
Abstract
IgA antibodies play an important role in humoral immunity. IgA is the predominant antibody in mucosal secretions and the second most prevalent in the serum. It occupies a unique position among human antibodies in that it can both trigger and suppress inflammatory responses, depending on the situation. Recent structural and functional studies have revealed details of the structure of IgA and its interaction with key cell-surface receptors. We look at the role IgA and IgA receptors (particularly FcαRI) play in the pathogenesis of diseases such as IgA nephropathy and other autoimmune conditions. Finally, we address the potential of IgA as a therapeutic tool to either trigger specific inflammatory responses to destroy target cells or suppress inflammatory responses in the case of autoimmune diseases, and the promise of mucosal vaccines for eliciting specific IgA responses to pathogens in mucosal environments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
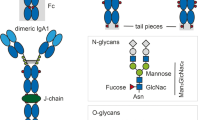
Immunoglobulin A (IgA) represents one of the five classes of human antibody (IgG, IgA, IgM, IgE, and IgD). Humans produce more IgA antibody per day than any other isotype; it is the second most prevalent antibody in the serum, while it is the predominant species in mucosal secretions [1]. There are several distinct forms of IgA: monomeric IgA, dimeric or polymeric IgA, and secretory IgA. Furthermore, there are two IgA subclasses in humans, IgA1 and IgA2. The IgA1 subclass contains extensive O-linked glycosylation in the hinge region connecting the Fc and Fab regions. In contrast, the IgA2 subclass has a truncated hinge region lacking O-linked glycosylation (see Fig. 1) [1]. The simple monomeric IgA form has two heavy and two light chains like all other antibody isotypes. Each IgA heavy chain features a C-terminal extension known as the tailpiece. The IgA tailpieces can form intermolecular disulfide bonds with a protein called the J chain, leading to the formation of dimeric (or occasionally polymeric) IgA (dIgA) [2]. In the serum, approximately 95% of IgA is in the monomeric form with the remaining 5% comprising dimeric or polymeric IgA [1]. A receptor called the polymeric Ig receptor (pIgR) expressed on the basolateral surface of mucosal epithelial cells specifically binds dIgA and transcytoses across the mucosal epithelial cell. During transcytosis, the ectodomain of pIgR forms a disulfide bond to dIgA, and the ectodomain is cleaved, releasing a covalent complex known as the secretory IgA (SIgA) [2].

Structural views of IgA1 and IgA2. Solution structure of (a) IgA1 and (b) IgA2 as determined by solution scattering [7, 8]. Note the extended conformation of the O-glycosylated hinge in IgA1, the compact conformation of IgA2, and the orientation of the tailpieces in both subclasses. (c) Front view and (d) side view of IgA1-Fc (Fcα) taken from the FcαRI:Fcα complex [9]. Note the location of the N-glycans, which are found on the external surface of the CH2 and CH3 domains and are highly solvent exposed, in contrast to N-glycans of IgG and IgE
Serum and secretory IgA have distinct immunological functions. Secretory IgA is responsible for immune exclusion in which SIgA binds to pathogens and prevents their adherence to and invasion of the mucosal epithelium. SIgA is not capable of triggering phagocytosis of pathogens [3, 4]. On the other hand, any pathogens that are able to breach the epithelial layer can be targeted by serum IgA [3]. In contrast to SIgA, serum IgA antibodies in immune complexes are very effective at initiating a wide range of inflammatory responses, including phagocytosis, antibody-dependent cellular cytotoxicity, oxidative burst, and cytokine release [1]. Inflammatory responses triggered by IgA are often mediated by the IgA-specific receptor FcαRI (CD89) [5], although Fcα/μR can also mediate similar responses to IgA and IgM immune complexes [6].
IgA structure in solution
Perkins and colleagues have used small-angle X-ray and neutron scattering to construct low-resolution models of both IgA1 and IgA2 in solution [7, 8]. These experiments are able to determine the relative orientation of domains within the intact antibodies in solution without the need for trapping the antibodies in crystalline form. Monomeric IgA1 is striking in that it adopts a roughly T-shaped conformation with long, extended hinge regions connecting the Fc and Fab regions (Fig. 1a) [8]. The extended nature of the hinge is due to its heavy O-glycosylation with five distinct O-linked glycan sites within the 23-residue hinge peptide. In contrast, IgA2 adopts a more compact structure [7], consistent with the 13-residue deletion in the hinge region that removes all potential O-glycosylation sites (Fig. 1b). The best-fit models from solution scattering data suggest that the tailpieces for both IgA1 and IgA2 are folded back toward the main body of the IgA Fc domains, rather than extending outward into the solution.
Crystal structure of IgA1-Fc
The crystal structure of an IgA1-Fc core fragment (called Fcα) lacking the hinge and tailpiece regions was recently solved in complex with FcαRI (Fig. 1c,d) [9]. This represented the first detailed view of the Fc portion of any IgA antibody. Although the overall structure resembles other IgG-Fc (Fcγ) and IgE-Fc (Fcε) structures, there are a few important differences. First, the CH2 domains of Fcα are tethered together by an unusual pair of disulfide bonds connecting residue C299 in the DE loop of each CH2 domain with C242 at the base of the hinge in the opposite heavy chain. It is interesting to note that C299 in Fcα corresponds to the conserved asparagine in Fcγ and Fcε to which the N-linked glycans are attached. In all Fc structures solved before Fcα, two N-linked glycan chains are found between the upper Fc domains. In contrast, on Fcα the N-linked glycans are linked to N263, found on the external surface of the CH2 domain. Unlike the N-glycans in Fcγ and Fcε that are primarily buried between the two heavy chains, the Fcα N-glycans are solvent-exposed (Fig. 1d). The electron density in the crystal structure of the complex was not very well-defined for the Fcα N-glycans, and only one of the two branching oligosaccharides chains emanating from the core oligosaccharide could be resolved. As described below in more detail, the N-glycans (and O-glycans) of IgA1 were implicated in the pathogenesis of IgA nephropathy. It is potentially significant that the N-glycans of IgA1, unlike IgG and IgE, are highly solvent accessible and poised to interact with different IgA receptors.
IgA receptors in humans
There are several known receptors for IgA, although few were characterized in much detail. The best-known IgA receptors include the polymeric Ig receptor (pIgR) [10], FcαRI [11], and Fcα/μR [6]. Other IgA receptors include the transferrin receptor (TfR) [12], a secretory component receptor (SCR) on eosinophils [13], an IgA2-specific receptor expressed on M cells within Peyer’s patches [14], and a receptor for SIgA and pIgA on natural killer cells [15]. Finally, there are a few lectin-like receptors that interact with specific glycan epitopes on IgA, including the asialoglycoprotein receptor (ASGPR) [16], galectin 1 [17], and an IgA1/IgD receptor on T cells [18].
pIgR is expressed on the basolateral surface of mucosal epithelial cells, and is specific for polymeric IgA or IgM. It is responsible for the transcytosis of polymeric IgA (and IgM) across the mucosal epithelium to form secretory IgA or IgM. pIgR has no inflammatory role and functions primarily in transcytosis, as far as is known [10]. FcαRI and Fcα/μR mediate inflammatory responses to IgA-immune complexes. FcαRI binds monomeric and polymeric IgA1 or IgA2, although it does not bind SIgA except in the presence of an integrin co-receptor [4, 19]. Clustering of FcαRI by IgA-immune complexes in the serum can trigger phagocytosis, respiratory burst, antibody-dependent cellular cytotoxicity and cytokine release [1]. Fcα/μR has sequence similarity to pIgR and is also specific for both IgA and IgM [6]. However, in terms of function, it more closely resembles FcαRI, as it activates phagocytosis of IgA-bound or IgM-bound antigens.
TfR, which is primarily responsible for iron homeostasis, was recently identified as a mesangial IgA1 receptor [12]. As described below, TfR was implicated in the pathogenesis of IgA nephropathy, along with FcαRI [20]. SCR is expressed on eosinophils and is specific for the secretory component of SIgA (secretory component is the ectodomain of pIgR that is covalently linked to pIgA). It triggers degranulation by eosinophils and may also be responsible for the ability of SIgA to degranulate basophils [13]. The M cell receptor is found within Peyer’s patches, regions within the gut-associated lymphoid tissue responsible for antigen sampling [14]. This receptor specifically binds IgA2 via the CH1 and CH2 regions and is thought to be involved in gut antigen sampling via SIgA. ASGPR is not precisely an IgA-specific receptor; instead, it is specific for N-glycans and O-glycans on a large number of serum proteins, including IgA. It is expressed in the liver, and is responsible for the clearance of IgA from the serum [16]. ASGPR primarily interacts with IgA2 and therefore may be responsible for the increased prevalence of IgA1 in the serum [21]. This review will focus primarily on the receptors pIgR and FcαRI, for which both structural and functional data are available. Fcα/μR is described more fully in another chapter in this issue.
pIgR structure and function
The polymeric immunoglobulin receptor (pIgR) is a glycosylated type I transmembrane protein that transports pIgs from the basolateral surface of mucosal epithelia into mucosal secretions. pIgR consists of a 620-residue extracellular region containing five Ig-like domains, a transmembrane helix, and a 103-residue cytoplasmic tail. Human pIgR binds and transports both pIgA and pIgM [22]. Binding of human pIgA to pIgR takes place in two steps. In the first step, a noncovalent interaction occurs between the N-terminal pIgR domain (D1) and the CH3, and possibly CH2, domains of one of the Fc regions of dIgA [23, 24]. In the second step, a disulfide bond is formed between Cys467 in the extracellular C-terminal domain (D5) of human pIgR and residue C311 in the CH2 domain of the second IgA molecule [25]. The pIg–pIgR complexes are then transcytosed to the apical surface of the mucosal epithelium where an unknown enzyme cleaves the extracellular domain of pIgR, releasing the pIg–pIgR complex from the membrane to form secretory Ig (SIg) [2]. The cleaved extracellular domain of human pIgR, also known as the secretory component (SC), is covalently attached via a disulfide bond to dIgA or noncovalently attached to pIgM [26, 27]. Free SC is also released into secretions [26].
Protection against microorganisms and bacterial toxins takes place at different stages of pIgR-mediated dIgA transport [1, 28]. First, SIgA can act via immune exclusion in the gut by binding and cross-linking microorganisms and inhibiting their adherence to the mucosal walls [1, 29, 30]. Second, SIgA can intercept and neutralize viral pathogens during transepithelial transport [31]. Third, dIgA can bind viruses and bacteria that have invaded the mucosal cell at the basolateral surface and pIgR can shuttle the antibody–antigen complex to the apical surface, removing the pathogen [32, 33]. Lastly, free SC in the absence of IgA can bind pathogens and bacterial toxins [34, 35].
The X-ray crystal structure of the N-terminal domain (D1) of human pIgR was recently solved (Fig. 2a) [36]. pIgR D1 folds as a V-type Ig-like domain with highly noncanonical conformations in the loops corresponding to the complementarity-determining regions (CDRs) in the epitope-binding sites of antibodies [36]. A single helical turn in CDR1 of D1 causes a hydrophobic residue (V29) that would typically be buried in Ig variable domains to instead become exposed to solvent, which is consistent with mutagenesis results implicating this region (T27, S28, N30, H32, and T33) as a dIgA binding site [37]. CDR2 consists of a very short, tightly turning loop containing a glutamic acid that may be required for IgM binding. It is interesting to note that only pIgR orthologs that bind pIgM (such as human and cow) have a charged residue in their CDR2 loops [22]. Unlike the CDR3 loop of antibody Ig domains, pIgR CDR3 folds back onto the body of D1, which precludes the dimerization mode observed for the VH–VL interface in antibody Fab fragments. Furthermore, binding experiments using surface plasmon resonance (SPR) spectroscopy showed that pIgR has significant affinity for J chain-containing dIgA and that the glycosylation state of the pIgR D1 did not affect binding [36]. Mutational studies have shown that D1 of pIgR interacts with residues in the DE and FG loops of the IgA CH3 domain [38, 39], and that the CDR2 loop is most important for this interaction, although both CDR1 and CDR3 loops also play a role [40].

Structural views of IgA receptors pIgR and FcαRI and the FcαRI:Fcα complex. (a) Crystal structure of the N-terminal domain of pIgR [36]. The CDR loops involved in binding dIgA are labeled. (b) Crystal structure of FcαRI, as seen in the FcαRI:Fcα complex [9]. Note the four N-linked glycosylation sites. (c) Crystal structure of the FcαRI:Fcα complex [9]. Two FcαRI molecules bind to a single Fcα dimer with D1 of each receptor contacting the CH2–CH3 interface on IgA1. (d) Close-up view of N-glycans in the FcαRI:Fcα complex. The IgA1 N-glycan (red) approaches within 8 Å of FcαRI but does not come into direct contact. In contrast, the N58 N-glycan of FcαRI (blue) does come into contact with IgA1
FcαRI inflammatory functions
The FcαR gene is found within the leukocyte receptor cluster on chromosome 19 [41]. This gene cluster includes genes encoding a number of different immune receptors, including the leukocyte imunoglobulin-like receptor (LIR) and killer cell immunoglobulin-like receptor (KIR) families, and also includes the collagen receptors GPVI and LAIR-1. Sequence analysis indicates that FcαRI and other receptors in the leukocyte receptor cluster contain one or more immunoglobulin-like (Ig) domains in their extracellular regions, similar to other Fc receptors found on chromosome 1, such as FcγRI, FcγRII, FcγRIII, and FcεRI. However, the receptors on chromosome 19 are only distantly related to those of chromosome 1. FcαRI is a glycosylated type I transmembrane receptor with two Ig-like domains in the extracellular region, a transmembrane helix, and a small cytoplasmic domain. An arginine in the transmembrane region of FcαRI forms a salt bridge with aspartate residues in two disulfide-linked FcR γ-chain co-receptors [42], which contain immunoreceptor tyrosine activation motifs (ITAM) capable of recruiting cytoplasmic signaling partners [43].
Clustering of the cell-surface FcαRI:γγ complex upon interaction with IgA-immune complexes leads to phosphorylation of the γ-chain and recruitment of SH2-containing downstream signaling molecules [1]. It seems likely that FcαRI signaling via the γ-chain resembles signaling by the T cell receptor via the ζ-chain in which orientation-independent receptor clustering, rather than formation of a specific oligomeric receptor conformation, activates the signaling cascade [44]. In addition to its role in signaling, the γ-chain governs the pathway of endocytosed FcαRI. Mutation of the intramembrane arginine in FcαRI abolishes the interaction with the γ-chain, and it alters the targeting of internalized FcαRI:IgA:antigen complexes [45]. Internalized complexes lacking γ-chain are directed to the recycling pathway and return IgA into the serum, whereas FcαRI:γ-chain:IgA:antigen complexes are targeted to the lysosome for degradation of the antigen [45]. It was also reported that a significant population of cell-surface FcαRI exists that is not associated with the γ-chain, suggesting that IgA catabolism or recycling by FcαRI is regulated by its interaction with the γ-chain [45]. Recent work showed that FcαRI is not simply tethered to γ-chain solely via intramembrane salt bridges, but that a more extensive interface exists between the transmembrane helices of FcαRI and the γ-chain dimer. Mutation of residues in these transmembrane regions of either FcαRI or γ-chain can abolish signaling [46, 47]. Clustering of FcαRI by IgA-immune complexes or anti-FcαRI monoclonal antibodies leads to shedding of the FcαRI ectodomain in a process that requires signaling via the γ-chain [48]. It is thought that this process may be responsible for fine-tuning the immune responses mediated through FcαRI. Furthermore, shedding of FcαRI by IgA1 was shown to have important implications on the development of IgAN symptoms, as described below [49].
FcαRI anti-inflammatory functions
Although secretory IgA is known to function by a passive mechanism and to have anti-inflammatory properties in the gut, serum IgA-immune complexes are able to trigger robust inflammatory responses via FcαRI [1]. However, it has long been noted that serum IgA itself has the capacity to diminish IgG-induced inflammatory responses. An intriguing recent report from Monteiro and colleagues [50] showed that the anti-inflammatory effects of serum IgA (in the absence of antigen) are also mediated through FcαRI. Thus, FcαRI appears to be unique in its ability to function both as an activating and inhibitory receptor. In vitro studies using rat mast cells transfected with FcαRI revealed that when bound by serum IgA or anti-FcαRI Fab, FcαRI was able to inhibit the activation of inflammatory responses via FcεRI or FcγRs. This inhibition required the presence of the γ-chain and involved the recruitment of SHP-1 to FcαRI, which blocked signaling via Syk, LAT, and ERK [50]. Furthermore, in vivo studies using a murine IgE-mediated asthma model further confirmed this inhibitory effect of FcαRI, suggesting that IgA may play a protective role in allergy by suppressing inflammatory responses [50]. These results were surprising because inhibitory functions are typically mediated via immunoreceptor tyrosine-based inhibitory motif (ITIM)-containing inhibitory receptors that co-aggregate with activating receptors. This is the first report of an ITAM receptor being able to transduce an inhibitory signal. FcαRI may therefore represent a new target for the design of anti-inflammatory therapies for asthma and allergy.
Crystal structure of FcαRI
The crystal structure of the human FcαRI ectodomain was solved alone and in complex with Fcα [9]. The FcαRI ectodomain is composed of two Ig-like domains oriented at approximately a 90° angle (Fig. 2b). Its overall structure closely resembles those of the related leukocyte Ig-like (LIRs) [51] and killer cell inhibitory receptors (KIRs) [52] and the platelet collagen receptor glycoprotein VI [53]. Although other Ig-like Fc receptors such as FcγRIIa, FcγRIIb, FcγRIII, and FcεRI also have tandem Ig-like domains oriented at an acute angle [54–57], the position of domain 1 (D1) is flipped over to the opposite side in FcαRI, LIRs, and KIRs relative to the other FcRs [9]. Ordered N-linked carbohydrate was observed at each of the four potential glycosylation sites in the IgA1-bound FcαRI ectodomain (in the free receptor, two N-glycans were observed in one molecule in the asymmetric unit, and the other two sites were observed in the second molecule).
Crystal structure of the FcαRI:Fcα complex
The structure of the FcαRI:Fcα complex showed that two FcαRI molecules bind a single Fcα dimer, consistent with prior analytical ultracentrifugation and SPR experiments (Fig. 2c) [9, 58]. Domain 1 of each FcαRI molecule interacts with residues in both the CH2 and CH3 domains of Fcα, as predicted by previous mutational work [59–63]. The binding interface is composed of a large hydrophobic patch surrounded by polar and charged residues that contribute potential hydrogen bonds [9]. The hydrophobic core in the binding interface consists of Fcα residues L257, L258, M433, L441, A442, F443 and the aliphatic portion of the R382 side chain packing against FcαRI residues Y35, L54, F56, G84, H85 and the aliphatic portion of K55. The surrounding residues that contribute hydrogen bonds include FcαRI R52, K55, R82, and Fcα R382. The binding interface observed in the crystal structure is consistent with prior mutational studies, although significant portions of the binding sites on both Fcα and FcαRI had not been previously identified.
Disease states linked to IgA
IgA nephropathy (IgAN)
IgA nephropathy (IgAN) is the most common form of glomerulonephritis, characterized by deposits of IgA1 in the glomerular mesangium. IgA1 deposition is followed by the infiltration of inflammatory immune cells, mesangial cell proliferation and mesangial matrix expansion [20]. IgAN, also called Berger’s disease [64], affects approximately 1.3% of the population worldwide [65] and leads to renal failure in 20–30% of cases within 10–20 years of onset. Although clinically well-characterized by the deposition of blood (hematuria) and protein (proteinuria) into the urine, it is often a silent disease, which may go undetected for many years [20]. In IgAN patients who have had kidney transplants due to renal failure, mesangial IgA deposition often recurs after several years [66], suggesting that IgAN is actually a blood-borne disease with effects localized to the kidney.
IgA1 and FcαRI aberrations implicated in IgA nephropathy
IgAN patients display several major abnormalities in the IgA system: (1) increased levels of serum IgA and IgA-immune complexes (IC) [20] (2) increased ratio of polymeric (pIgA) to monomeric (mIgA) [67, 68] and (3) the generation of abnormally glycosylated IgA1 [69]. The enhanced levels (twofold to threefold) of serum IgA cannot fully explain IgA deposition in the kidneys of IgAN patients because AIDS and IgA myeloma are often associated with increased plasma levels of IgA yet do not typically cause mesangial IgA deposits [20]. The higher proportion of polymeric IgA1 in IgAN patient serum could have important implications given that mesangial deposits in IgAN are primarily composed of polymeric IgA1 [70]. A large number of alterations in IgA1 O-glycosylation and N-glycosylation were reported, including oversialylation [71] or undersialylation and undergalactosylation [72] of the O-glycans, and oversialylation [73] or truncation [74] of the N-glycans. Furthermore, an overall decrease in the number of O-glycosylation sites in the IgA1 hinge region was reported in IgA1 [72].
Abnormal glycosylation events potentially alter a number of molecular events in IgAN. Oversialylated IgA1 has a reduced affinity for ASPGR, which would reduce clearance by the liver and therefore lead to higher serum IgA1 levels [75]. Undersialylation and undergalactosylation lead to the aggregation of IgA1 in a hinge-specific manner [76], and undergalactosylation of IgA1 leads to increased adhesion to extracellular matrix proteins [77]. Anti-IgA antibodies recognize epitopes on underglycosylated IgA1, and injection of enzymatically deglycosylated IgA1 in rats leads to mesangial deposits and recruitment of neutrophils to the mesangium [78]. Finally, IgA1 from IgAN patients was reported to show altered affinity for IgA receptors: either increased [79] or decreased [80] binding to FcαRI-expressing cells, and increased binding to transferrin receptor (TfR)-expressing cells [81].
An interesting feature of the IgA1 structure is the accessibility of its glycan moieties. The solution structure of intact IgA1 revealed that the O-glycosylated hinge region is highly extended (Fig. 1a), similar to other O-glycosylated mucin-like peptides such as the CD8 stalk [82]. Furthermore, the N-glycans on the IgA1 CH2 domains are almost completely solvent exposed, unlike the CH2 or CH3 N-glycans on IgG or IgE, respectively (Fig. 1d). The exposed nature of these glycans could have important implications for their accessibility to lectin-like receptors. Furthermore, the propensity of undergalactosylated IgA1 to aggregate [76] is consistent with the exposed nature of the hinge region containing the affected O-glycans.
Less is known about potential FcαRI aberrations in IgAN. Impaired endocytosis of FcαRI in IgAN was described [83], leading to increased IgA recycling and higher serum IgA levels. Furthermore, FcαRI with increased N-glycosylation levels and potential FcαRI splice variants were reported in IgAN patients [84], although their significance in the pathogenesis of IgAN is unknown. The crystal structure of FcαRI complexed with Fcα revealed that one N-glycan attached to N58 of FcαRI does come into contact with Fcα (Fig. 2d) [9], suggesting that alterations in the glycosylation profile of FcαRI could play a role in modulating the FcαRI:IgA interaction.
Role of the FcαRI–IgA interaction in a mouse model of IgA nephropathy
In a study by Monteiro and colleagues, circulating soluble FcαRI ectodomain bound to IgA1 was found in 40% of IgAN patients but not in patients with other diseases of the immune system or kidney [49]. To determine the role that FcαRI plays in the pathogenesis of IgAN, they generated several transgenic mouse lines expressing human FcαRI on monocytes and macrophages (mice do not express an FcαRI homolog) [49]. The transgenic mice developed essentially all the symptoms of IgAN, including mesangial IgA deposits, infiltration of macrophages in the mesangial and interstitial areas, hematuria, and proteinuria. Rag2−/− SCID mice (expressing no FcαRI or IgA) injected with serum from the FcαRI-transgenic mice also developed IgAN symptoms, indicating that the cause of disease was blood-borne. Depletion of FcαRI from the transgenic mouse serum before injection into the Rag2−/− SCID mice led to less pronounced IgAN symptoms. Finally, an FcαRI-transgenic SCID mouse (expressing FcαRI but no IgA) did not develop IgAN symptoms unless IgAN patient-derived human IgA1 was injected. These elegant experiments revealed that soluble FcαRI and IgA together were required for the development of IgAN-like symptoms. It should be pointed out that mouse IgA is predominantly polymeric (unlike human IgA1) and has fewer N-glycosylation sites than human IgA1.
Interaction of the transferrin receptor with the IgA1 immune complexes in the glomerulus
An IgA-specific receptor was reported on kidney mesangial cells [85]. This receptor was distinct from other known IgA receptors such as FcαRI, pIgR, or ASGPR because these are not expressed on mesangial cells [86, 87]. Recently, Moura et al. demonstrated that, unexpectedly, TfR is a mesangial IgA receptor [12]. TfR specifically binds IgA1 but not IgA2 with clear preference for polymeric IgA1. Deletion of either O-glycans or N-glycans of IgA1 abolished binding to TfR [81]. It is interesting to note that TfR is overexpressed on mesangial cells in patients with IgAN relative to healthy controls [88], and IgAN patient-derived IgA1 bound more tightly to TfR-expressing cells [81]. These results suggest that TfR may be responsible for trapping IgA1 in the mesangium, leading to the well-known mesangial IgA1 deposits that are hallmarks of IgAN.
Model for IgA1-receptor interactions in IgAN
In a current model for IgAN, the altered glycosylation or polymerization state of circulating IgA1 leads to higher affinity for, and increased shedding of, the FcαRI ectodomain, resulting in tightly associated circulating FcαRI:IgA1 immune complexes. These circulating FcαRI:IgA1 complexes are bound by mesangial cell TfR, leading to IgA1 deposition in the mesangium [20]. Although the role of FcαRI is unclear, it may be that the presence of soluble FcαRI in circulating IgA1 immune complexes favors their deposition due to either the increased size of the complex or due to the N-glycans of FcαRI itself binding to lectin-like mesangial receptors.
N-glycans in the FcαRI:Fcα crystal structure
Given the aberrant N-glycosylation and O-glycosylation of IgA1 in IgAN patients, and the reported abnormalities in N-glycosylation of FcαRI in some cases, the contribution of glycans to the FcαRI:Fcα binding interface is of significant interest. The O-glycosylated hinge is located far from the FcαRI binding site on IgA1, and thus the O-glycans would not be expected to affect the binding of FcαRI. In the crystal structure of the FcαRI:Fcα complex described above, N-glycans were observed at all potential N-linked glycosylation sites of both FcαRI and Fcα in the complex [9]. Of the four sites on FcαRI, the N-glycan attached to FcαRI N58 extends toward the CH3 domain of Fcα and forms two potential hydrogen bonds with E348 of Fcα (Fig. 2d, see the blue N-glycans) The crystallized FcαRI ectodomain was expressed in insect cells, yielding high-mannose N-glycans. Therefore, the complex N-glycans found on the natural human receptor might interact more extensively with IgA1. Another interesting feature of FcαRI glycosylation is that the two N-glycans on the C-terminal domain extend outward from the complex by up to 28 Å (Fig. 2c), resulting in an overall width of the FcαRI:Fcα complex of 193 Å. The large dimensions of the complex, combined with the long polysaccharides extending out on either end, could contribute to the trapping of circulating soluble FcαRI:IgA1 complexes in the mesangium of the kidney in IgAN, particularly if lectin-like receptors are involved.
In the FcαRI:Fcα crystal structure, the N-glycans of Fcα do not contact FcαRI, although they approach within 8 Å of the receptor (Fig. 2d, see the red N-glycans) [9]. The CHO cell-expressed Fcα that was crystallized has complex carbohydrate similar to that found on human proteins, so it should represent a close approximation to the N-glycans on normal human IgA1. However, the electron density was rather weak for the N-glycans and as a result, only one branch of the biantennary oligosaccharide was visible. It is therefore possible that IgA1 N-glycans could play a role in interacting with FcαRI in vivo, and further structural work is needed to address this issue.
Allergy and asthma
Various studies have indicated that mucosal and systemic production of allergen-specific IgA occurs in patients with asthma and/or allergic rhinitis [89–91]. At sites of allergic inflammation where basophils and eosinophils play an active role, mucous secretions contain large amounts of SIgA [92]. In vitro studies using immobilized SIgA and mIgA showed that SIgA but not mIgA can induce histamine release from human basophils [93]. SIgA-mediated basophil degranulation may therefore play an important role in the exacerbation of allergic inflammation at the mucosal surface. Eosinophils can also trigger allergic responses in asthma via respiratory burst, which produces superoxide anions and reactive oxygen species [94]. In a recent study, IgA—particularly the secretory form—was shown to potently induce NADPH oxidase activity and eosinophil degranulation in an FcαRI-dependent process [95]. Furthermore, when incubated with SIgA in vitro, blood eosinophils release large amounts of eosinophil cationic protein, eosinophil peroxidase, eosinophil derived neurotoxin, and various cytokines [96].
Conversely, despite the ability of SIgA to activate eosinophils, there is evidence that IgA can also suppress inflammatory responses in asthma and allergy [97]. Chronic obstructive pulmonary disease was associated with a decrease in secretory IgA levels, apparently due to the downregulation of pIgR on bronchial epithelial cells [97]. Children with low IgA levels showed higher likelihood of asthma, otitis media and allergy, and the severity of allergic responses was elevated relative to children with intermediate or high IgA levels [98]. Furthermore, pretreatment of mice with allergen-specific monoclonal IgA prevented airway hyperresponsiveness and the infiltration of eosinophils upon allergen challenge [99].
Ankylosing spondylitis (AS)
Ankylosing spondylitis (AS) is a chronic inflammatory form of arthritis that affects the spinal joints. Patients with AS have elevated levels of serum monomeric IgA1 [100, 101]. AS may represent an abnormal IgA response to bacterial infection, as elevated jejeunal SIgA levels can be reduced by treatment with the antibiotic sulfasalazine [102]. Furthermore, increased levels of IgA1 and IgA2 specific to Klebsiella pneumoniae were reported in AS patients [103]. Recently, FcαRI expression was found to be downregulated in blood phagocytes of AS patients with increased serum IgA [104]. The increased levels of serum IgA and IgA-IC could be explained by altered FcαRI-mediated endocytosis and increased recycling of IgA towards the cell surface [100]. It is interesting to note that AS patients who also display IgA deficiency were shown to have particularly severe AS [100]. This suggests that IgA and IgA-IC may not be directly involved in the pathogenesis of AS and that instead, increased serum IgA may actually serve a protective role against joint inflammation.
Celiac disease
Celiac disease is a gastrointestinal disorder triggered by immune responses to wheat proteins, particularly gliadins that are modified by the tissue enzyme transglutaminase in genetically susceptible individuals. IgA and IgG antibodies to gliadin show cross-reactivity to tissue transglutaminase, inducing inflammatory responses that eventually cause a loss of villi within the intestinal epithelium, leading to poor nutrient absorption [105]. In addition, IgA antibodies to a variety of food allergens were found in patients with celiac disease [106]. IgA autoantibodies in celiac disease are often associated with dermatitis herpetiformis in which IgA is deposited in affected skin [107]. Although IgA antibodies can be helpful in the diagnosis of celiac disease, the link between IgA and pathogenesis is unclear, as the disease appears to be primarily T cell-mediated [105]. Furthermore, it was shown that IgA deficiency is associated with a tenfold risk of celiac disease, suggesting that, as for ankylosing spondylitis, IgA may play a protective role [108].
Other disease states with elevated or aberrant IgA populations
A number of diseases, including alcoholic cirrhosis, AIDS, IgA myeloma, Sjögren’s syndrome and Henoch–Schönlein purpura, exhibit elevated levels of serum IgA. However, the relevance of increased IgA to disease pathogenesis is unknown. In alcoholic cirrhosis and AIDS the increased IgA levels may be associated with aberrant expression of FcαRI and defective FcαRI-mediated endocytosis [79, 109]. In the autoimmune disease Sjögren’s syndrome, characterized by lymphoid cell infiltration of lacrimal and salivary glands, the N-glycans of monomeric IgA1 are oversialylated [110], which could presumably affect the FcαRI–IgA interaction and alter recycling of IgA. Furthermore, in Henoch–Schönlein purpura (HSP), a form of systemic vasculitis, IgA deposits form in affected blood vessels [111]. In HSP patients who have renal involvement, undersialylation [112] and undergalactosylation [113] of IgA1 O-glycans were reported, leading to IgA1 deposition in the glomerulus, in a manner similar to IgAN. Indeed, HSP and IgAN may actually represent different degrees of the same disease with HSP being the more severe form and IgAN being only the renal manifestation of HSP.
Therapeutic approaches based on IgA-receptor interactions
Anti-tumor immunotherapy
Antibody-based immunotherapy has become an effective treatment for a number of different cancers. Therapeutic monoclonal antibodies (mAb) can induce tumor cell death by a variety of mechanisms. Some act by simply cross-linking antigens on tumor cells, leading to apoptosis, cell cycle arrest or the inhibition of cell proliferation [114]. Others act indirectly via FcR-mediated effector functions of immune cells such as phagocytosis, Ab-dependent cellular cytotoxicity and enhanced presentation of tumor antigens [115].
A number of studies showed that therapeutic antibodies able to trigger immune responses via FcαRI show exciting potential. These antibodies can either be intact IgA or bispecific antibodies (BsAb) that recognize both the ectodomain of FcαRI and the tumor antigen of interest [116]. In a comparison of hapten-directed antibodies of different human isotypes, IgA2 was found to be more effective than IgG isotypes at recruiting neutrophils and inducing tumor cell death [116]. Similarly, an IgA1 antibody against EpCAM proved more effective than IgG1 at recruiting neutrophils to kill EpCAM-positive, solid tumor cells [117]. A comparison of bispecific antibodies targeting FcαRI or FcγRs in humans and transgenic mice showed that FcαRI was the most effective FcR at triggering tumor cell killing and enhanced migration of neutrophils into tumors [118]. Upon granulocyte colony-stimulating factor (G-CSF) treatment, immature neutrophils are mobilized from the bone marrow and mediate tumor cell lysis via FcαRI but are incapable of tumor cell killing via the FcγR [118]. Since neutrophils constitute the most abundant cytotoxic cells in humans, improved neutrophil recruitment may enhance tumor cell lysis in whole blood, especially when tumor cells are complement-resistant or when neutrophils were primed by G-CSF or granulocyte-macrophage colony-stimulating factor (GM-CSF) [119].
Anti-inflammatory therapy
FcαRI has the unique ability to mediate either activating or inhibitory inflammatory responses depending on the nature of its interactions with IgA [50]. Monoclonal Fabs targeting the FcαRI ectodomain or engineered IgA constructs could therefore play a valuable therapeutic role in inhibiting inflammatory processes in autoimmune disorders or allergy and asthma. For example, leukocyte infiltration, a key characteristic of asthma, was markedly inhibited by treatment with anti-FcαRI mAb in a murine model of IgE-triggered asthma [50]. The key advantage of this approach is that the inhibitory response mediated via FcαRI extends to both IgG-specific and IgE-specific receptors, suggesting the potential for widespread anti-inflammatory effects.
Mucosal vaccines
In the gut, secretory IgA plays an important role in immune exclusion and pathogen clearance, as described above. However, there is also evidence that specific mechanisms are in place for sampling gut antigens complexed with SIgA. Antigen sampling occurs in discrete patches of organized gut-associated lymphoid tissue (GALT) such as the Peyer’s patches of the distal ileum [120, 121]. Specialized cells called microfold or M cells in the Peyer’s patches mediate transcytosis of antigens and microbes across the gut epithelial layer and delivery to lymphoid and dendritic cells found in a pocket in the basolateral surface of the M cell. Upon antigen capture, immature dendritic cells then migrate from the GALT to lymphoid organs for antigen presentation to naïve T cells [120, 121]. Thus, both systemic and mucosal immune responses can be generated by mucosal antigens, and the GALT represents a potential avenue for vaccination. A particularly attractive possibility is the development of oral vaccines that can lead to both mucosal and systemic responses.
There are a couple of important aspects related to IgA when discussing mucosal vaccination. First, vaccines capable of inducing a strong pathogen-specific SIgA response in the mucosae could be very effective at preventing infection because for most pathogens, the initial adherence and invasion occur via the respiratory, genitourinary or gastrointestinal tissues. Furthermore, it was demonstrated that protection against reinfection by various pathogens is better correlated with levels of specific SIgA antibodies than with serum antibodies [122]. A prime example of this is the oral polio vaccine in which efficacy was correlated with the production of mucosal anti-polio IgA [123]. Intranasal vaccination with capsular polysaccharides against S. pneumonaie, influenza and adenoviruses was also found to elicit a protective SIgA response [122, 124]. However, despite the promise of this type of approach, mucosal vaccines are limited due to the difficulty in eliciting a response to nonreplicating agents, probably because of their rapid clearance/degradation in the gut [97, 120]. A second avenue of investigation involves using SIgA itself as a carrier for inducing immunity via the gut. M cells express a receptor for SIgA that can transcytose SIgA or SIgA-complexed antigens [14]. Targeting this receptor could be an effective approach for developing an oral vaccine. For example, Corthésy et al. created antigenized SIgA by inserting a linear epitope from Shigella flexneri invasin B into the secretory component and reconstituting into SIgA [125]. This antigenized SIgA was able to immunize mice, as shown by the production of both serum and mucosal antibodies specific for invasin B.
Conclusions
Recent studies have provided significant insights into IgA-mediated processes in health and human disease. Structural data on intact IgA1 and IgA2 in solution have highlighted unusual aspects of these antibodies, such as the extended O-glycosylated hinge of IgA1 and the compact conformation of IgA2. Furthermore, crystallographic studies on pIgR, FcαRI, and the FcαRI:IgA1-Fc complex have revealed the nature of the IgA-binding sites and have revealed highly solvent-exposed N-glycans in IgA1 with possible implications for IgA nephropathy. Functional studies have elucidated the intriguing dual nature of IgA in which it is capable of both activating and inhibiting inflammatory responses via FcαRI. This duality is observed in many of the disease states in which IgA was implicated. Additional research will be needed to better understand how FcαRI can distinguish between activating and inhibitory signals and to develop new strategies for the control of these processes for therapeutic use. Finally, recent results have provided exciting glimpses into the potential uses of IgA and its receptors for novel therapies to fight cancer, ameliorate autoimmune diseases, or immunize patients in a straightforward manner through the use of oral vaccines.
References
Monteiro RC, van de Winkel JG (2003) IgA Fc receptors. Annu Rev Immunol 177–204
Norderhaug IN, Johansen FE, Schjerven H, Brandtzaeg P (1999) Regulation of the formation and external transport of secretory immunoglobulins. Crit Rev Immunol 19:481–508
van Egmond M, van Garderen E, van Spriel AB, Damen CA, van Amersfoort ES, van Zandbergen G, van Hattum J, Kuiper J, van de Winkel JG (2000) FcaRI-positive liver Kupffer cells: reappraisal of the function of immunoglobulin A in immunity. Nat Med 6:680–685
Vidarsson G, van Der Pol WL, van Den Elsen JM, Vile H, Jansen M, Duijs J, Morton HC, Boel E, Daha MR, Corthesy B, van De Winkel JG (2001) Activity of human IgG and IgA subclasses in immune defense against Neisseria meningitidis serogroup B. J Immunol 166:6250–6256
Monteiro RC, Kubagawa H, Cooper MD (1990) Cellular distribution, regulation, and biochemical nature of an Fc alpha receptor in humans. J Exp Med 171:597–613
Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T, Eyre HJ, Sutherland GR, Endo Y, Fujita T, Miyabayashi T, Sakano S, Tsuji T, Nakayama E, Phillips JH, Lanier LL, Nakauchi H (2000) Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat Immunol 1:441–446
Boehm MK, Woof JM, Kerr MA, Perkins SJ (1999) The Fab and Fc fragments of IgA1 exhibit a different arrangement from that in IgG: a study by X-ray and neutron solution scattering and homology modelling. J Mol Biol 286:1421–1447
Keown MB, Ghirlando R, Young RJ, Beavil AJ, Owens RJ, Perkins SJ, Sutton BJ, Gould HJ (1995) Hydrodynamic studies of a complex between the Fc fragment of human IgE and a soluble fragment of the Fc epsilon RI alpha chain. Proc Natl Acad Sci USA 92:1841–1845
Herr AB, Ballister ER, Bjorkman PJ (2003) Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature 423:614–620
Mostov KE (1994) Transepithelial transport of immunoglobulins. Annu Rev Immunol 12:63–84
Maliszewski CR, March CJ, Schoenborn MA, Gimpel S, Shen L (1990) Expression cloning of a human Fc receptor for IgA. J Exp Med 172:1665–1672
Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, Cooper MD, Monteiro RC (2001) Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med 194:417–425
Lamkhioued B, Gounni AS, Gruart V, Pierce A, Capron A, Capron M (1995) Human eosinophils express a receptor for secretory component. Role in secretory IgA-dependent activation. Eur J Immunol 25:117–125
Mantis NJ, Cheung MC, Chintalacharuvu KR, Rey J, Corthesy B, Neutra MR (2002) Selective adherence of IgA to murine Peyer’s patch M cells: evidence for a novel IgA receptor. J Immunol 169:1844–1851
Mota G, Manciulea M, Cosma E, Popescu I, Hirt M, Jensen-Jarolim E, Calugaru A, Galatiuc C, Regalia T, Tamandl D, Spittler A, Boltz-Nitulescu G (2003) Human NK cells express Fc receptors for IgA which mediate signal transduction and target cell killing. Eur J Immunol 33:2197–2205
Mestecky J, McGhee JR (1987) Immunoglobulin A (IgA): molecular and cellular interactions involved in IgA biosynthesis and immune response. Adv Immunol 40:153–245
Sangeetha SR, Appukuttan PS (2005) IgA1 is the premier serum glycoprotein recognized by human galectin-1 since T antigen (Galbeta1→3GalNAc−) is far superior to non-repeating N-acetyl lactosamine as ligand. Int J Biol Macromol 35:269–276
Rudd PM, Fortune F, Patel T, Parekh RB, Dwek RA, Lehner T (1994) A human T-cell receptor recognizes ‘O’-linked sugars from the hinge region of human IgA1 and IgD. Immunology 83:99–106
van Spriel AB, Leusen JH, van Egmond M, Dijkman HB, Assmann KJ, Mayadas TN, van de Winkel JG (2001) Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood 97:2478–2486
Monteiro RC, Moura IC, Launay P, Tsuge T, Haddad E, Benhamou M, Cooper MD, Arcos-Fajardo M (2002) Pathogenic significance of IgA receptor interactions in IgA nephropathy. Trends Mol Med 8:464–468
Rifai A, Fadden K, Morrison SL, Chintalacharuvu KR (2000) The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J Exp Med 191:2171–2182
Socken DJ, Underdown BJ (1978) Comparison of human, bovine and rabbit secretory component-immunoglobulin interactions. Immunochemistry 15:499–506
Frutiger S, Hughes GJ, Hanly WC, Kingzette M, Jaton JC (1986) The amino-terminal domain of rabbit secretory component is responsible for noncovalent binding to immunoglobulin A dimers. J Biol Chem 261:16673–16681
Geneste C, Iscaki S, Mangalo R, Pillot J (1986) Both Fc alpha domains of human IgA are involved in in vitro interaction between secretory component and dimeric IgA. Immunol Lett 13:221–226
Fallgreen-Gebauer E, Gebauer W, Bastian A, Kratzin HD, Eiffert H, Zimmermann B, Karas M, Hilschmann N (1993) The covalent linkage of secretory component to IgA. Structure of sIgA. Biol Chem Hoppe Seyler 374:1023–1028
Brandtzaeg P (1975) Human secretory immunoglobulin M. An immunochemical and immunohistochemical study. Immunology 29:559–570
Mostov KE, Kraehenbuhl JP, Blobel G (1980) Receptor-mediated transcellular transport of immunoglobulin: synthesis of secretory component as multiple and larger transmembrane forms. Proc Natl Acad Sci USA 77:7257–7261
Hamburger AE, Bjorkman PJ, Herr AB (2006) Structural insights into antibody-mediated mucosal immunity. Curr Top Microbiol Immunol 308:173–204
Lamm ME (1997) Interaction of antigens and antibodies at mucosal surfaces. Annu Rev Microbiol 51:311–340
Brandtzaeg P, Baekkevold ES, Farstad IN, Jahnsen FL, Johansen FE, Nilsen EM, Yamanaka T (1999) Regional specialization in the mucosal immune system: what happens in the microcompartments? Immunol Today 20:141–151
Mazanec MB, Kaetzel CS, Lamm ME, Fletcher D, Nedrud JG (1992) Intracellular neutralization of virus by immunoglobulin A antibodies. Proc Natl Acad Sci USA 89:6901–6905
Kaetzel CS, Robinson JK, Chintalacharuvu KR, Vaerman JP, Lamm ME (1991) The polymeric immunoglobulin receptor (secretory component) mediates transport of immune complexes across epithelial cells: a local defense function for IgA. Proc Natl Acad Sci USA 88:8796–8800
Lamm ME, Robinson JK, Kaetzel CS (1992) Transport of IgA immune complexes across epithelial membranes: new concepts in mucosal immunity. Adv Exp Med Biol 327:91–94
Dallas SD, Rolfe RD (1998) Binding of Clostridium difficile toxin A to human milk secretory component. J Med Microbiol 47:879–888
de Oliveira IR, de Araujo AN, Bao SN, Giugliano LG (2001) Binding of lactoferrin and free secretory component to enterotoxigenic Escherichia coli. FEMS Microbiol Lett 203:29–33
Hamburger AE, West AP Jr, Bjorkman PJ (2004) Crystal structure of a polymeric immunoglobulin binding fragment of the human polymeric immunoglobulin receptor. Structure 12:1925–1935
Coyne RS, Siebrecht M, Peitsch MC, Casanova JE (1994) Mutational analysis of polymeric immunoglobulin receptor/ligand interactions. Evidence for the involvement of multiple complementarity determining region (CDR)-like loops in receptor domain I. J Biol Chem 269:31620–31625
White KD, Capra JD (2002) Targeting mucosal sites by polymeric immunoglobulin receptor-directed peptides. J Exp Med 196:551–555
Hexham JM, White KD, Carayannopoulos LN, Mandecki W, Brisette R, Yang YS, Capra JD (1999) A human immunoglobulin (Ig)A calpha3 domain motif directs polymeric Ig receptor-mediated secretion. J Exp Med 189:747–752
Roe M, Norderhaug IN, Brandtzaeg P, Johansen FE (1999) Fine specificity of ligand-binding domain 1 in the polymeric Ig receptor: importance of the CDR2-containing region for IgM interaction. J Immunol 162:6046–6052
Wende H, Colonna M, Ziegler A, Volz A (1999) Organization of the leukocyte receptor cluster (LRC) on human chromosome 19q13.4. Mamm Genome 10:154–160
Feng J, Garrity D, Call ME, Moffett H, Wucherpfennig KW (2005) Convergence on a distinctive assembly mechanism by unrelated families of activating immune receptors. Immunity 22:427–438
Morton HC, van den Herik-Oudijk IE, Vossebeld P, Snijders A, Verhoeven AJ, Capel PJ, van de Winkel JG (1995) Functional association between the human myeloid immunoglobulin A Fc receptor (CD89) and FcR γ chain. Molecular basis for CD89/FcR γ chain association. J Biol Chem 270:29781–29787
Cochran JR, Cameron TO, Stone JD, Lubetsky JB, Stern LJ (2001) Receptor proximity, not intermolecular orientation, is critical for triggering T-cell activation. J Biol Chem 276:28068–28074
Launay P, Patry C, Lehuen A, Pasquier B, Blank U, Monteiro RC (1999) Alternative endocytic pathway for immunoglobulin A Fc receptors (CD89) depends on the lack of FcRgamma association and protects against degradation of bound ligand. J Biol Chem 274:7216–7225
Wines BD, Trist HM, Ramsland PA, Hogarth PM (2006) A common site of the Fc receptor gamma subunit interacts with the unrelated immunoreceptors FcalphaRI and FcepsilonRI. J Biol Chem 281:17108–17113
Bakema JE, de Haij S, den Hartog-Jager CF, Bakker J, Vidarsson G, van Egmond M, van de Winkel JG, Leusen JH (2006) Signaling through mutants of the IgA receptor CD89 and consequences for Fc receptor gamma-chain interaction. J Immunol 176:3603–3610
van Zandbergen G, Westerhuis R, Mohamad NK, van de Winkel JG, Daha MR, van Kooten C (1999) Crosslinking of the human Fc receptor for IgA (FcalphaRI/CD89) triggers FcR gamma-chain-dependent shedding of soluble CD89. J Immunol 163:5806–5812
Launay P, Grossetete B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, Patey-Mariaud de Serre N, Lehuen A, Monteiro RC (2000) Fcalpha receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med 191:1999–2009
Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, Henin D, Benhamou M, Pretolani M, Blank U, Monteiro RC (2005) Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity 22:31–42
Chapman TL, You I, Joseph IM, Bjorkman PJ, Morrison SL, Raghavan M (1999) Characterization of the interaction between the herpes simplex virus type I Fc receptor and immunoglobulin G. J Biol Chem 274:6911–6919
Fan QR, Mosyak L, Winter CC, Wagtmann N, Long EO, Wiley DC (1997) Structure of the inhibitory receptor for human natural killer cells resembles haematopoietic receptors. Nature 389:96–100
Horii K, Kahn ML, Herr AB (2006) Structural basis for platelet collagen responses by the immune-type receptor glycoprotein VI. Blood 108:936–942
Garman SC, Kinet JP, Jardetzky TS (1998) Crystal structure of the human high-affinity IgE receptor. Cell 95:951–961
Maxwell KF, Powell MS, Hulett MD, Barton PA, McKenzie IF, Garrett TP, Hogarth PM (1999) Crystal structure of the human leukocyte Fc receptor, FcγRIIa. Nat Struct Biol 6:437–442
Sondermann P, Huber R, Oosthuizen V, Jacob U (2000) The 3.2-A crystal structure of the human IgG1 Fc fragment-FcgammaRIII complex. Nature 406:267–273
Sondermann P, Huber R, Jacob U (1999) Crystal structure of the soluble form of the human Fcgamma-receptor IIb: a new member of the immunoglobulin superfamily at 1.7 A resolution. EMBO J 18:1095–1103
Herr AB, White CL, Milburn C, Wu C, Bjorkman PJ (2003) Bivalent binding of IgA1 to FcalphaRI suggests a mechanism for cytokine activation of IgA phagocytosis. J Mol Biol 327:645–657
Carayannopoulos L, Hexham JM, Capra JD (1996) Localization of the binding site for the monocyte immunoglobulin (Ig) A-Fc receptor (CD89) to the domain boundary between Calpha2 and Calpha3 in human IgA1. J Exp Med 183:1579–1586
Pleass RJ, Anderson CM, Dunlop JI, Woof JM (1997) Probing the Fc alpha R binding site on IgA by mutagenesis of the IgA Fc region. Biochem Soc Trans 25:328S
Pleass RJ, Dunlop JI, Anderson CM, Woof JM (1999) Identification of residues in the CH2/CH3 domain interface of IgA essential for interaction with the human fcalpha receptor (FcalphaR) CD89. J Biol Chem 274:23508–23514
Wines BD, Hulett MD, Jamieson GP, Trist HM, Spratt JM, Hogarth PM (1999) Identification of residues in the first domain of human Fc alpha receptor essential for interaction with IgA. J Immunol 162:2146–2153
Wines BD, Sardjono CT, Trist HM, Lay CS, Hogarth PM (2001) The interaction of Fc alpha RI with IgA and its implications for ligand binding by immunoreceptors of the leukocyte receptor cluster. J Immunol 166:1781–1789
Berger J, Hinglais N (1968) [Intercapillary deposits of IgA–IgG]. J Urol Nephrol (Paris) 74:694–695
Savoldi S (2001) Berger’s disease. In: F Scolari (ed) Orphanet encyclopedia (updated: January 2004)
Berger J, Yaneva H, Nabarra B, Barbanel C (1975) Recurrence of mesangial deposition of IgA after renal transplantation. Kidney Int 7:232–241
Valentijn RM, Radl J, Haaijman JJ, Vermeer BJ, Weening JJ, Kauffmann RH, Daha MR, van Es LA (1984) Circulating and mesangial secretory component-binding IgA-1 in primary IgA nephropathy. Kidney Int 26:760–766
Novak J, Julian BA, Tomana M, Mesteck J (2001) Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol 21:310–327
Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J (1999) Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest 104:73–81
Tomino Y, Sakai H, Miura M, Endoh M, Nomoto Y (1982) Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clin Exp Immunol 49:419–425
Leung JC, Tang SC, Chan DT, Lui SL, Lai KN (2002) Increased sialylation of polymeric lambda-IgA1 in patients with IgA nephropathy. J Clin Lab Anal 16:11–19
Odani H, Hiki Y, Takahashi M, Nishimoto A, Yasuda Y, Iwase H, Shinzato T, Maeda K (2000) Direct evidence for decreased sialylation and galactosylation of human serum IgA1 Fc O-glycosylated hinge peptides in IgA nephropathy by mass spectrometry. Biochem Biophys Res Commun 271:268–274
Linossier MT, Palle S, Berthoux F (2003) Different glycosylation profile of serum IgA1 in IgA nephropathy according to the glomerular basement membrane thickness: normal versus thin. Am J Kidney Dis 41:558–564
Amore A, Cirina P, Conti G, Brusa P, Peruzzi L, Coppo R (2001) Glycosylation of circulating IgA in patients with IgA nephropathy modulates proliferation and apoptosis of mesangial cells. J Am Soc Nephrol 12:1862–1871
Basset C, Devauchelle V, Durand V, Jamin C, Pennec YL, Youinou P, Dueymes M (1999) Glycosylation of immunoglobulin A influences its receptor binding. Scand J Immunol 50:572–579
Iwase H, Ohkawa S, Ishii-Karakasa I, Hiki Y, Kokubo T, Sano T, Tanaka A, Toma K, Kobayashi Y, Hotta K (1999) Study of the relationship between sticky human serum IgA1 and its O-glycan glycoform. Biochem Biophys Res Commun 261:472–477
Kokubo T, Hiki Y, Iwase H, Tanaka A, Toma K, Hotta K, Kobayashi Y (1998) Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J Am Soc Nephrol 9:2048–2054
Sano T, Hiki Y, Kokubo T, Iwase H, Shigematsu H, Kobayashi Y (2002) Enzymatically deglycosylated human IgA1 molecules accumulate and induce inflammatory cell reaction in rat glomeruli. Nephrol Dial Transplant 17:50–56
Grossetete B, Launay P, Lehuen A, Jungers P, Bach JF, Monteiro RC (1998) Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: evidence for a negative regulatory role of serum IgA. Kidney Int 53:1321–1335
van Zandbergen G, van Kooten C, Mohamad NK, Reterink TJ, de Fijter JW, van de Winkel JG, Daha MR (1998) Reduced binding of immunoglobulin A (IgA) from patients with primary IgA nephropathy to the myeloid IgA Fc-receptor, CD89. Nephrol Dial Transplant 13:3058–3064
Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, Novak J, Vrtovsnik F, Haddad E, Chintalacharuvu KR, Monteiro RC (2004) Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J Am Soc Nephrol 15:622–634
Merry AH, Gilbert RJ, Shore DA, Royle L, Miroshnychenko O, Vuong M, Wormald MR, Harvey DJ, Dwek RA, Classon BJ, Rudd PM, Davis SJ (2003) O-glycan sialylation and the structure of the stalk-like region of the T cell co-receptor CD8. J Biol Chem 278:27119–27128
Monteiro RC, Grossetete B, Nguyen AT, Jungers P, Lehuen A (1995) Dysfunctions of Fc alpha receptors by blood phagocytic cells in IgA nephropathy. Contrib Nephrol 111:116–122
Toyabe S, Kuwano Y, Takeda K, Uchiyama M, Abo T (1997) IgA nephropathy-specific expression of the IgA Fc receptors (CD89) on blood phagocytic cells. Clin Exp Immunol 110:226–232
Barratt J, Greer MR, Pawluczyk IZ, Allen AC, Bailey EM, Buck KS, Feehally J (2000) Identification of a novel Fcalpha receptor expressed by human mesangial cells. Kidney Int 57:1936–1948
Westerhuis R, Van Zandbergen G, Verhagen NA, Klar-Mohamad N, Daha MR, van Kooten C (1999) Human mesangial cells in culture and in kidney sections fail to express Fc alpha receptor (CD89). J Am Soc Nephrol 10:770–778
Leung JC, Tsang AW, Chan DT, Lai KN (2000) Absence of CD89, polymeric immunoglobulin receptor, and asialoglycoprotein receptor on human mesangial cells. J Am Soc Nephrol 11:241–249
Haddad E, Moura IC, Arcos-Fajardo M, Macher MA, Baudouin V, Alberti C, Loirat C, Monteiro RC, Peuchmaur M (2003) Enhanced expression of the CD71 mesangial IgA1 receptor in Berger disease and Henoch–Schonlein nephritis: association between CD71 expression and IgA deposits. J Am Soc Nephrol 14:327–337
Peebles RS Jr, Hamilton RG, Lichtenstein LM, Schlosberg M, Liu MC, Proud D, Togias A (2001) Antigen-specific IgE and IgA antibodies in bronchoalveolar lavage fluid are associated with stronger antigen-induced late phase reactions. Clin Exp Allergy 31:239–248
Nahm DH, Kim HY, Park HS (1998) Elevation of specific immunoglobulin A antibodies to both allergen and bacterial antigen in induced sputum from asthmatics. Eur Respir J 12:540–545
Terada N, Terada Y, Shirotori K, Ishikawa K, Togawa K, Konno A (1996) Immunoglobulin as an eosinophil degranulation factor: change in immunoglobulin level in nasal lavage fluid after antigen challenge. Acta Otolaryngol 116:863–867
Peebles RS Jr, Liu MC, Lichtenstein LM, Hamilton RG (1995) IgA, IgG and IgM quantification in bronchoalveolar lavage fluids from allergic rhinitics, allergic asthmatics, and normal subjects by monoclonal antibody-based immunoenzymetric assays. J Immunol Methods 179:77–86
Iikura M, Yamaguchi M, Fujisawa T, Miyamasu M, Takaishi T, Morita Y, Iwase T, Moro I, Yamamoto K, Hirai K (1998) Secretory IgA induces degranulation of IL-3-primed basophils. J Immunol 161:1510–1515
Seminario MC, Gleich GJ (1994) The role of eosinophils in the pathogenesis of asthma. Curr Opin Immunol 6:860–864
Pleass RJ, Lang ML, Kerr MA, Woof JM (2006) IgA is a more potent inducer of NADPH oxidase activation and degranulation in blood eosinophils than IgE. Mol Immunol 1401–1408
Abu-Ghazaleh RI, Fujisawa T, Mestecky J, Kyle RA, Gleich GJ (1989) IgA-induced eosinophil degranulation. J Immunol 142:2393–2400
Pilette C, Durham SR, Vaerman JP, Sibille Y (2004) Mucosal immunity in asthma and chronic obstructive pulmonary disease: a role for immunoglobulin A? Proc Am Thorac Soc 1:125–135
Ludviksson BR, Eiriksson TH, Ardal B, Sigfusson A, Valdimarsson H (1992) Correlation between serum immunoglobulin A concentrations and allergic manifestations in infants. J Pediatr 121:23–27
Schwarze J, Cieslewicz G, Joetham A, Sun LK, Sun WN, Chang TW, Hamelmann E, Gelfand EW (1998) Antigen-specific immunoglobulin-A prevents increased airway responsiveness and lung eosinophilia after airway challenge in sensitized mice. Am J Respir Crit Care Med 158:519–525
Montenegro V, Monteiro RC (1999) Elevation of serum IgA in spondyloarthropathies and IgA nephropathy and its pathogenic role. Curr Opin Rheumatol 11:265–272
Veys EM, van Leare M (1973) Serum IgG, IgM, and IgA levels in ankylosing spondylitis. Ann Rheum Dis 32:493–496
Feltelius N, Hvatum M, Brandtzaeg P, Knutson L, Hallgren R (1994) Increased jejunal secretory IgA and IgM in ankylosing spondylitis: normalization after treatment with sulfasalazine. J Rheumatol 21:2076–2081
Hunter T, Harding GK, Kaprove RE, Schroeder ML (1981) Fecal carriage of various Klebsiella and Enterobacter species in patients with active ankylosing spondylitis. Arthritis Rheum 24:106–108
Montenegro V, Chiamolera M, Launay P, Goncalves CR, Monteiro RC (2000) Impaired expression of IgA Fc receptors (CD89) by blood phagocytic cells in ankylosing spondylitis. J Rheumatol 27:411–417
Brandtzaeg P (2006) The changing immunological paradigm in coeliac disease. Immunol Lett 105:127–139
Woof JM, Kerr MA (2006) The function of immunoglobulin A in immunity. J Pathol 208:270–282
Fry L (2002) Dermatitis herpetiformis: problems, progress and prospects. Eur J Dermatol 12:523–531
Collin P, Maki M, Keyrilainen O, Hallstrom O, Reunala T, Pasternack A (1992) Selective IgA deficiency and coeliac disease. Scand J Gastroenterol 27:367–371
Silvain C, Patry C, Launay P, Lehuen A, Monteiro RC (1995) Altered expression of monocyte IgA Fc receptors is associated with defective endocytosis in patients with alcoholic cirrhosis. Potential role for IFN-gamma. J Immunol 155:1606–1618
Basset C, Durand V, Jamin C, Clement J, Pennec Y, Youinou P, Dueymes M, Roitt IM (2000) Increased N-linked glycosylation leading to oversialylation of monomeric immunoglobulin A1 from patients with Sjogren’s syndrome. Scand J Immunol 51:300–306
Saulsbury FT (2001) Henoch–Schonlein purpura. Curr Opin Rheumatol 13:35–40
Saulsbury FT (1997) Alterations in the O-linked glycosylation of IgA1 in children with Henoch–Schonlein purpura. J Rheumatol 24:2246–2249
Allen AC, Willis FR, Beattie TJ, Feehally J (1998) Abnormal IgA glycosylation in Henoch–Schonlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant 13:930–934
Glennie MJ, Johnson PW (2000) Clinical trials of antibody therapy. Immunol Today 21:403–410
von Mehren M, Adams GP, Weiner LM (2003) Monoclonal antibody therapy for cancer. Annu Rev Med 54:343–369
Valerius T, Stockmeyer B, van Spriel AB, Graziano RF, van den Herik-Oudijk IE, Repp R, Deo YM, Lund J, Kalden JR, Gramatzki M, van de Winkel JG (1997) FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood 90:4485–4492
Huls G, Heijnen IA, Cuomo E, van der Linden J, Boel E, van de Winkel JG, Logtenberg T (1999) Antitumor immune effector mechanisms recruited by phage display-derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res 59:5778–5784
Otten MA, Rudolph E, Dechant M, Tuk CW, Reijmers RM, Beelen RH, van de Winkel JG, van Egmond M (2005) Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol 174:5472–5480
Dechant M, Vidarsson G, Stockmeyer B, Repp R, Glennie MJ, Gramatzki M, van de Winkel JG, Valerius T (2002) Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood 100:4574–4580
Neutra MR, Kozlowski PA (2006) Mucosal vaccines: the promise and the challenge. Nat Rev Immunol 6:148–158
Niedergang F, Kweon MN (2005) New trends in antigen uptake in the gut mucosa. Trends Microbiol 13:485–490
Ogra PL (2003) Mucosal immunity: some historical perspective on host–pathogen interactions and implications for mucosal vaccines. Immunol Cell Biol 81:23–33
Onorato IM, Modlin JF, McBean AM, Thoms ML, Losonsky GA, Bernier RH (1991) Mucosal immunity induced by enhance-potency inactivated and oral polio vaccines. J Infect Dis 163:1–6
Tamura S, Kurata H, Funato H, Nagamine T, Aizawa C, Kurata T (1989) Protection against influenza virus infection by a two-dose regimen of nasal vaccination using vaccines combined with cholera toxin B subunit. Vaccine 7:314–320
Corthesy B, Kaufmann M, Phalipon A, Peitsch M, Neutra MR, Kraehenbuhl JP (1996) A pathogen-specific epitope inserted into recombinant secretory immunoglobulin A is immunogenic by the oral route. J Biol Chem 271:33670–33677
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gomes, M.M., Herr, A.B. IgA and IgA-specific receptors in human disease: structural and functional insights into pathogenesis and therapeutic potential. Springer Semin Immun 28, 383–395 (2006). https://doi.org/10.1007/s00281-006-0048-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-006-0048-x