
Abstract
Purpose
Several retrospective studies have shown that the antitumor efficacy of capecitabine-containing chemotherapy decreases when co-administered with a proton pump inhibitor (PPI). Although a reduction in capecitabine absorption by PPIs was proposed as the underlying mechanism, the effects of PPIs on capecitabine pharmacokinetics remain unclear. We prospectively examined the effects of rabeprazole on the pharmacokinetics of capecitabine and its metabolites.
Methods
We enrolled patients administered adjuvant capecitabine plus oxaliplatin (CapeOX) for postoperative colorectal cancer (CRC) patients and metastatic CRC patients receiving CapeOX with/without bevacizumab. Patients receiving a PPI before registration were allocated to the rabeprazole group, and the PPI was changed to rabeprazole (20 mg/day) at least 1 week before the initiation of capecitabine treatment. On day 1, oral capecitabine (1000 mg/m2) was administered 1 h after rabeprazole intake. Oxaliplatin (and bevacizumab) administration on day 1 was shifted to day 2 for pharmacokinetic analysis of the first capecitabine dose. Plasma concentrations of capecitabine, 5′-deoxy-5-fluorocytidine, 5′-deoxy-5-fluorouridine, and 5-fluorouracil were analyzed by high-performance liquid chromatography. Effects of rabeprazole on inhibition of cell proliferation by each capecitabine metabolite were examined with colon cancer cells (COLO205 and HCT116).
Results
Five and 9 patients enrolled between September 2017 and July 2018 were allocated to rabeprazole and control groups, respectively. No significant effects of rabeprazole on area under the plasma concentration–time curve divided by capecitabine dose for capecitabine and its three metabolites were observed. Rabeprazole did not affect the proliferation inhibition of colon cancer cells by the respective capecitabine metabolites.
Conclusion
Rabeprazole does not affect capecitabine pharmacokinetics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Capecitabine is a prodrug that is metabolized to its active form, 5-fluorouracil (5-FU), in three enzymatic steps. After oral administration, capecitabine is first metabolized by the liver or intestinal carboxylesterase to form 5′-deoxy-5-fluorocytidine (5′-DFCR). It is then converted to 5′-deoxy-5-fluorouridine (5′-DFUR) by cytidine deaminase expressed in the liver, and further to 5-FU by thymidine phosphorylase (TP), which is preferentially located in tumor tissues [1, 2]. Capecitabine is widely used globally to treat a variety of solid tumors including colorectal cancer (CRC), gastroesophageal cancer, and breast cancer. It is used as monotherapy or in combination therapy with other anticancer drugs. Especially, combination chemotherapy of capecitabine with oxaliplatin (CapeOX) has been proven to be highly effective for adjuvant treatment of CRC as well as for the treatment of metastatic CRC [3,4,5].
TRIO-013/Logic study is a randomized phase III trial that examined the effects of administering CapeOX plus lapatinib in patients with HER2-positive advanced gastroesophageal adenocarcinoma [6]. Although the addition of lapatinib to CapeOX did not increase overall survival (OS), a subset analysis of the study revealed that co-administrating proton pump inhibitor (PPI) decreased the antitumor efficacy of CapeOX [7]. In the placebo arm of the TRIO-013/Logic study, PPI-treated patients had poorer median progression-free survival (PFS), OS, and disease control rate than patients who were not treated with a PPI. A multivariate analysis also showed the poorer PFS and OS in patients with PPI treatment. In patients treated with CapeOX and lapatinib, multivariate analysis demonstrated a significantly shorter OS in PPI-treated patients; nonetheless, PPIs had less effect on PFS. In another retrospective study investigating the effect of PPIs on the efficacy of adjuvant capecitabine monotherapy in patients with early-stage (Stage I–III) CRC, those who received both capecitabine and PPI had significantly poorer 5-year recurrence-free survival than those treated with capecitabine alone [8]. Regarding the underlying mechanism for the poorer clinical outcome of capecitabine-containing chemotherapy in patients who received PPI, it has been proposed that the PPI-induced elevation in gastric pH may reduce capecitabine’s dissolution in the digestive tract which may result in a decreased absorption of capecitabine via epithelial cells in the small intestine. This eventually lowers the efficacy of capecitabine chemotherapy by decreasing the plasma concentrations of capecitabine and its metabolites, and any further tumor concentration of 5-FU [7, 8].
To date, the effects of PPI treatment on the pharmacokinetics of capecitabine and its three metabolites has not been evaluated. Alternatively, PPIs can alter the activity of factors involved in the pharmacodynamics of capecitabine, leading to decrease in tumor cell death; this can result in a poor efficacy of capecitabine treatment.
Based on this background, we prospectively examined the effects of a PPI, rabeprazole, on the pharmacokinetics of capecitabine and its three metabolites in patients with postoperative CRC who were treated with adjuvant CapeOX, or those with metastatic CRC who administered CapeOX with or without bevacizumab. The effects of rabeprazole on the inhibition of proliferation by the three capecitabine metabolites were also assessed in colon cancer cell lines.
Patients and methods
Chemicals
Capecitabine, 5′-DFUR, and 5-FU were obtained from FUJIFILM Wako Pure Chemical Corporation (Tokyo, Japan). 5′-DFCR and rabeprazole sodium salt were purchased from Tokyo Chemical Industry (Tokyo, Japan), and 2′-deoxy-2′,2′-difluorouridine (dFdU) was obtained from Toronto Research Chemicals (Toronto, Canada). All chemicals and solvents were of the highest grade commercially available.
Patients
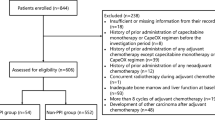
Eligible patients were 20 years or older with histologically confirmed postoperative or metastatic CRC, and indicated for treatment with a CapeOX containing regimen for an adjuvant or metastatic setting. Eligible patients also had an Eastern Cooperative Oncology Group performance status of ≤ 1, a life expectancy of 3 months or longer for metastatic CRC and no history of chemotherapy within 2 weeks. Each patient was confirmed to have adequate bone marrow function (neutrophil count ≥ 1500/μL; hemoglobin ≥ 9.0 g/dL; platelet count ≥ 75,000/μL), liver function (total bilirubin ≤ 1.5 × the upper limit of normal [ULN]; alanine transaminase and aspartate transaminase ≤ 2.0 × ULN; and alkaline phosphatase ≤ 2.5 × ULN), and renal function (a creatinine clearance ≥ 51 mL/min as calculated by the Cockcroft–Gault equation) within 14 days of the initiation of CapeOX treatment.
Study design
This was a prospective study of patients with stage III or high-risk stage II CRC who were fully recovered after curative resection and were candidates for the administration of CapeOX as an adjuvant chemotherapy or those with metastatic CRC who were candidates for the administration of CapeOX with or without bevacizumab therapy, at Showa University Hospital. Our main objective was to examine whether or not PPI treatment with capecitabine affects the pharmacokinetics of capecitabine and its metabolites. We selected 20 mg rabeprazole as PPI to be administered with capecitabine based on the reasons described below.
The principal cytochrome P450 (CYP) isoforms involved in the metabolism of PPIs are CYP2C19 and CYP3A4. Of these two, genetic polymorphisms in CYP2C19 can affect its activity in the liver and, in turn, the metabolic and pharmacokinetic profiles of PPIs. Thus, the acid inhibitory effects of PPIs are dependent on CYP2C19 genotype status [9]. However, unlike other PPIs, rabeprazole is predominantly metabolized via a non-enzymatic reduction and metabolized partially by CYP2C19 [9], which is the least affected by CYP2C19 polymorphism among PPIs. Although, the median 24-h intragastric pH values after a single oral dose of 20 mg were significantly different among the three different genotypes [10], intragastric pH after 8 days of repeated dosing with 20 mg rabeprazole was not significantly affected by the CYP2C19 genotype status [11].
Patients who received PPI therapy before registration were assigned to the rabeprazole group. If these patients were treated with a PPI other than rabeprazole, the PPI was changed to rabeprazole (20 mg/day) at least 1 week prior to the initiation of capecitabine treatment. Patients who did not receive a PPI before the registration were assigned to the control group. Therapeutic drugs which are known to inhibit carboxylesterase were discontinued at least 1 week prior to the initiation of capecitabine treatment.
The study protocol was approved by the Institutional Review Board of Showa University. All the patients provided written informed consent to use their peripheral blood samples and medical information for research purposes. The study was registered at University Hospital Medical Information Network-Clinical Trials Registry Japan (UMIN000031182).
Treatment
On day 1, oral capecitabine (1000 mg/m2) was administered twice daily. Oxaliplatin (and bevacizumab for metastatic CRC) administration on day 1 in the first cycle was shifted to day 2 for the pharmacokinetic analysis of the first capecitabine dose. CapeOX consisted of a 2-h intravenous infusion of oxaliplatin (130 mg/m2) on day 2 and oral capecitabine (1000 mg/m2) twice daily on days 2–14 [3, 5]. For patients with metastatic CRC, if necessary, a 7.5 mg/kg bevacizumab was also administered as a 90-min intravenous infusion before oxaliplatin treatment on day 2 [3]. CapeOX was repeated every 3 weeks without the shift of oxaliplatin and bevacizumab from the second cycle.
Blood sampling for pharmacokinetic analysis
Blood samples for the pharmacokinetic analysis of capecitabine and its three metabolites were obtained on the first day of the first cycle. For patients in the rabeprazole group, the drug was administered at 8 a.m. on day 1, while the first dose of capecitabine was administered at 9 a.m. Blood samples were taken immediately before rabeprazole administration and 1, 1.5, 2, 3, 4, 6, 7, and 9 h after rabeprazole administration. For patients in the control group, the first dose of capecitabine was administered at 9 a.m. on day 1. Blood samples were taken immediately before capecitabine administration and 0.5, 1, 2, 3, 5, 6, and 8 h after capecitabine administration. The samples were centrifuged immediately, and stored at − 80 °C until analysis.
Determination of capecitabine, 5′-DFCR, 5′-DFUR and 5-FU concentration
Plasma concentrations of capecitabine, 5′-DFCR, and 5′-DFUR were measured using a reverse-phase high-performance liquid chromatography (HPLC) method, with modifications [12]. Briefly, 150 μL of plasma sample was mixed with 450 µL of acetonitrile containing 2.2 μM dFdU (internal standard) in a vortex mixer. The mixture was then centrifuged at 16,000×g for 10 min at 4 °C. The supernatant was transferred to another tube and evaporated to dryness at 45 °C in an SPD1010 SpeedVac System (Thermo Fisher Scientific, Waltham, MA, USA). The residue was reconstituted in 200 µL of 10 mM sodium phosphate buffer (pH 4.8), and a 100-µL portion was injected into an HPLC system (Hitachi Chromaster; a 5110 pump, a 5210 autosampler, a 5310 column oven, and a 5430 diode array detector; Hitachi, Tokyo, Japan), equipped with a C30 analytical column (Develosil C30-UG-5, 150 × 4.6 mm; Nomura Chemical, Seto, Japan). HPLC was carried out at 30 °C at a flow rate of 1.0 mL/min. The mobile phase consisted of 10 mM sodium phosphate buffer (pH 4.8) for solvent A, and 80% methanol solution for solvent B. A 50-min run was carried out at 97% (0–9 min), followed by a linear gradient of 97–0% (9–35 min), 0% (35–40 min), and 97% (40–50 min) for solvent A. The eluent was monitored at 265 nm to detect 5′-DFCR and 5′-DFUR, and at 315 nm for capecitabine. Quantifications of capecitabine, 5′-DFCR, and 5′-DFUR were achieved by comparing the respective peak areas on a chromatogram to that of the internal standard, dFdU. The quantification limits were 0.156 μM. The respective intra- and inter-assay coefficients of variation were 16.6 and 17.0% for capecitabine, 8.54 and 2.92% for 5′-DFCR, and 6.62 and 7.63% for 5′-DFUR.
Since a clear separation between the 5-FU peak and that for the unidentified chemical(s) contained in plasma could not be obtained using the above method, the following modified method was used to measure the concentrations of 5-FU: 150-μL of plasma sample was mixed with 150 µL of 5% perchloric acid containing 10 μM dFdU (internal standard) in a vortex mixer. The mixture was then centrifuged at 16,000×g for 10 min at 4 °C, and a 100-µL portion of the supernatant was injected into the HPLC system. HPLC was carried out by using the same condition presented above, except for the solvent gradient. A 35-min run was carried out at 97% (0–9 min), followed by a linear gradient of 97–56% (9–20 min), 0% (20–25 min), and 97% (25–35 min) for solvent A. Quantifications of 5-FU was achieved by comparing the peak area on a chromatogram to that of the internal standard. The quantification limit was 0.125 μM, and the intra- and inter-assay coefficients of variation were 8.01 and 7.85%, respectively.
Pharmacokinetic parameters
The plasma concentration–time data of capecitabine, 5′-DFCR, 5′-DFUR, and 5-FU were analyzed by using a standard non-compartmental method with WinNonlin, version 6.4 software (Pharsight, Mountain View, CA, USA). Area under the plasma concentration–time curve (AUC, µM∙h) of capecitabine, 5′-DFCR, 5′-DFUR and 5-FU from time zero to the last sampling time was calculated with the linear trapezoidal rule (to peak plasma concentration) and linear-log trapezoidal rule (to the last quantifiable concentration). Capecitabine oral clearance (CL/F, L/h) was obtained by dividing the single capecitabine dose (μmol/body, calculated based on the molecular weight, 359.35) by the AUC, with extrapolation to infinity (Dose/AUC).
Toxicity evaluation
Clinical and laboratory adverse events were classified based on the Common Terminology Criteria for Adverse Events, version 4.0. All patients who received at least one cycle of chemotherapy with CapeOX were deemed assessable for toxicity.
CYP2C19 genotyping
Genomic DNA was extracted from 200 µL of peripheral blood stored at − 80 °C using a QIAamp Blood Kit (QIAGEN GmbH, Hilden, Germany).
Two polymorphisms (CYP2C19*2, rs4244285; CYP2C19*3, rs4986893) were examined by direct DNA sequencing. CYP2C19 gene fragments containing these polymorphic sites were amplified via polymerase chain reaction (PCR) using the methods of Morais et al. [13, 14] with some modifications. The following primers were used for PCR: CYP2C19*2, forward 5′-TAT AGT TTT AAA TTA CAA CCA GAG GTT GGC-3′ and reverse 5′-CTC CAA AAT ATC ACT TTC CAT AAA AGC AAG-3′; CYP2C19*3, forward 5′-TGT GAT CTG CTC CAT TAT TTT CCA G-3′ and reverse 5′-AAA GAC TGT AAG TGG TTT CTC AGG AAG-3′. PCR was carried out using the following cycling conditions: initial denaturation, 95 °C for 10 s; annealing, 55 °C for 30 s; and extension, 72 °C for 30 s (number of cycles = 35). PCR products were purified and sequenced.
Helicobacter pylori infection and atrophic gastritis evaluations
Using an enzyme immunoassay, we examined Helicobacter pylori (H. pylori) infection by measuring the serum immunoglobulin G antibodies to H. pylori [15]. Atrophic gastritis was evaluated by medical oncologists based on the concentrations of serum pepsinogen I and II measured with latex agglutination (BML, Tokyo, Japan).
Cell lines and cultures
COLO205 and HCT116 colon cancer cell lines were used. COLO205 cells were gifted from Prof. Kiyoshi Nose (Showa University, Tokyo Japan) and cultured with RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Thermo Scientific, Waltham, MA, USA), and maintained in a 5% CO2 incubator at 37 °C under humidified conditions. HCT116 cells were purchased from RIKEN Bio Resource Center (Ibaraki, Japan) and cultured with DMEM supplemented with 10% fetal bovine serum and maintained in a 5% CO2 incubator at 37 °C under humidified conditions.
Cell proliferation assay
COLO205 cells (5 × 103/well) and HCT116 cells (3 × 103/well) were seeded in 96-well plates and incubated for 24 h, followed by continuous exposure to the indicated concentrations of rabeprazole, 5′-DFCR, 5′-DFUR, and 5-FU for 72 h. Cell Counting Kit-8 (CCK) (Dojindo Molecular Technologies, Kumamoto, Japan) was added to each well, and cells were incubated for additional 4 h [16]. The absorbance was measured with Appliskan Multimode Microplate Reader (Thermo Scientific, Waltham, MA, USA) at 490 nm.
Statistical analysis
Fisher’s exact test or Chi-square test was used to compare patient characteristics between the rabeprazole and control groups, whereas the Wilcoxon test was used to compare the pharmacokinetic parameters of capecitabine and its three metabolite between the rabeprazole and control groups. Differences in cell viability with or without the addition of each capecitabine metabolite were analyzed by Tukey–Kramer HSD test. Two-tailed P values less than 0.05 were considered statistically significant. All analyses were performed using the JMP software, version 14.0 (SAS Institute, Cary, NC, USA).
Results
Patient characteristics
From September 2017 to July 2018, 14 patients were assessed for their eligibility to participate in this study. Five patients who received PPI and 9 patients who did not receive PPI were allocated to the rabeprazole and control groups, respectively. Table 1 shows the characteristics of patients enrolled in this study. No differences in age and gender distribution were observed in the rabeprazole and control groups. Renal and liver functions were normal in all patients, and did not differ between the rabeprazole and control groups. All patients with metastatic CRC received CapeOX or CapeOX plus bevacizumab as first line chemotherapy. Two and one patients in the rabeprazole and control groups, respectively, were administered CapeOX plus bevacizumab. The distribution of CYP2C19 genotypes did not significantly differ between the two groups.
Effect of rabeprazole on the pharmacokinetics of capecitabine and its three metabolites
Figure 1 depicts the pharmacokinetic profiles of capecitabine, 5′-DFCR, 5′-DFUR, and 5-FU in the rabeprazole and control groups. Inter-individual variability was observed in the pharmacokinetic profiles of these compounds; however, as shown in Table 2, there were no significant effects of rabeprazole on the AUC, CL/F, time to maximum plasma concentration (Tmax), maximum plasma concentration (Cmax), and elimination half-life (t1/2) of capecitabine. In addition, rabeprazole did not affect the pharmacokinetic parameters of 5′-DFCR, 5′-DFUR and 5-FU (Table 2). Because the single capecitabine dose was dependent on the body surface area of patients (1500 or 1800 mg; Table 1), AUC values of capecitabine and its three metabolites were divided by the dose. The effects of rabeprazole on the pharmacokinetic parameter were then examined, but we found no significant effects of rabeprazole on the AUC divided by capecitabine dose (Fig. 2).

Plasma concentration–time profiles of capecitabine, 5′-DFCR, 5′-DFUR, and 5-FU. Plasma concentration–time profile of each compound was derived from patients in the rabeprazole and control groups. Each color in the profile for the rabeprazole group shown in a, c, e, and g represents the same patient. Each color in the profile for the control group depicted in b, d, f, and h represents the same patient

AUC of a capecitabine, b 5′-DFCR, c 5′-DFUR, and d 5-FU divided by the capecitabine dose in the rabeprazole and control groups. Bars represent the average of AUC/dose
Toxicity
Hand-foot syndrome and diarrhea are frequently observed in patients treated with capecitabine [3, 5, 17, 18]. These adverse reactions were observed in patients in the control group. Five patients (56%) had grade 1 hand-foot syndrome, and 3 patients (33%) suffered from grade 1 and 2 diarrhea (2, grade 1; 1, grade 2). Grade 1 neuropathy, possibly due to oxaliplatin treatment was observed in 60 and 78% of patients in the rabeprazole and control groups, respectively. Grade 3 anorexia was observed in one patient in the rabeprazole group. Although one patient in the rabeprazole group had grade 3 hypertension, this adverse event was considered to be due to the co-administration of bevacizumab.
Effects of rabeprazole on the inhibition of cell proliferation by capecitabine metabolites
Effects of rabeprazole on the inhibition of cell proliferation by 5′-DFCR, 5′-DFUR and 5-FU were examined with colon cancer cells (COLO205 and HCT116). COLO205 cells were exposed to the half-maximal inhibitory concentration (IC50) of 5′-DFCR (1000 µM), 5′-DFUR (127 µM), or 5-FU (3.1 µM) for proliferation of COLO205 cells [2]. HCT116 cells were exposed to the IC50 of 5′-DFCR (830 µM), 5′-DFUR (39 µM), or 5-FU (3.7 µM) to examine proliferation of HCT116 cells [2]. A 1 µM concentration of rabeprazole was added to these cells; this value is approximately equal to the average maximum plasma concentration in clinical practice (20 mg/day of rabeprazole) [10, 11]. As shown in Fig. 3, we did not observe any significant effects of rabeprazole on the inhibition of cell proliferation by 5′-DFCR, 5′-DFUR, and 5-FU.

Effects of rabeprazole on the inhibition of proliferation of a COLO205 cells and b HCT11 cells by 5′-DFCR, 5′-DFUR, and 5-FU. White bars represent the data obtained without the addition of rabeprazole, and black bars indicate data obtained by adding 1 µM rabeprazole. Absorbance for the control experiments without the addition of rabeprazole is defined as 100%. Data are expressed as mean ± SD (N = 6–9)
Discussion
Several retrospective studies have demonstrated that co-administrating a PPI with capecitabine decreases the antitumor efficacy or reduces the survival advantage of capecitabine monotherapy or CapeOX in patients with gastroesophageal adenocarcinoma or CRC in adjuvant or metastatic setting [7, 8, 19]. Hence, authors of these reports propose that PPI-induced elevation of gastric pH may reduce the dissolution of capecitabine in the digestive tract which may result in decreased capecitabine absorption via epithelial cells in the small intestine. Our present prospective pharmacokinetic study revealed that co-administrating PPI, rabeprazole, with capecitabine did not significantly affect the pharmacokinetics of capecitabine as well as its three metabolites (Table 2 and Fig. 2), indicating that pharmacokinetic drug–drug interaction does not exist between capecitabine and rabeprazole. Retrospective studies without pharmacokinetic analyses of capecitabine could have limitations to examine such pharmacokinetics-related drug–drug interactions. This is because one might not be able to correctly evaluate the time of PPI intake, particularly in relation to the time of capecitabine intake was not necessarily able to be correctly evaluated [7, 8, 19]. As discussed by Hussaarts et al. [20], this is of importance because PPIs show a delayed onset of action and only reach their maximum elevation in intragastric pH around 3–4 h after administration. To add, the elevation of intragastric pH by PPIs is only present for approximately 12 h of duration, which theoretically makes it possible to combine these agents with either fewer or larger effects on the exposure to capecitabine.
Helicobacter pylori infection has been reported to frequently induce elevation in gastric pH levels [21]. The infection is also known to cause atrophic gastritis, which in turn decreases gastric acid secretion especially in elderly patients [22], and this may elevate gastric pH. Owing to this, H. pylori infection and atrophic gastritis might display the similar effects to rabeprazole intake. In the present study, we thus assessed the H. pylori infection and the presence or absence of atrophic gastritis. All 5 patients in the rabeprazole had neither H. pylori infection nor atrophic gastritis, while 4 patients in the control patients had both disorders, 4 had neither, and 1 had H. pylori infection alone. AUC values of capecitabine and its 3 metabolites divided by capecitabine dose in the 5 patients administered rabeprazole and the 4 patients in the control with both H. pylori infection and atrophic gastritis did not differ from the 4 control patients without H. pylori infection and atrophic gastritis, respectively (Supplementary Tables 1 and 2, respectively). This result supports our finding that the elevation of gastric pH by the intake of rabeprazole was associated, to a minor extent, with the pharmacokinetics of capecitabine and its three metabolites.
Although the therapeutic drugs known to inhibit carboxylesterase were discontinued at least 1 week prior to the initiation of capecitabine treatment, 2 patients in the control group were, respectively, administered candesartan for hypertension at 0.5 h prior to capecitabine intake and oseltamivir, prophylactically, for influenza at 1 h prior to the capecitabine treatment; these drugs are known substrates of carboxylesterase 1 [23, 24]. To our knowledge, information regarding the ability of these drugs to inhibit the catalytic activity of carboxylesterase 1 does not exist. Therefore, we evaluated the effects of these drugs on carboxylesterase-mediated transformation of capecitabine to its metabolites by calculating the ratio of AUC/dose for capecitabine to that for metabolites calculated by adding the values for 5′-DFCR, 5′-DFUR, and 5-FU (metabolic ratio). The ratio for the 7 patients who were not taking any medication was 0.175 ± 0.089, but a higher ratio was found for the patient who received candesartan (0.275). The patient administered oseltamivir had a ratio of 0.124, which is within the average ± SD for the 7 patients. These results suggest that candesartan and oseltamivir have minimal effects on the carboxylesterase-mediated capecitabine metabolism in the control patients of our study.
We also found that the inhibition by 5′-DFCR, 5′-DFUR or 5-FU against the proliferation of colon cancer cells, COLO205 and HCT116, was not affected by the addition of clinically relevant concentration of rabeprazole (Fig. 3). Taking all of our findings into account, the poorer effectiveness of the capecitabine-related regimen in patients administered PPI observed in previous retrospective studies are necessary to be prospectively confirmed with pharmacokinetic analysis.
In conclusion, we clarified that the co-administration of rabeprazole with capecitabine did not affect the pharmacokinetics of capecitabine and its 3 metabolites, 5′-DFCR, 5′-DFUR, and 5-FU. We also found that rabeprazole does not affect the inhibition of colon cancer cell proliferation by the addition of the respective capecitabine metabolites.
References
Reigner B, Blesch K, Weidekamm E (2001) Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet 40:85–104
Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H (1998) Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer 34:1274–1281
Cassidy J, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Saltz L (2008) Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J Clin Oncol 26:2006–2012
Haller DG, Tabernero J, Maroun J, de Braud F, Price T, Van Cutsem E, Hill M, Gilberg F, Rittweger K, Schmoll HJ (2011) Capecitabine plus oxaliplatin compared with fluorouracil and folinic acid as adjuvant therapy for stage III colon cancer. J Clin Oncol 29:1465–1471
Schmoll HJ, Cartwright T, Tabernero J, Nowacki MP, Figer A, Maroun J, Price T, Lim R, Van Cutsem E, Park YS, McKendrick J, Topham C, Soler-Gonzalez G, de Braud F, Hill M, Sirzen F, Haller DG (2007) Phase III trial of capecitabine plus oxaliplatin as adjuvant therapy for stage III colon cancer: a planned safety analysis in 1,864 patients. J Clin Oncol 25:102–109
Hecht JR, Bang YJ, Qin SK, Chung HC, Xu JM, Park JO, Jeziorski K, Shparyk Y, Hoff PM, Sobrero A, Salman P, Li J, Protsenko SA, Wainberg ZA, Buyse M, Afenjar K, Houe V, Garcia A, Kaneko T, Huang Y, Khan-Wasti S, Santillana S, Press MF, Slamon D (2016) Lapatinib in combination with capecitabine plus oxaliplatin in human epidermal growth factor receptor 2-positive advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma: TRIO-013/LOGiC—a randomized phase III trial. J Clin Oncol 34:443–451
Chu MP, Hecht JR, Slamon D, Wainberg ZA, Bang YJ, Hoff PM, Sobrero A, Qin S, Afenjar K, Houe V, King K, Koski S, Mulder K, Hiller JP, Scarfe A, Spratlin J, Huang YJ, Khan-Wasti S, Chua N, Sawyer MB (2017) Association of proton pump inhibitors and capecitabine efficacy in advanced gastroesophageal cancer: secondary analysis of the TRIO-013/LOGiC randomized clinical trial. JAMA Oncol 3:767–773
Sun J, Ilich AI, Kim CA, Chu MP, Wong GG, Ghosh S, Danilak M, Mulder KE, Spratlin JL, Chambers CR, Sawyer MB (2016) Concomitant administration of proton pump inhibitors and capecitabine is associated with increased recurrence risk in early stage colorectal cancer patients. Clin Colorectal Cancer 15:257–263
Ishizaki T, Horai Y (1999) Review article: cytochrome P450 and the metabolism of proton pump inhibitors–emphasis on rabeprazole. Aliment Pharmacol Ther 13(Suppl 3):27–36
Horai Y, Kimura M, Furuie H, Matsuguma K, Irie S, Koga Y, Nagahama T, Murakami M, Matsui T, Yao T, Urae A, Ishizaki T (2001) Pharmacodynamic effects and kinetic disposition of rabeprazole in relation to CYP2C19 genotypes. Aliment Pharmacol Ther 15:793–803
Shirai N, Furuta T, Moriyama Y, Okochi H, Kobayashi K, Takashima M, Xiao F, Kosuge K, Nakagawa K, Hanai H, Chiba K, Ohashi K, Ishizaki T (2001) Effects of CYP2C19 genotypic differences in the metabolism of omeprazole and rabeprazole on intragastric pH. Aliment Pharmacol Ther 15:1929–1937
Tabata T, Katoh M, Tokudome S, Hosakawa M, Chiba K, Nakajima M, Yokoi T (2004) Bioactivation of capecitabine in human liver: involvement of the cytosolic enzyme on 5′-deoxy-5-fluorocytidine formation. Drug Metab Dispos 32:762–767
De Morais SM, Wilkinson GR, Blaisdell J, Meyer UA, Nakamura K, Goldstein JA (1994) Identification of a new genetic defect responsible for the polymorphism of (S)-mephenytoin metabolism in Japanese. Mol Pharmacol 46:594–598
de Morais SM, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA (1994) The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 269:15419–15422
Shimatani T, Moriwaki M, Xu J, Tazuma S, Inoue M (2006) Acid-suppressive effects of rabeprazole: comparing 10 mg and 20 mg twice daily in Japanese Helicobacter pylori-negative and -positive CYP2C19 extensive metabolisers. Dig Liver Dis 38:802–808
Wang X, Liu C, Wang J, Fan Y, Wang Z, Wang Y (2017) Proton pump inhibitors increase the chemosensitivity of patients with advanced colorectal cancer. Oncotarget 8:58801–58808
Hamzic S, Kummer D, Milesi S, Mueller D, Joerger M, Aebi S, Amstutz U, Largiader CR (2017) Novel genetic variants in carboxylesterase 1 predict severe early-onset capecitabine-related toxicity. Clin Pharmacol Ther 102:796–804
Walko CM, Lindley C (2005) Capecitabine: a review. Clin Ther 27:23–44
Wong GG, Ha V, Chu MP, Dersch-Mills D, Ghosh S, Chambers CR, Sawyer MB (2018) Effects of proton pump inhibitors on FOLFOX and CapeOx regimens in colorectal cancer. Clin Colorectal Cancer. https://doi.org/10.1016/j.clcc.2018.11.001
Hussaarts K, van Leeuwen RWF, Mathijssen RHJ (2018) Factors affecting the association of proton pump inhibitors and capecitabine efficacy in advanced gastroesophageal cancer. JAMA Oncol 4:263–264
Haruma K, Mihara M, Okamoto E, Kusunoki H, Hananoki M, Tanaka S, Yoshihara M, Sumii K, Kajiyama G (1999) Eradication of Helicobacter pylori increases gastric acidity in patients with atrophic gastritis of the corpus-evaluation of 24-h pH monitoring. Aliment Pharmacol Ther 13:155–162
Haruma K, Kamada T, Kawaguchi H, Okamoto S, Yoshihara M, Sumii K, Inoue M, Kishimoto S, Kajiyama G, Miyoshi A (2000) Effect of age and Helicobacter pylori infection on gastric acid secretion. J Gastroenterol Hepatol 15:277–283
Ishizuka T, Yoshigae Y, Murayama N, Izumi T (2013) Different hydrolases involved in bioactivation of prodrug-type angiotensin receptor blockers: carboxymethylenebutenolidase and carboxylesterase 1. Drug Metab Dispos 41:1888–1895
Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L, Akhlaghi F, Yan B (2006) Anti-influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J Pharmacol Exp Ther 319:1477–1484
Acknowledgements
We would like to thank Ms. Mayu Kato for her various assistance in the clinical pharmacokinetic study, and also to thank Editage for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We have no conflict of interest to declare.
Ethical approval
The study protocol was approved by the Institutional Review Board of Showa University. All patients gave written informed consent for the use of their peripheral blood samples and medical information for research purposes. The study was registered on University Hospital Medical Information Network-Clinical Trials Registry Japan (UMIN000031182).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sekido, M., Fujita, Ki., Kubota, Y. et al. Rabeprazole intake does not affect systemic exposure to capecitabine and its metabolites, 5′-deoxy-5-fluorocytidine, 5′-deoxy-5-fluorouridine, and 5-fluorouracil. Cancer Chemother Pharmacol 83, 1127–1135 (2019). https://doi.org/10.1007/s00280-019-03837-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-019-03837-y