
Abstract
Purpose
To characterize the cardiovascular safety profile of regorafenib in patients with advanced cancer.
Methods
Patients received regorafenib 160 mg/day for 21 days followed by a 7-day break. The primary endpoint was the change from baseline in QTcF at the regorafenib t max (Day 21, Cycle 1 or 2) and changes in left ventricular ejection fraction (LVEF) from baseline on Cycle 2, Day 21. Secondary objectives were pharmacokinetics, safety, anti-tumor activity and effects on electrocardiogram intervals. QT intervals were corrected using the methods of Fridericia (QTcF) and Bazett (QTcB). LVEF was assessed by multigated acquisition scanning.
Results
Fifty-three patients were enrolled, and all received at least one dose of regorafenib 160 mg. Twenty-five patients received regorafenib for 21 days without dose reduction. The mean change from baseline in QTcF at t max was (−)2 ms (90 % CI −8, 3). No patient experienced a change from baseline in QTcF > 60 ms, and two had QTcF changes between 30 and 60 ms. No patient had a QTcF or QTcB > 480 ms. In 27 patients who received at least 80 mg of regorafenib, the mean change from baseline in LVEF% ± SD was 1.7 ± 7.8. In 14 patients without a dose reduction, the mean change from baseline in LVEF% was (−)0.1 ± 8.6 at Cycle 2, Day 21. Four patients experienced a LVEF decrease between 10 and 20 %.
Conclusion
The effects of regorafenib on the QT/QTc interval and LVEF were modest and unlikely to be of clinical significance in the setting of advanced cancer therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regorafenib is a small molecule inhibitor of a number of protein kinases, including those involved in tumor angiogenesis (VEGFR-1, VEGFR-2 and VEGFR-3, TIE2), oncogenesis (KIT, RET, RAF1, BRAF, BRAFV600E) and maintenance of the tumor microenvironment (PDGFR, FGFR) [1]. This drug has a similar chemical structure to sorafenib, with the substitution of a hydrogen atom and a fourth fluorine atom in the central aromatic ring [2, 3]. As a result, in preclinical studies, regorafenib appears to be pharmacologically more potent [1].
The US Food and Drug Administration (FDA) and other regulatory agencies have indicated that an evaluation of the QT/QTc interval is required for all new molecular entities (International Conference on Harmonization) [4]. Preclinical and phase I/II trials of regorafenib have shown no significant cardiovascular toxicity [5, 6]. However, none of these trials were specifically designed to evaluate cardiovascular safety. Therefore, this trial was conducted to evaluate the effects of multiple doses of regorafenib on cardiac function, specifically the QT/QTc interval and left ventricular ejection fraction (LVEF).
Materials and methods
Trial design
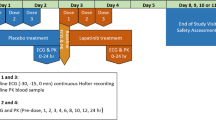
This prospective, multicenter open-label trial was designed to evaluate the cardiovascular safety of regorafenib by assessing the effect on the QT/QTc interval and LVEF. Secondary objectives were to evaluate the pharmacokinetics, safety and anti-tumor activity of regorafenib in patients with advanced solid tumors. Another secondary objective was to evaluate the effect of regorafenib on ECG intervals other than QT/QTc, including QRS and PR. Regorafenib was administered at a dose of 160 mg once daily for the first 21 days followed by a 7-day break in each 28-day cycle until the occurrence of disease progression or toxicity requiring discontinuation of drug, consent withdrawal, or death. In the event toxicities required dose reductions, the pre-defined adjusted dose levels were 120 mg and 80 mg.
Toxicities were graded using the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Tumor response to therapy was evaluated as a secondary endpoint using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Evaluation of tumor response was performed at baseline and within the last 7 days of Cycle 2. Following Cycle 2, tumor response was evaluated every three cycles (last 7 days of Cycles 5, 8, and so on) and at the end of treatment visit.
The trial protocol was approved by the institutional review boards at all the participating centers, and all patients gave written informed consent prior to entering the trial. The study was conducted according to good clinical practice and the Declaration of Helsinki and its amendments. The trial was registered at www.ClinicalTrials.gov as NCT01339104.
Patient population
Patients who were at least 18 years of age and had histologically or cytologically confirmed solid tumors with measurable disease or non-measurable disease according to RECIST 1.1 were eligible for this trial. All patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, adequate hematological (hemoglobin ≥ 9 g/dL, absolute neutrophil count ≥1500/mm3), renal [serum creatinine ≤1.5 times upper limit normal (ULN)] and hepatic (bilirubin ≤1.5 times ULN, alkaline phosphatase ≤4 times ULN, INR ≤ 1.5 ULN) function, and have a life expectancy of at least 3 months. The LVEF was required to be ≥50 % at the baseline multigated acquisition (MUGA) scan, and the QTc interval was to be ≤470 ms at screening. Patients with a history of cardiac disease, including congestive heart failure >NYHA class II, active coronary disease, myocardial infarction within the past 6 months, or concomitant QT prolonging drugs and type 1A or 3 antiarrhythmics, were excluded from the study. Other exclusion criteria included any ventricular tachyarrhythmia, long QT syndrome, required use of a pacemaker, atrial fibrillation, left bundle branch block, uncontrolled hypertension (diastolic BP ≥ 90 mmHg or systolic BP ≥ 140), dehydration of NCI-CTCAE grade >1, active clinically serious infection, symptomatic brain metastases or meningeal brain tumor, and any condition that was unstable or could have jeopardized the safety or compliance of the subject in the study. Patients treated with prior anthracyclines or any other anticancer therapy within 4 weeks, prior to the first dose of study drug, were also excluded.
Pharmacodynamic evaluation
The primary pharmacodynamic ECG variable was the change in QT/QTc at the t max of regorafenib on Day 21 of Cycle 1 or Cycle 2 from the average of the six baseline QT/QTc intervals from ECGs collected pre-dose on Cycle 1, Day (−)1 and corrected for heart rate using the method of Fridericia (QTcF). Secondary analysis included the change in QT/QTc at the t max of regorafenib corrected using Bazett’s formula (QTcB). For the secondary analysis, the effects of the metabolites of regorafenib on QT/QTc were analyzed in the same manner. Measurements of the LVEF during regorafenib treatment by MUGA scanning were taken at baseline (within 7 days prior to Cycle 1, Day 1) and on Cycle 2, Day 21. From Cycle 3 onwards, LVEF assessments were done every three cycles starting at Cycle 5 (last 7 days of Cycles 5, 8, and so on) and at the end of treatment visit.
Pharmacokinetic evaluation
Blood samples (5 mL) for the determination of plasma concentration of regorafenib were collected on Cycle 1, Day 21 over 24 h post-dose. The following pharmacokinetic parameters were calculated for regorafenib and its major human metabolites M-2 and M-5: AUC (0–t last)ss was the area under the plasma concentration versus time curve from time zero to the last sample time (t last) at steady state, where t last was at approximately 24 h on Day 21, Cycle 1 or Cycle 2; C max,ss was the maximum plasma concentration at steady state, Day 21, Cycle 1 or Cycle 2; and t max,ss was the time to maximum concentration at steady state, Day 21, Cycle 1 or Cycle 2.
Statistical methods
Quantitative pharmacodynamic data were analyzed by descriptive statistics using the arithmetic mean, standard deviation, median, minimum and maximum. Qualitative data are presented in frequency tables.
The pharmacodynamic effect of regorafenib on ECG parameters was analyzed assuming a normal distribution of the data. For the primary analysis, the individual QTcF at the t max of regorafenib on Cycle 1, Day 21 was subtracted from the average of six baseline QTcF intervals from the ECGs collected pre-dose on Cycle 1, Day 1, and 90 % confidence intervals (CIs) were calculated.
For the secondary analysis, the effects of the metabolites of regorafenib were analyzed in the same manner. For the analysis of time-matched changes from baseline adjusted for differences at 0 h, a linear regression was performed and 90 % CIs were derived. In a further exploratory analysis, the average and the mean of all post-dose QTc intervals were subtracted from the mean of the baseline QTc intervals. In addition, all analyses were done for other ECG parameters such as the QRS and PR intervals and using Bazett’s method for heart rate correction.
Frequencies were calculated in subgroups defined according to whether the change from baseline in QT and QTc intervals was ≤30, >30–60 or >60 ms and whether the absolute QT and QTc values were ≤450, >450–480, >480–500 and >500 ms.
The pharmacodynamic effect of regorafenib on LVEF was analyzed assuming normally distributed data (90 % CIs for the change from baseline in the LVEF on Cycle 2, Day 21). Pharmacokinetic data are presented for patients who received 160 mg regorafenib daily with no dose interruptions or modifications. Geometric mean concentrations were calculated for each of the sampling points. For pharmacokinetic parameters, the geometric mean, percent coefficient of variation, median and range are presented.
For the analysis of time-matched change from baseline, an ANCOVA (analysis of covariance) was performed and 90 % CIs were derived.
Results
Fifty-three patients with advanced cancer were enrolled, and all received at least one dose of regorafenib 160 mg. The clinical characteristics of these patients are given in Table 1. There were 27 females and 26 males, and the mean age [standard deviation (SD)] was 59.8 (9.2) years.
Pharmacodynamic evaluation
QT/QTc
Thirty patients were evaluable for ECG analysis and a total of 25 received 160 mg of regorafenib for 21 days without dose modification prior to the post-treatment ECG evaluations on Day 21 of Cycle 1 or Cycle 2.
Table 2 displays the results for the 25 patients without dose modification for the QTcF, QTcB, and uncorrected QT intervals at baseline and at the t max of regorafenib and the changes from baseline at t max. Mean changes from baseline QTcF and QTcB at t max were (−)2 ms (90 % CI −8, 3) and (−)4 ms (90 % CI −10, 1), respectively.
A sensitivity analysis of the primary endpoint was performed in the 30 patients with valid Holter ECG assessments. Twenty-nine of these 30 patients were included in the pharmacokinetic analysis (one patient was excluded because 4 tablets were returned from the Cycle 1 allotment of 84 tablets). Following one cycle of regorafenib, mean changes from baseline of (−)2 ms (90 % CI −6, 3) and (−)3 ms (90 % CI −8, 2) at the regorafenib t max were observed for QTcF and QTcB, respectively. The outcome was similar in patients valid for Holter ECG assessments who did not have dose modifications.
Secondary QT/QTc analysis
In the 25 patients valid for Holter ECG who did not have dose modifications and were valid for pharmacokinetic analysis, mean changes from baseline QTcF and QTcB at t max for the M-2 metabolite were (−)3 ms (90 % CI −9, 3) and (−)5 ms (90 % CI −12, 1), respectively. Mean changes from baseline at t max for the M-5 metabolite were (−)3 ms (90 % CI −8, 3) and (−)4 ms (90 % CI −11, 2), respectively.
In the 29 patients valid for Holter ECG and pharmacokinetic analysis, mean changes from baseline QTcF and QTcB at t max for the M-2 metabolite were (−)2 ms (90 % CI −8, 4) and (−)3 ms (90 % CI −9, 3), respectively. Mean changes from baseline at t max for the M-5 metabolite were (−)2 ms (90 % CI −7, 4) and (−)2 ms (90 % CI −8, 4), respectively.
Maximum change in QT/QTc from baseline
Analysis of the 25 patients who did not have dose modifications and were valid for Holter ECG assessment showed median maximum increases from baseline QTcF and QTcB of 3 ms (90 % CI 0, 9) and 2 ms (90 % CI −1, 7), respectively. Similar results were observed in all patients valid for Holter ECG assessments.
Categorical evaluation of QTc
Thirty patients with valid Holter ECG data were analyzed. Holter ECG evaluations were made over a 24-h period on Day 21 of Cycle 1 or Cycle 2, at the steady state of regorafenib and compared with baseline values. Changes in QTc were divided into three subclasses: ≤30, >30–60, >60 ms.
No patient showed a change in QTcF of >60 ms. The maximum increase from baseline in QTcF was 57 ms (from a baseline of 396 ms) at 2 h post-dose. Two patients had changes in QTcF from baseline in the >30–60 ms range.
No patient experienced a change in QTcB from baseline of >60 ms. The maximum change in QTcB from baseline was an increase of 55 ms (from a baseline of 418 ms) at 2 h post-dose. Two patients had changes in QTcB from baseline in the >30–60 ms range.
Three patients had changes in uncorrected QTc from baseline of >60 ms. The maximum increase was a 76 ms change from a baseline value of 403 ms at 5 h post-dose.
None of the patients had a baseline QTcF > 450 ms, and none had a QTcF evaluation of >480 ms during the Holter post-ECG monitoring visit. Five patients had absolute QTcF evaluations of >450 ms. Five patients had an absolute baseline QTcB between 450 and 480 ms, but none had an absolute baseline QTcB > 480 ms during the post-treatment Holter ECG monitoring visit. Ten patients had absolute QTcB evaluations between 450 and 480 ms.
Additional ECG parameters
Changes in the QRS interval were evaluated in the 25 patients who had no dose modifications and in 24 patients for the PR interval (baseline not evaluable). The mean change from baseline at the t max of regorafenib in patients with no dose modification showed small decreases in the QRS interval (−1 ms) and in the PR interval (−4 ms). Similar results were observed in the 30 patients valid for Holter ECG analysis, with no change in the QRS interval and (−)4 ms change in the PR interval. The maximum changes from baseline in QRS and PR were evaluated over the 24-h measurement period, where the baseline QRS and PR were calculated as the average of the baseline values of the ECGs recorded over 24 h on Cycle 1, Day −1. Median maximum changes from baseline were 4 ms for the QRS interval and 7 ms for the PR interval. Similar results were observed for 30 patients evaluable for Holter ECG analysis, with median maximum changes from baseline of 4 ms for the QRS interval and 8 ms for the PR interval.
LVEF
Fifteen patients had two full cycles of regorafenib at the 160 mg dose level without dose reductions prior to the post-treatment evaluation. In general, there was almost no change from baseline in LVEF% in the first 2 cycles and only minor variations after additional cycles (Online Resource 1). The mean change (±SD) from baseline in LVEF% at Cycle 2, Day 21 was (−)0.1 ± 8.6 in 14 patients (n = 1 had no measurement at Cycle 2, Day 21) (Table 3). The individual changes from baseline in LVEF% at Cycle 2, Day 21 for all patients evaluable for LVEF analysis who did not have a dose reduction are shown in Fig. 1.

Individual changes from baseline in LVEF% at Cycle 2, Day 21 in 14 of 15 patients valid for LVEF analysis without dose reductions. One patient did not have an LVEF measurement on Cycle 2, Day 21
All patients evaluable for LVEF analysis with baseline and any post-treatment LVEF data and who were on a minimum of 80 mg of the drug were also evaluated (n = 27). The mean change from baseline (±SD) to any first post-baseline measurement (any time after the first dose of regorafenib) was 1.7 ± 7.8 LVEF% in these 27 patients, whereas the mean change in LVEF% from baseline to the Cycle 2, Day 21 visit was 1.4 ± 8.4 (n = 22). The mean LVEF remained essentially steady after subsequent cycles of regorafenib (i.e., after Cycle 2, Day 21), with the mean change from baseline ranging from (−)3.5 ± 5.8 LVEF% to 4.7 ± 5.5 LVEF% through the Cycle 20, Day 1 assessment (Online Resource 2). After 4 cycles of therapy, the mean LVEF change was (−)2.4 ± 6.6 (n = 20). Long-term data collected at Cycle 20, Day 1 revealed a mean LVEF change of 4.7 ± 5.5 (n = 3). At the end of the study period, the mean change from baseline was 1.0 ± 8.0 LVEF% (n = 14).
Of all subjects valid for the safety analysis who had a post-baseline LVEF value (n = 41), 4 (9 %) had an increase in LVEF > 10 %. Four patients experienced an LVEF decrease between 10 and 20 %. For two of these patients, LVEF was measured as <50 % during the trial. The LVEF normalized subsequently during ongoing therapy with regorafenib without dose reductions in two patients. In the other two patients, the observed decrease in LVEF could be explained by either the onset of atrial fibrillation or initiation of clonidine therapy. Furthermore, one additional patient had a baseline LVEF of 54 %, which decreased to 48 % on Day 21 of Cycle 2 (which was also the end of treatment visit).
Pharmacokinetic evaluation
The pharmacokinetic parameters of regorafenib, and the major metabolites M-2 and M-5, were calculated in the subset of patients who received 160 mg daily with no dose modifications (n = 25). The data for patients treated with 160 mg are given in Table 4.
AUC (0–t last)ss was analyzed, but not AUC (0–24) as the half-life could not be calculated accurately in this study, and therefore, any extrapolation could lead to errors in the estimation of AUC (0–24). For all subjects, the time deviations of the last sampling point (t last) were not greater than 1 h at the 24-h time point. Therefore, AUC (0–t last)ss closely approximates AUC (0–24)ss. The variability in PK parameters was higher for the M-2 and M-5 metabolites compared with regorafenib, as displayed in Table 4. The mean AUC (0–t last)ss and mean C max,ss of regorafenib were slightly greater than the corresponding values for both metabolites, although the maximal AUC and C max values for individual patients were seen with the metabolites, reflecting the high PK variability associated with both M-2 and M-5.
The mean concentration–time profile for regorafenib and its metabolites showed the highest concentration occurring at time zero with a secondary peak for regorafenib and M-2 at 3–5 h post-dose and steadily increasing concentrations for M-5 (Fig. 2). The ranges of individual AUC (0–t last)ss and C max values for regorafenib and its metabolites in steady state were wide, as observed in other studies. More variability was observed for the M-2 and M-5 metabolites, than for regorafenib.

Mean plasma concentration–time profile for regorafenib and metabolites M-2 and M-5 on Cycle 1, Day 21 for patients receiving 160 mg regorafenib daily in patients valid for Holter ECG without dose modification and pharmacokinetic analysis (n = 25)
Safety
The overall mean (±SD) time on regorafenib therapy (including dose interruptions) was 127 (±125) days with a median treatment of 92 days (range 1–490) for the 53 subjects valid for safety analysis.
The most common treatment-emergent drug-related adverse events (all grades) were as follows: mucositis (n = 21, 39.6 %), hand–foot skin reaction (n = 18, 34.0 %), diarrhea and fatigue (n = 17, 32.1 %, each), anorexia (n = 14, 26.4 %), hypertension (n = 12, 22.6 %), hoarseness and nausea (n = 9 each, 17.0 %) and pain in an extremity (n = 8, 15.1 %).
The most common treatment-emergent drug-related grade ≥3 adverse events occurring in ≥2 patients were as follows: hand–foot skin reaction, hypophosphatemia, hypertension (n = 5 each, 9.4 %); diarrhea, hyponatremia (n = 3 each, 5.7 %); hyperbilirubinemia and hypokalemia (n = 2 each, 3.8 %). There were 36 patients (67.9 %) who had a treatment modification due to treatment-emergent adverse events, including dose interruptions (n = 31, 58.5 %), dose reductions (n = 16, 30.2 %) and permanent discontinuation (n = 10, 18.9 %). The most frequent treatment-emergent adverse events that led to dose interruption were hand–foot syndrome (n = 9, 17.0 %), diarrhea (n = 5, 9.4 %), hyponatremia and hypertension (n = 4 each, 7.5 %), atrial fibrillation, hyperbilirubinemia and vomiting (n = 3 each, 5.7 %). The most frequent treatment-related adverse events that led to dose reduction were hand–foot syndrome (n = 6, 11.3 %), fatigue, anorexia, hypertension and diarrhea (n = 2 each, 3.8 %). The most frequent treatment-emergent adverse event leading to permanent discontinuation of regorafenib was hyperbilirubinemia (n = 2, 3.8 %).
A total of 24 patients (45.3 %) experienced at least one treatment-emergent serious adverse event, and in four of these patients (7.5 %) the events were study drug-related (n = 1 grade 3 diarrhea; n = 1 grade 5 small intestine perforation; hyperbilirubinemia, n = 1 grade 2 and n = 1 grade 3). There were no episodes of Torsades de Pointes, sudden death, ventricular fibrillation, ventricular flutter, syncope or seizure during the trial. Seven patients (13.2 %) died within 30 days of treatment discontinuation. The cause of death was reported as progressive disease in six patients (11.3 %) and an adverse event associated with clinical progression in the other patient.
Efficacy
Forty-five patients were evaluable for efficacy, with four patients (8.9 %) achieving a partial response and 30 (66.7 %) stable disease. Seven patients (15.6 %) had progressive disease.
Discussion
A number of studies have reported cardiac adverse events associated with tyrosine kinase inhibitors with angiogenic targets, particularly sunitinib. These cardiac events have included QT/QTc interval prolongation, decrease in LVEF, hypertension, myocardial infarction and congestive cardiac failure [7–12]. A cardiovascular safety trial of the multikinase inhibitor, sorafenib, documented modest changes in ECG, LVEF, blood pressure and heart rate in patients with advanced cancer [13]. Additionally, a randomized, double-blind, placebo-controlled trial of repeated oral doses of pazopanib in patients with solid tumors revealed a concentration-dependent decrease in heart rate and a small concentration-independent prolongation of the QTcF interval [14]. A recent trial-level meta-analysis of 6548 patients treated within randomized phase II and III trials comparing arms with and without a FDA-approved VEGFR TKI reported that these can be associated with QTc prolongation albeit of low clinical significance [15]. Notably, regorafenib was not included in this analysis because QTc prolongation was not listed as an adverse event in any of the trials.
Our trial was specifically designed to evaluate the cardiovascular effects of regorafenib in a population of patients with advanced cancer. Overall, the effects of regorafenib on the cardiovascular parameters of QTc interval and LVEF observed in the current trial were modest and unlikely to be of clinical significance in the setting of advanced cancer treatment.
References
Wilhelm SM, Dumas J, Adnane L et al (2011) Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer 129:245–255
Wilhelm SM, Carter C, Tang L et al (2004) BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109
Fabian MA, Biggs WH 3rd, Treiber DK et al (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 23:329–336
International Conference on Harmonisation (2005) Guidance on E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Fed Regist 70:61134-61135
Strumberg D, Scheulen ME, Schultheis B et al (2012) Regorafenib (BAY 73-4506) in advanced colorectal cancer: a phase I study. Br J Cancer 106:1722–1727
Mross K, Frost A, Steinbild S et al (2012) A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res 18:2658–2667
Khakoo AY, Kassiotis CM, Tannir N et al (2008) Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer 112:2500–2508
Machiels JP, Bletard N, Pirenne P, Jacquet L, Bonbled F, Duck L (2008) Acute cardiac failure after sunitinib. Ann Oncol 19:597–599
Schmidinger M, Zielinski CC, Vogl UM et al (2008) Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 26:5204–5212
Chu TF, Rupnick MA, Kerkela R et al (2007) Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 370:2011–2019
Telli ML, Witteles RM, Fisher GA, Srinivas S (2008) Cardiotoxicity associated with the cancer therapeutic agent sunitinib malate. Ann Oncol 19:1613–1618
Di Lorenzo G, Autorino R, Bruni G et al (2009) Cardiovascular toxicity following sunitinib therapy in metastatic renal cell carcinoma: a multicenter analysis. Ann Oncol 20:1535–1542
Tolcher AW, Appleman LJ, Shapiro GI et al (2011) A phase I open-label study evaluating the cardiovascular safety of sorafenib in patients with advanced cancer. Cancer Chemother Pharmacol 67:751–764
Heath EI, Infante J, Lewis LD et al (2013) A randomized, double-blind, placebo-controlled study to evaluate the effect of repeated oral doses of pazopanib on cardiac conduction in patients with solid tumors. Cancer Chemother Pharmacol 71:565–573
Ghatalia P, Je Y, Kaymakcalan MD, Sonpavde G, Choueiri TK (2014) QTc interval prolongation with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Br J Cancer 112:296–305
Acknowledgments
This trial was sponsored by Bayer HealthCare Pharmaceuticals. We acknowledge the assistance of Ann Contijoch, Bayer Healthcare Pharmaceuticals, in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
R.L. Jones serves as an advisory board member for Immune Design, Daiichi Sankyo, Merck, and Blueprint Medicines. He also reports conducting trials sponsored by Bayer HealthCare Pharmaceuticals, Eisai, Novartis, Johnson & Johnson, Pharmamar, Infinity Pharmaceuticals, Threshold Pharmaceuticals, and Morphotek. D.C. Smith has received research funding from Bayer HealthCare Pharmaceuticals. K. Diefenbach, J. Lettier, and O. Boix are employees of Bayer HealthCare Pharmaceuticals; K. Diefenbach and O Boix also report stock ownership in Bayer HealthCare Pharmaceuticals. A.C. Lockhart has received research funding from Amgen, Bayer, Daiichi Sankyo, EMD Serono, Genentech/Roche, Lilly, Millennium Takeda, Novartis, Sanofi, Teva, and Zenyaku Kogyo. K.N. Moore serves as an advisory board member for Astrazeneca, Immunogen, Genentech, Amgen, and Advaxis. J.C. Bendell and C.L. O'Bryant report no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jones, R.L., Bendell, J.C., Smith, D.C. et al. A phase I open-label trial evaluating the cardiovascular safety of regorafenib in patients with advanced cancer. Cancer Chemother Pharmacol 76, 777–784 (2015). https://doi.org/10.1007/s00280-015-2827-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2827-3