronic intermittent hypoxic rats. J Appl Physiol 90(5):2007–2013 discussion 2000
Phillips SA et al (2004) Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol 286(1):H388–H393. doi:10.1152/ajpheart.00683.2003
Lefebvre B et al (2006) Functional assessment of vascular reactivity after chronic intermittent hypoxia in the rat. Respir Physiol Neurobiol 150(2–3):278–286. doi:10.1016/j.resp. 2005.05.020
Ip MS et al (2000) Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am J Respir Crit Care Med 162(6):2166–2171
Lavie L et al (2003) Plasma levels of nitric oxide and L-arginine in sleep apnea patients: effects of nCPAP treatment. J Mol Neurosci 21(1):57–63. doi:10.1385/JMN:21:1:57
Teramoto S et al (2003) Oxygen administration improves the serum level of nitric oxide metabolites in patients with obstructive sleep apnea syndrome. Sleep Med 4(5):403–407. doi:10.1016/S1389-9457(03)00102-3
Ohike Y et al (2005) Amelioration of vascular endothelial dysfunction in obstructive sleep apnea syndrome by nasal continuous positive airway pressure—possible involvement of nitric oxide and asymmetric NG, NG-dimethylarginine. Circ J 69(2):221–226. doi:10.1253/circj.69.221
Duchna HW et al (2005) Long-term effects of nasal continuous positive airway pressure on vasodilatory endothelial function in obstructive sleep apnea syndrome. Sleep Breath 9(3):97–103. doi:10.1007/s11325-005-0024-z
Lattimore JL et al (2006) Treatment of obstructive sleep apnoea leads to improved microvascular endothelial function in the systemic circulation. Thorax 61(6):491–495. doi:10.1136/thx.2004.039164
Noda A et al (2007) Continuous positive airway pressure improves daytime baroreflex sensitivity and nitric oxide production in patients with moderate to severe obstructive sleep apnea syndrome. Hypertens Res 30(8):669–676. doi:10.1291/hypres.30.669
Ozkan Y et al (2008) Circulating nitric oxide (NO), asymmetric dimethylarginine (ADMA), homocysteine, and oxidative status in obstructive sleep apnea–hypopnea syndrome (OSAHS). Sleep Breath 12(2):149–154. doi:10.1007/s11325-007-0148-4
Mason RP (2006) Nitric oxide mechanisms in the pathogenesis of global risk. J Clin Hypertens (Greenwich) 8(8 Suppl 2):31–38. doi:10.1111/j.1524-6175.2006.05838.x quiz 40
Belaidi E et al (2009) Major role for HIF-1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. J Am Coll Cardiol 53(15):1309–1317
Mulvany MJ et al (1996) Vascular remodeling. Hypertension 28(3):505–506
Drager LF et al (2007) Effects of continuous positive airway pressure on early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 176(7):706–712. doi:10.1164/rccm.200703-500OC
Arnaud C et al. (2008) Intermittent hypoxia induces inflammatory vascular remodeling in C57bl6 mice. In: American Thoracic Society 2008 International Conference. Am J Resp Crit Care Med 177, Meeting abstracts, 2008, Toronto, Canada
Belaidi E et al. (2008) Dual endothelin-1 receptor antagonism prevents chronic intermittent hypoxia-induced cardiovascular alterations in rat. In: European Respiratory Society 2008 International Conference. Eur Resp J 32(suppl 52), Meeting abstract, 2008, Berlin, Germany
Arnaud C et al. (2009) Intermittent hypoxia induces inflammatory vascular and myocardial remodeling: role of the endothelin system. In: American Thoracic Society 2009 International Conference. Am J Respir Crit Care Med 179, Meeting Abstracts, 2009, San Diego, USA
Allahdadi KJ, Walker BR, Kanagy NL (2005) Augmented endothelin vasoconstriction in intermittent hypoxia-induced hypertension. Hypertension 45(4):705–709. doi:10.1161/01.HYP.0000153794.52852.04
Lefebvre B et al (2008) Leukotriene B4: early mediator of atherosclerosis in obstructive sleep apnoea? Eur Respir J 32(1):113–120. doi:10.1183/09031936.00137107
Chapman MJ, Sposito AC (2008) Hypertension and dyslipidaemia in obesity and insulin resistance: pathophysiology, impact on atherosclerotic disease and pharmacotherapy. Pharmacol Ther 117(3):354–373. doi:10.1016/j.pharmthera.2007.10.004
Newman AB et al (2005) Progression and regression of sleep-disordered breathing with changes in weight: the sleep heart health study. Arch Intern Med 165(20):2408–2413. doi:10.1001/archinte.165.20.2408
Peled N et al (2007) The association of OSA with insulin resistance, inflammation and metabolic syndrome. Respir Med 101(8):1696–1701. doi:10.1016/j.rmed.2007.02.025
Kono M et al (2007) Obstructive sleep apnea syndrome is associated with some components of metabolic syndrome. Chest 131(5):1387–1392. doi:10.1378/chest.06-1807
Meslier N et al (2003) Impaired glucose-insulin metabolism in males with obstructive sleep apnoea syndrome. Eur Respir J 22(1):156–160. doi:10.1183/09031936.03.00089902
Strohl KP et al (1994) Insulin levels, blood pressure and sleep apnea. Sleep 17(7):614–618
Vgontzas AN et al (2000) Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab 85(3):1151–1158. doi:10.1210/jc.85.3.1151
Ip MS et al (2002) Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med 165(5):670–676
McArdle N et al (2007) Metabolic risk factors for vascular disease in obstructive sleep apnea: a matched controlled study. Am J Respir Crit Care Med 175(2):190–195. doi:10.1164/rccm.200602-270OC
Punjabi NM et al (2002) Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med 165(5):677–682
Stoohs RA, Facchini F, Guilleminault C (1996) Insulin resistance and sleep-disordered breathing in healthy humans. Am J Respir Crit Care Med 154(1):170–174
Sharma SK et al (2007) Obesity, and not obstructive sleep apnea, is responsible for metabolic abnormalities in a cohort with sleep-disordered breathing. Sleep Med 8(1):12–17. doi:10.1016/j.sleep. 2006.06.014
Barcelo A et al (2008) Insulin resistance and daytime sleepiness in patients with sleep apnoea. Thorax 63(11):946–950. doi:10.1136/thx.2007.093740
Polotsky VY et al (2009) Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med 179(3):228–234. doi:10.1164/rccm.200804-608OC
Muniyappa R et al (2007) Cardiovascular actions of insulin. Endocr Rev 28(5):463–491. doi:10.1210/er.2007-0006
Nigro J et al (2006) Insulin resistance and atherosclerosis. Endocr Rev 27(3):242–259. doi:10.1210/er.2005-0007
Polotsky VY et al (2003) Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol 552(Pt 1):253–264. doi:10.1113/jphysiol.2003.048173
Martin SS, Qasim A, Reilly MP (2008) Leptin resistance: a possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J Am Coll Cardiol 52(15):1201–1210. doi:10.1016/j.jacc.2008.05.060
Dubey L, Hesong Z (2006) Role of leptin in atherogenesis. Exp Clin Cardiol 11(4):269–275
Beltowski J (2006) Leptin and atherosclerosis. Atherosclerosis 189(1):47–60. doi:10.1016/j.atherosclerosis.2006.03.003
Sierra-Johnson J et al (2008) Effect of altitude on leptin levels, does it go up or down? J Appl Physiol 105(5):1684–1685. doi:10.1152/japplphysiol.01284.2007
Grosfeld A et al (2002) Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. J Biol Chem 277(45):42953–42957. doi:10.1074/jbc.M206775200
Barcelo A et al (2005) Neuropeptide Y and leptin in patients with obstructive sleep apnea syndrome: role of obesity. Am J Respir Crit Care Med 171(2):183–187. doi:10.1164/rccm.200405-579OC
Borgel J et al (2006) Obstructive sleep apnoea and its therapy influence high-density lipoprotein cholesterol serum levels. Eur Respir J 27(1):121–127. doi:10.1183/09031936.06.00131304
Saarelainen S, Lahtela J, Kallonen E (1997) Effect of nasal CPAP treatment on insulin sensitivity and plasma leptin. J Sleep Res 6(2):146–147
Chin K et al (1999) Changes in intra-abdominal visceral fat and serum leptin levels in patients with obstructive sleep apnea syndrome following nasal continuous positive airway pressure therapy. Circulation 100(7):706–712
Ip MS et al (2000) Serum leptin and vascular risk factors in obstructive sleep apnea. Chest 118(3):580–586. doi:10.1378/chest.118.3.580
Shimizu K et al (2002) Plasma leptin levels and cardiac sympathetic function in patients with obstructive sleep apnoea–hypopnoea syndrome. Thorax 57(5):429–434. doi:10.1136/thorax.57.5.429
Okamoto Y et al (2006) Adiponectin: a key adipocytokine in metabolic syndrome. Clin Sci (Lond) 110(3):267–278. doi:10.1042/CS20050182
Gualillo O, Gonzalez-Juanatey JR, Lago F (2007) The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. Trends Cardiovasc Med 17(8):275–283. doi:10.1016/j.tcm.2007.09.005
Lago F et al (2007) Adipokines as emerging mediators of immune response and inflammation. Nat Clin Pract Rheumatol 3(12):716–724. doi:10.1038/ncprheum0674
Makino S et al (2006) Obstructive sleep apnoea syndrome, plasma adiponectin levels, and insulin resistance. Clin Endocrinol (Oxf) 64(1):12–19. doi:10.1111/j.1365-2265.2005.02407.x
Tokuda F et al (2008) Serum levels of adipocytokines, adiponectin and leptin, in patients with obstructive sleep apnea syndrome. Intern Med 47(21):1843–1849. doi:10.2169/internalmedicine.47.1035
Wolk R et al (2005) Plasma levels of adiponectin, a novel adipocyte-derived hormone, in sleep apnea. Obes Res 13(1):186–190. doi:10.1038/oby.2005.24
Zhang XL et al (2004) Serum adiponectin level in patients with obstructive sleep apnea hypopnea syndrome. Chin Med J (Engl) 117(11):1603–1606
Zhang XL et al (2006) Serum adiponectin levels in adult male patients with obstructive sleep apnea hypopnea syndrome. Respiration 73(1):73–77. doi:10.1159/000088690
Masserini B et al (2006) Reduced levels of adiponectin in sleep apnea syndrome. J Endocrinol Invest 29(8):700–705
Zhang XL et al (2007) Effect of continuous positive airway pressure treatment on serum adiponectin level and mean arterial pressure in male patients with obstructive sleep apnea syndrome. Chin Med J (Engl) 120(17):1477–1481
Nakagawa Y et al (2008) Nocturnal reduction in circulating adiponectin concentrations related to hypoxic stress in severe obstructive sleep apnea–hypopnea syndrome. Am J Physiol Endocrinol Metab 294(4):E778–E784. doi:10.1152/ajpendo.00709.2007
Kanbay A et al (2008) Comparison of serum adiponectin and tumor necrosis factor-alpha levels between patients with and without obstructive sleep apnea syndrome. Respiration 76(3):324–330. doi:10.1159/000134010
Lam JC et al (2008) Hypoadiponectinemia is related to sympathetic activation and severity of obstructive sleep apnea. Sleep 31(12):1721–1727
Chen B et al (2006) Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem Biophys Res Commun 341(2):549–556. doi:10.1016/j.bbrc.2006.01.004
Hosogai N et al (2007) Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56(4):901–911. doi:10.2337/db06-0911
Wang B, Wood IS, Trayhurn P (2007) Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch 455(3):479–492. doi:10.1007/s00424-007-0301-8
Ye J et al (2007) Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 293(4):E1118–E1128. doi:10.1152/ajpendo.00435.2007
Magalang UJ et al (2008) Intermittent hypoxia suppresses adiponectin secretion by adipocytes. Exp Clin Endocrinol Diabetes 117:129–134
Yamamoto Y et al (2008) Resistin is closely related to systemic inflammation in obstructive sleep apnea. Respiration 76(4):377–385. doi:10.1159/000141866
Wu H et al (2007) T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 115(8):1029–1038. doi:10.1161/CIRCULATIONAHA.106.638379
Thalmann S, Meier CA (2007) Local adipose tissue depots as cardiovascular risk factors. Cardiovasc Res 75(4):690–701. doi:10.1016/j.cardiores.2007.03.008
Henrichot E et al (2005) Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol 25(12):2594–2599. doi:10.1161/01.ATV.0000188508.40052.35
McGill HC Jr et al (2002) Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 105(23):2712–2718. doi:10.1161/01.CIR.0000018121.67607.CE
Fantuzzi G, Mazzone T (2007) Adipose tissue and atherosclerosis: exploring the connection. Arterioscler Thromb Vasc Biol 27(5):996–1003. doi:10.1161/ATVBAHA.106.131755
Ohman MK et al (2008) Visceral adipose tissue inflammation accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation 117(6):798–805. doi:10.1161/CIRCULATIONAHA.107.717595
Vela D et al (2007) The role of periadventitial fat in atherosclerosis. Arch Pathol Lab Med 131(3):481–487
Newman AB et al (2001) Relation of sleep-disordered breathing to cardiovascular disease risk factors: the Sleep Heart Health Study. Am J Epidemiol 154(1):50–59. doi:10.1093/aje/154.1.50
Robinson GV et al (2004) Circulating cardiovascular risk factors in obstructive sleep apnoea: data from randomised controlled trials. Thorax 59(9):777–782. doi:10.1136/thx.2003.018739
Lavie L (2003) Obstructive sleep apnoea syndrome—an oxidative stress disorder. Sleep Med Rev 7(1):35–51. doi:10.1053/smrv.2002.0261
Tan KC et al (2005) HDL dysfunction in obstructive sleep apnea. Atherosclerosis 184:377–382
Lavie L, Vishnevsky A, Lavie P (2004) Evidence for lipid peroxidation in obstructive sleep apnea. Sleep 27(1):123–128
Li J et al (2005) Intermittent hypoxia induces hyperlipidemia in lean mice. Circ Res 97(7):698–706. doi:10.1161/01.RES.0000183879.60089.a9
Li J et al (2007) Hyperlipidemia and lipid peroxidation are dependent on the severity of chronic intermittent hypoxia. J Appl Physiol 102(2):557–563. doi:10.1152/japplphysiol.01081.2006
Lattimore JD et al (2005) Repetitive hypoxia increases lipid loading in human macrophages—a potentially atherogenic effect. Atherosclerosis 179(2):255–259. doi:10.1016/j.atherosclerosis.2004.11.010
Savransky V et al (2007) Chronic intermittent hypoxia induces atherosclerosis. Am J Respir Crit Care Med 175(12):1290–1297. doi:10.1164/rccm.200612-1771OC
Savransky V et al (2008) Dyslipidemia and atherosclerosis induced by chronic intermittent hypoxia are attenuated by deficiency of stearoyl coenzyme A desaturase. Circ Res 103(10):1173–1180. doi:10.1161/CIRCRESAHA.108.178533
McNicholas WT, Bonsignore MR (2007) Sleep apnoea as an independent risk factor for cardiovascular disease: current evidence, basic mechanisms and research priorities. Eur Respir J 29(1):156–178. doi:10.1183/09031936.00027406
Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444(7121):860–867. doi:10.1038/nature05485
Shamsuzzaman AS et al (2002) Elevated C-reactive protein in patients with obstructive sleep apnea. Circulation 105(21):2462–2464. doi:10.1161/01.CIR.0000018948.95175.03
Yokoe T et al (2003) Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 107(8):1129–1134. doi:10.1161/01.CIR.0000052627.99976.18
Yao M et al (2006) The relationship between sleep-disordered breathing and high-sensitivity C-reactive protein in Japanese men. Sleep 29(5):661–665
Taheri S et al (2007) Correlates of serum C-reactive protein (CRP)—no association with sleep duration or sleep disordered breathing. Sleep 30(8):991–996
Guilleminault C, Kirisoglu C, Ohayon MM (2004) C-reactive protein and sleep-disordered breathing. Sleep 27(8):1507–1511
Kohler M et al (2009) Effects of continuous positive airway pressure on systemic inflammation in patients with moderate to severe obstructive sleep apnoea: a randomised controlled trial. Thorax 64(1):67–73. doi:10.1136/thx.2008.097931
Carpagnano GE et al (2002) Increased 8-isoprostane and interleukin-6 in breath condensate of obstructive sleep apnea patients. Chest 122(4):1162–1167. doi:10.1378/chest.122.4.1162
Ohga E et al (1999) Increased levels of circulating ICAM-1, VCAM-1, and L-selectin in obstructive sleep apnea syndrome. J Appl Physiol 87(1):10–14
Ohga E et al (2003) Effects of obstructive sleep apnea on circulating ICAM-1, IL-8, and MCP-1. J Appl Physiol 94(1):179–184
Schulz R et al (2000) Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med 162(2 Pt 1):566–570
Dyugovskaya L et al (2008) Delayed neutrophil apoptosis in patients with sleep apnea. Am J Respir Crit Care Med 177(5):544–554. doi:10.1164/rccm.200705-675OC
Dyugovskaya L et al (2005) Activated CD8+ T-lymphocytes in obstructive sleep apnoea. Eur Respir J 25(5):820–828. doi:10.1183/09031936.05.00103204
Dyugovskaya L, Lavie P, Lavie L (2005) Lymphocyte activation as a possible measure of atherosclerotic risk in patients with sleep apnea. Ann N Y Acad Sci 1051:340–350. doi:10.1196/annals.1361.076
van Buul JD, Hordijk PL (2004) Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol 24(5):824–833. doi:10.1161/01.ATV.0000122854.76267.5c
Tzima E et al (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437(7057):426–431. doi:10.1038/nature03952
Greenberg H et al (2006) Chronic intermittent hypoxia activates nuclear factor-kappaB in cardiovascular tissues in vivo. Biochem Biophys Res Commun 343(2):591–596. doi:10.1016/j.bbrc.2006.03.015
Minoguchi K et al (2004) Elevated production of tumor necrosis factor-alpha by monocytes in patients with obstructive sleep apnea syndrome. Chest 126(5):1473–1479. doi:10.1378/chest.126.5.1473
Paigen B et al (1985) Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 57(1):65–73. doi:10.1016/0021-9150(85)90138-8
Stewart-Phillips JL, Lough J (1991) Pathology of atherosclerosis in cholesterol-fed, susceptible mice. Atherosclerosis 90(2–3):211–218. doi:10.1016/0021-9150(91)90117-L
Hofker MH, van Vlijmen BJ, Havekes LM (1998) Transgenic mouse models to study the role of APOE in hyperlipidemia and atherosclerosis. Atherosclerosis 137(1):1–11. doi:10.1016/S0021-9150(97)00266-9
Jun JC et al. (2008) Intermittent hypoxia accelerates aortic atherosclerosis in ApoE deficient mice. In: American Thoracic Society 2008 International Conference. Am J Resp Crit Care Med 177, Meeting abstract, 2008, Toronto, Canada
Arnaud C et al. (2008) Chronic intermittent hypoxia accelerates atherosclerotic plaque formation in ApoE knockout mice independently of cholesterol levels. In: European Respiratory Society 2008 International Conference. Eur Resp J 32(suppl 52), Meeting abstract, 2008, Berlin, Germany.
Acknowledgments
This work was supported by a grant from AGIR@dom to CA and MD. CA is recipient of fellowship from the Fondation pour la Recherche Médicale (France).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Arnaud, C., Dematteis, M., Pepin, JL. et al. Obstructive sleep apnea, immuno-inflammation, and atherosclerosis. Semin Immunopathol 31, 113–125 (2009). https://doi.org/10.1007/s00281-009-0148-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-009-0148-5