
Abstract
Cisplatin is a widely used chemotherapeutic drug for the treatment of various cancers. However, the ototoxicity severely limited its maximum dose. The present study was designed to evaluate the effect of Ginkgolide B (GB), a major component of Ginkgo biloba extracts, on cisplatin-induced ototoxicity and to elucidate the molecular mechanism in vitro and in vivo. In HEI-OC1 auditory cells, GB concentration-dependently inhibited the reduction of cell viability and increase in apoptosis exerted by cisplatin. Cisplatin-activated mitochondrial apoptotic molecular events were significantly inhibited by GB. In addition, GB notably suppressed the increase in NOX2 and p47phox expression and the decrease in nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1) expression in cisplatin-exposed cells. Inhibition of Nrf2 using SiRNA and blockage of HO-1 by zinc protoporphyrin IX (ZnPP) suppressed the protective effects of GB. Moreover, GB prevented cisplatin-induced reduction of Akt phosphorylation and LY294002, an inhibitor of PI3 K/Akt signaling, blocked the anti-apoptotic effect of GB in cisplatin-treated cells. Furthermore, the protective effect of GB was tested in cisplatin-exposed rats. GB treatment markedly protected animals against cisplatin-induced hearing loss and vestibular dysfunction. Inhibition of Akt and HO-1 significantly suppressed the improvement in hearing loss and vestibular dysfunction in GB-treated rats. We demonstrate that GB decreases ROS generation through reducing NOX2 expression and enhancing activity through Akt–Nrf2–HO-1 pathway, resulting in inhibition of mitochondrial apoptosis and final reduction of cisplatin-induced ototoxicity in vitro and in vivo. Our findings have gained an insight into the mechanism of GB-exerted protective effect against cisplatin-induced ototoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cisplatin (cis-diammine-dichloroplatinum (II), CDDP) is a widely used chemotherapeutic agent for the treatment of several neoplastic diseases, such as head and neck cancers [31]. However, the clinical use of cisplatin is limited by several severe side effects, including neurotoxicity, nephrotoxicity, myelotoxicity, gastrointestinal toxicity, and ototoxicity [13, 17, 40, 42]. It is noted that hearing loss is a major dose-limiting side effect of cisplatin, resulting in bilateral, irreversible, and progressive sensorineural hearing loss and a severe decrease in quality of life of cancer patients [32]. It is estimated that up to 93 % patients who receive cisplatin chemotherapy may experience ear-related symptoms, with no effective treatment available for cisplatin-induced ototoxicity [36]. Thereof, ototoxicity represents a major limitation to effective CDDP chemotherapy [28]. Cisplatin mainly damages the outer and inner hair cells, targeting Corti, and induces degeneration of spiral ganglion neurons, leading to irreversible hearing loss. In addition to that, the ears play a pivotal role in the regulation of balance. Thus, cisplatin can also induce vestibulotoxicity [2]. The occurrence of oxidative stress and apoptosis has been reported to be involved in cisplatin-induced ototoxicity [9, 38].
Extracts of Ginkgo biloba leaves are widely used herbal products and/or dietary supplements in patients with neurodegenerative, vascular, and audiovestibular disorders [3, 34, 41]. It has been shown that G. biloba extracts exhibit potent antioxidant and anti-apoptotic activity, which is involved in the pharmacological activities [15, 43]. Moreover, G. biloba extracts were shown to be able to protect against cisplatin-induced ototoxicity [7, 14, 34]. Ginkgolide B (GB) is one of the major components of G. biloba extracts [19] that possesses many pharmacological properties, including anti-inflammatory effect, anti-tumor effect, anti-apoptotic effect, and antioxidant activity [4, 18, 44, 45]. However, the effect of GB on cisplatin-induced ototoxicity is unclear.
The present study was designed to examine the potential protective effect of GB on cisplatin-induced ototoxicity and to elucidate the possible molecular mechanisms. The results showed that GB decreased ROS generation through reducing NOX2 expression and enhancing Nrf2–HO-1 antioxidant pathway via activation of Akt signaling, resulting in inhibition of mitochondrial apoptosis and final reduction of cisplatin-induced ototoxicity in vitro and in vivo.
Materials and methods
Chemicals and materials
β-actin, tubulin-α, cytochrome c, caspsae 3, caspsae 9, PARP, Nrf2, HO-1, NOX2, and P47phox antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). GB was obtained from the Research Center of Traditional Chinese Medicine (Wuhan, China) and dissolved in DMSO. DHE, MitoSOX, and Rhodamine 123 were purchased from Invitrogen. Cisplatin, zinc protoporphyrin IX, and LY294002 were procured from Sigma. All of the other chemicals used were of the highest grade available commercially.
Cell culture and treatment
House Ear Institute-Organ of Corti 1 (HEI-OC1) cells are conditionally immortalized mice cochlear cells that have been previously characterized [9]. HEI-OC1 cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM; GIBCO/BRL) containing 10 % fetal bovine serum (FBS; GIBCO/BRL) and 50 U/mL interferon-γ (Genzyme, Cambridge, MA, USA) without antibiotics. To evaluate the effect of GB on cisplatin-induced cytotoxicity, HEI-OC1 cells were incubated with cisplatin in the presence or absence of indicated concentrations of GB with or without specific inhibitors for 24 h.
Cell viability
HEI-OC1 cells were seeded in 96-well plates, with 1 × 104 cells in each well. After incubation for 24 h, cells were incubated with 1–100 μM cisplatin, or 1–200 μM GB, or 100 μM cisplatin with or without 1–50 μM GB in the presence or absence of indicated inhibitors for another 24 h. Cell viability was determined by the 3-(4,5-dimethylthiazoyl-2-yl) 2,5-diphenyltetrazolium bromide (MTT) assay.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
Cells were seeded in special dishes for confocal observation. After 60–70 % confluence, the cells were incubated with 1–100 μM cisplatin in the presence or absence of indicated concentrations of GB with without specific inhibitors. After the treatment, the cells were washed with phosphate-buffered saline (PBS), fixed in 4 % paraformaldehyde, and incubated with 3 % H2O2 in methanol for 10 min at room temperature to block endogenous peroxidase activity. After PBS washing, dishes were incubated with the TUNEL reaction mixture for 1 h at 37 °C and then stained with DAPI for 15 min at room temperature. Finally, cells were observed under a confocal microscope and TUNEL-positive cells were counted under a 200× magnification field. Results were shown as folds of TUNEL-positive cells as control. In situ TUNEL assay was conducted according to the manufacturer’s instructions (Roche).
Real-time polymerase chain reaction (qRT-PCR)
After the treatment, total RNA was isolated from HEI-OC1 cells according to the manufacturer’s instructions (TIANGEN, China). The concentration of total RNA was determined by spectrophotometry. Then, RNA was reverse-transcribed to cDNA using a cDNA synthesis kit (Takara). The samples were analyzed by RT-PCR using the BIORAD RT-PCR System for quantitative evaluation. One microliter of cDNA was amplified with SYBR Premix Ex Taq (TaKaRa). PCR was performed with the following mouse primers: Bax: forward: 5′ CCC ACC AGC TCT GAA CAG 3′; backward: 5′ TCA GCT TCT TGG TGG ACG 3′. Bcl-2: forward: 5′ ACG GTG GTG GAG GAA CTC 3′; backward: 5′ ACC CAG CCT CCG TTA TCC 3′. Nrf2: forward: 5′ CCA CAT TCC CAA ACA AGA 3′; backward: 5′ GGG CAG TGA AGA CTG AAC 3′. HO-1: forward: 5′ TCA GAA GGG TCA GGT GTC 3′; backward: 5′ CTC AGG GAA GTA GAG TGG 3′.
Mitochondrial membrane potential (MMP)
Cells were seeded in special dishes for confocal observation. After the treatment, the cells were washed with PBS and incubated with Rhodamine 123 (Rho123) at 37 °C for 30 min. After that, fluorescence was observed under a confocal microscope.
Oxygen consumption rate
After the treatment, oxygen consumption rate was analyzed to evaluate mitochondrial function. Briefly, cultured cells were trypsinized, washed in PBS, and then resuspended in Dulbecco’s phosphate-buffered saline (dPBS). Then, oxygen consumption rate was measured with a Clark oxygen electrode and expressed as percentage of baseline.
Western blot
After the treatment, the cells were collected and lysed with cell lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1 % Triton X-100, 1 mM EDTA, 10 mM NaF, 1 mM Na3VO4, and protease inhibitor cocktail) on ice for 30 min. After centrifugation at 20,000×g for 20 min at 4 °C, the protein contents were determined by BCA assay kit (Pierce, USA). After boiling for 5 min in a 2 × SDS loading buffer, 20 μg of total protein was subjected to SDS-PAGE and transferred onto a PVDF membrane. Then, the membrane was blocked and probed with indicated primary antibodies overnight at 4 °C. After washing for four times, the membrane was incubated in the appropriate horseradish peroxidase-conjugated secondary antibody at 37 °C for 30 min. The protein bands were visualized using chemiluminescent reagents according to the manufacturer’s instructions and quantified using an image analyzer Quantity One System (Bio-Rad, Richmond, CA).
Caspase 3 and caspase 9 activities
After the experiment, the activities of caspase 9 and caspase 3 were assessed using commercial assay kits (Beyotime Institute of Biotechnology, China) according to the manufacturer’s instructions.
ROS determination
Cells were seeded in special dishes for confocal observation. After the experiment, cells were washed with PBS, stained with DHE (10 μM), a superoxide probe, or MitoSOX (500 nM), a mitochondrial superoxide-specific probe, for 30 min at 37 °C, and then observed under a confocal microscope.
Transfection and reporter gene assay
pGL6-ARE-luciferase (Beyotime Institute of Biotechnology, China) and Renilla TK (Promega) plasmids were transfected into cells using a TurboFect transfection reagent (Thermo Scientific). After the experiments, cells were harvested in passive lysis buffer (Promega) and reporter assay was performed using the dual-luciferase reporter assay system (Promega). Firefly luciferase activity was normalized to Renilla luciferase and shown as a ratio of relative light units.
Chromatin immunoprecipitations (CHIP)
After the experiment, CHIP was conducted using a Thermo Scientific Pierce Agarose ChIP Kit according to the manufacture’s instruction. Cells were cross-linked using formaldehyde (1 %) and then were harvested and sheared by sonication. The cell lysate was immunoprecipitated with Nrf2 antibodies. DNA was isolated from the immunoprecipitated chromatin, and PCR was performed to examine the presence of HO-1 gene promoter.
Animal treatment
The Animal Care and Use Committee of The Second Affiliated Hospital of Xi’an Jiaotong University approved the surgical procedures in accordance with the guidelines regarding the care and use of animals for experimental procedures. Sprague-Dawley rats (male, age 7 weeks, 200–250 g) were purchased from Animal Centre of Xi’an Jiaotong University. The rats were maintained in a temperature-controlled environment with a 12-h light/dark cycle and had free access to water and food.
The rats were randomly divided into five groups (eight per group): control, cisplatin, cisplatin + GB, cisplatin + GB + ZnPP, cisplatin + GB + LY294002. Rats in cisplatin group were given an intraperitoneal injection of cisplatin (16 mg/kg). The last three groups were intraperitoneally injected with GB (10 mg/kg), and zinc protoporphyrin IX (ZnPP, 10 mg/kg) or LY294002 (10 mg/kg) immediately after being given cisplatin. Rats in control group were given 2 % DMSO instead. Cisplatin was dissolved in saline at a concentration of 0.15 mg/mL and was administered intraperitoneally at a dose of 16 mg/kg. Auditory and vestibular functions of rats were evaluated 72 h after the administration of the cisplatin. The auditory brain stem response (ABR) test was conducted with the Biosig 32 system as previously described [8]. Each rat’s hearing status was assessed with an ABR test both before and 72 h after the administration of the cisplatin. Vestibular function was evaluated by tail-hanging test and swimming test [34]. In brief, the rats were lifted by the tail and kept hanging at a height of 30 cm for a 15 s interval and the number of head rotations was counted. For the swimming test, a stainless steel pool (length, 28 cm; width, 45 cm; and depth, 25 cm) was filled with body-temperature water at a depth of 19 cm. Rats were lifted by their tail and dropped into the center of the pool from a height of 20 cm. The time in seconds between contact with the water and separation from the water to the platform was counted.
Statistical analysis
All statistical analysis was performed by GraphPad Prism software. Results were expressed as the mean ± SD. Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by the Newmane Keuls multiple-comparison post hoc test. p value <0.05 was considered statistically significant.
Results
Protective effects of GB against cisplatin-induced cytotoxicity in HEI-OC1 cells
In the current study, HEI-OC1 cells were used to evaluate the protective effect of GB on cisplatin-induced cytotoxicity. HEI-OC1 cells were incubated with 1–100 μM cisplatin for 24 h, and cell viability was determined by MTT. The results showed that cisplatin exhibited concentration-dependent toxicity on cells and 100 μM cisplatin decreased cell viability to less than 50 % of that of control (Fig. 1a). The potential toxicity of GB on HEI-OC1 cells were assessed after 24 h exposure, and we showed that >100 μM GB significantly reduced HEI-OC1 cell viability (Fig. 1b). Thus, 100 μM cisplatin and less than 100 μM GB were used throughout the experiment [18]. In Fig. 1c, we figured out that 1–50 μM GB could notably inhibit cisplatin-induced decrease in cell viability. The results demonstrated that GB protected against cisplatin-induced cytotoxicity.

Effect of GB on cisplatin-induced cytotoxicity in HEI-OC1 cells. a Cells were incubated with 1–100 μM cisplatin for 24 h, and then cell viability was detected by MTT. b Cells were incubated with 1–200 μM GB for 24 h, and then cell viability was detected by MTT. c Cells were incubated with 100 μM cisplatin in the presence or absence of 1–50 μM GB for 24 h, and then cell viability was detected by MTT. Results were expressed as folds of control. *p < 0.05, compared with control; # p < 0.05, compared with cisplatin group
Protective effects of GB against cisplatin-induced mitochondrial apoptosis in HEI-OC1 cells
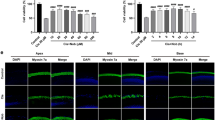
To evaluate whether anti-apoptotic effect of GB was involved in the protective effects against cisplatin-induced cytotoxicity, TUNEL assay was conducted to examine the apoptotic cells after the treatment of cisplatin in the presence or absence of GB. As shown in Fig. 2a, >10 μM cisplatin increased the percentage of TUNEL-positive cells in a concentration-dependent manner, which was consistent with the results of cell viability. As expected, 1–50 μM GB remarkably blocked 100 μM cisplatin-induced apoptosis, as evidenced by reduction of TUNEL-positive cells (Fig. 2b).

Effect of GB on cisplatin-induced mitochondrial apoptosis in HEI-OC1 cells. Cells were incubated with 1–100 μM cisplatin (a), or 100 μM cisplatin with or without 1–50 μM GB (b) for 24 h, and then cell apoptosis was detected by TUNEL assay. Cells were incubated with 100 μM cisplatin with or without 1–50 μM GB for 24 h, and then mRNA expression of Bax (c) and Bcl-2 (d) was measured by qRT-PCR. mRNA level was shown as folds of control. e Cells were incubated with 100 μM cisplatin with or without 1–50 μM GB for 24 h, and then MMP was detected by Rhodamine 123 staining. f Cells were incubated with 100 μM cisplatin with or without 1–50 μM GB for 24 h, and oxygen consumption rate was determined by a Clark oxygen electrode. Cells were incubated with 100 μM cisplatin with or without 1–50 μM GB for 24 h, and cytochrome c release in cytoplasm and cleavage of caspase 3, caspase 9, and PARP was determined by Western blot (f, h). Caspase 3 and caspase 9 activities were determined by commercial kits (i, j). *p < 0.05, compared with control; # p < 0.05, compared with cisplatin group
To further explore the mechanism of GB-exhibited inhibition of apoptosis, mitochondrial apoptotic pathway was determined. mRNA expression of Bax and Bcl-2 was detected by qRT-PCR, and the results showed that cisplatin decreased Bax expression and increased Bcl-2 expression, which was significantly inhibited by GB in a concentration-dependent manner (Fig. 2c, d). Then, mitochondrial membrane potential (MMP) was evaluated by Rhodamine (Rho123) staining. We found that Rho123 fluorescence was reduced by cisplatin and 50 μM GB evidently inhibited the decrease in Rho123 fluorescence in the presence of cisplatin, indicating that GB could protect against cisplatin-damaged MMP (Fig. 2e). Moreover, mitochondrial function was assessed by the determination of oxygen consumption rate using a Clark electrode. The results showed that exposure to cisplatin notably decreased oxygen-consuming activity, which could be inhibited when GB was in the culture, implicating that GB could protect against cisplatin-induced mitochondrial dysfunction (Fig. 2f). In the next step, we determined the cytoplasmic release of cytochrome c (Cyto C) through the detection of Cyto C protein expression in cytoplasm extraction. As shown in Fig. 2g, Cyto C expression in cytoplasm was dramatically increased by cisplatin. The administration of GB significantly inhibited the increase in cytoplasmic Cyto C induced by cisplatin (Fig. 2g). Next, Cyto C-exerted apoptotic cascades were examined. We found that cisplatin could promote the cleavage of caspase 3, caspase 9, and poly (ADP-ribose) polymerase (PARP), which event was blocked by the treatment of GB (Fig. 2h). Furthermore, cisplatin-enhanced caspase 3 and caspase 9 activities were inhibited by GB treatment (Fig. 2i, j). These results suggested that anti-apoptotic effect of GB, at least partly, contributed to the protective effect against cisplatin-induced cytotoxicity.
Protective effects of GB against cisplatin-induced oxidative stress in HEI-OC1 cells
In the next step, we examined the effect of cisplatin on ROS generation and antioxidant defenses in the presence or absence of GB in HEI-OC1 cells. DHE was used to detect superoxide level in a whole cell. We found that cisplatin could significantly increase cellular ROS generation, as reflected by enhancement of DHE fluorescence (Fig. 3a). Treatment of GB notably inhibited the fluorescence of DHE, implicating a regulatory role of GB in cisplatin-induced ROS generation (Fig. 3a). Moreover, MitoSOX, a mitochondrial superoxide-specific probe, was used to detect mitochondrial ROS generation. As reflected in Fig. 3b, in cisplatin-treated cells, MitoSOX fluorescence was increased markedly, indicating that cisplatin promoted ROS production in mitochondria. Next, antioxidant defense and ROS-generating enzymes were examined. Expression of nuclear factor erythroid 2-related factor 2 (Nrf2), heme oxygenase-1 (HO-1), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) 2 and p47phox was determined by qRT-PCR and Western blot. As shown in Fig. 3c, d, in cells treated by cisplatin, mRNA and protein expressions of Nrf2 and HO-1 were decreased, which event was inhibited by GB treatment in a concentration-dependent manner. Cisplatin increased NOX2 and p47phox expression, which may be involved in the generation of ROS induced by cisplatin. In the presence of GB, cisplatin-induced NOX2 and p47phox expression was significantly inhibited, which was concentration-dependent (Fig. 3c, d). Furthermore, Nrf2 activity was examined by the transfection of ARE-luciferase plasmid. ARE-luciferase activity in cisplatin-exposed cells was decreased notably (Fig. 3e). GB treatment significantly inhibited cisplatin-induced reduction of ARE-luciferase activity (Fig. 3e), further implicating the regulatory role of GB on Nrf2 transcription activity. We also searched for direct evidence of Nrf2-exerted regulation of HO-1 using CHIP assay. As shown in Fig. 3f, cisplatin resulted in a significant reduction of Nrf2-binding of HO-1 promoter. As expected, GB notably inhibited cisplatin-induced decrease in HO-1 promoter-binding by Nrf2 (Fig. 3f). These results indicated that promotion of Nrf2–HO-1-regulated antioxidant defense and decrease in NOX were involved in the inhibition of ROS generation induced by cisplatin.

Effect of GB on cisplatin-induced redox imbalance in HEI-OC1 cells. Cells were incubated with 100 μM cisplatin with or without 1–50 μM GB for 24 h. Cells were incubated with DHE (10 μM) (a) or MitoSOX (500 nM) (b) at 37 °C for 30 min, and fluorescence was observed under confocal microscope. mRNA expression of Nrf2, HO-1, NOX2, and p47phox was determined by qRT-PCR (c), and results were shown as folds of control. Protein expression of Nrf2, HO-1, NOX2, and p47phox was determined by Western blot. Representative blot was shown. ARE-luciferase activity was determined with STOP&GLO (Promega, Madison, WI), and firefly luciferase activity was normalized to Renilla luciferase (e). Binding of Nrf2 to the HO-1 promoter was assessed by CHIP assay. *p < 0.05, compared with control; # p < 0.05, compared with cisplatin group
Nrf2–HO-1-regulated antioxidant defense is involved in the protective effects of GB against cisplatin-induced cytotoxicity in HEI-OC1 cells
To test whether Nrf2-regulated HO-1 antioxidant defense was involved in the protective effects of GB against cisplatin-induced cytotoxicity, cells were transfected with SiRNA of Nrf2. As reflected in Fig. 4a, SiNrf2 could effectively inhibit Nrf2 expression in HEI-OC1 cells. Then, cells transfected with SiNrf2 were exposed to cisplatin in the presence of GB. In Fig. 4b, we showed that the inhibition of apoptosis by GB in cisplatin-treated cells was significantly blocked by SiNrf2, implicating the role of Nrf2 in GB-exerted protective effect. Moreover, zinc protoporphyrin IX (ZnPP), an inhibitor of HO-1, was used to examine the role of HO-1 in the protective effect of GB. In Fig. 4c, the results showed that in the presence of ZnPP, GB-induced inhibition of apoptosis in cisplatin-treated cells was significantly blocked. These results demonstrated that Nrf2–HO-1-regulated antioxidant defense is involved in the protective effects of GB against cisplatin-induced cytotoxicity in HEI-OC1 cells.

Role of Nrf2 and HO-1 in the protective effect of GB against cisplatin-induced cytotoxicity in HEI-OC1 cells. Cells were transfected with Nrf2 SiRNA and then exposed to 100 μM cisplatin with or without 50 μM GB for 24 h. Interference of Nrf2 and HO-1 was examined by Western blot (a). Apoptosis was examined by TUNEL assay (b). Cells were exposed to 100 μM cisplatin with or without 50 μM GB in the presence or absence of ZnPP, an HO-1 specific inhibitor, for 24 h. Apoptosis was examined by TUNEL assay (c). *p < 0.05
Nrf2–HO-1-regulated antioxidant defense is involved in the protective effects of GB against cisplatin-induced cytotoxicity in HEI-OC1 cells
In the next step, we detected the possible mediators of GB-exerted regulation of Nrf2/HO-1. Akt phosphorylation was devaluated by Western blot. As shown in Fig. 5a, incubation of cells with cisplatin resulted in notable reduction of Akt phosphorylation. GB could concentration-dependently inhibit cisplatin-inhibited phosphorylation of Akt (Fig. 5a). To test whether Akt activation was involved in GB-exerted regulation of Nrf2 and HO-1, we examined the effect of LY294002, an inhibitor of PI3 K/Akt signaling, on Nrf2 and HO-1 expression. In Fig. 5b, we found that in the presence of LY294002, the inhibition of cisplatin-induced reduction of Nrf2 and HO-1 was significantly blocked. Moreover, we also evaluated the effect of LY294002 on the protective effect of GB against apoptosis. As shown in Fig. 5c, inhibition of Akt by LY294002 significantly weakened the protective effect of GB against apoptosis, as reflected by increased percentage of cell apoptosis, compared with that of cisplatin + GB-treating cells. These results demonstrated that Akt signaling was involved in GB-exhibited regulation of Nrf2/HO-1 and the protective effect of GB against cisplatin-induced cytotoxicity.

Role of Akt signal in the protective effect of GB against cisplatin-induced cytotoxicity in HEI-OC1 cells. Cells were exposed to 100 μM cisplatin with or without 50 μM GB in the presence or absence of LY294002, a PI3 K/Akt inhibitor, for 24 h. Phosphorylation of Akt (a) and expression of Nrf2 and HO-1 (b) were determined by Western blot. Representative blot was shown. Apoptosis was examined by TUNEL assay (c). *p < 0.05
Protective effects of GB against cisplatin-induced ototoxicity in vivo
After the in vitro evaluation of GB, we further explored the protective effects of GB against cisplatin-induced ototoxicity in vivo. Rats were given an intraperitoneal injection of cisplatin (16 mg/kg), or injected with GB (10 mg/kg) with or without the injection of ZnPP or LY294002 immediately after being given cisplatin. Auditory and vestibular functions of rats were evaluated by auditory brain stem response (ABR) test. Vestibular function was evaluated by tail-hanging test and swimming test. We found that cisplatin resulted in a notable increase in hearing threshold at 16 and 32 kHz, which was inhibited by the treatment of GB (Fig. 6a, b). Both ZnPP and LY294002 could block GB-induced inhibition of hearing threshold increase in cisplatin-treated rats. Moreover, the administration of GB significantly inhibited cisplatin-induced vestibular dysfunction, as evidenced by decreased head rotations in the tail-hanging test and time intervals in the swimming test, compared with that of cisplatin-treated rats (Fig. 6c). As expected, ZnPP and LY294002 inhibited GB-exerted protective effects on vestibular function, as reflected by increase in head rotations in the tail-hanging test and time intervals in the swimming test, compared with that of cisplatin + GB-treated rats (Fig. 6d). These results demonstrated that GB could protect against cisplatin-induced ototoxicity in vivo. Based on the above results, it was noted that Akt-activated HO-1 defense was involved in the protective effect of GB against in vivo ototoxicity induced by cisplatin.

Protective effect of GB against cisplatin-induced ototoxicity in rats. Rats were intraperitoneally injected with cisplatin (16 mg/kg), with or without intraperitoneal injection of GB (10 mg/kg), and zinc protoporphyrin IX (ZnPP, 10 mg/kg) or LY294002 (10 mg/kg) immediately after being given cisplatin. a, b Hearing threshold (dB) at 16 and 32 kHz before (pre) and after (72 h) the administration (eight rats). c, d Number of head rotations in the tail-hanging test and average swimming time in the swimming test before (pre) and after (72 h) the administration (eight rats). *p < 0.05
Discussion
Ototoxicity severely limited the use of chemotherapeutic agent cisplatin. In the present study, we examined the effect of GB, a major component of G. biloba extracts, on cisplatin-induced ototoxicity. The results showed that GB could protect against ototoxicity induced by cisplatin in vitro and in vivo.
Apoptosis is a form of programmed cell death that is related to various physiological and pathophysiological conditions. Mitochondria play an important role in the regulation of apoptosis induced by intracellular stimuli [16]. Mitochondrial integrity, in a degree, determines whether apoptosis would occur. The integrity of mitochondria is tightly controlled by the pro- and anti-apoptotic Bcl-2 family of proteins [11, 21, 46]. Anti-apoptotic proteins, such as Bcl-2, maintain mitochondrial integrity, while the pro-apoptotic members of the family destroy the integrity of mitochondria, resulting in the release of apoptogenic proteins from mitochondria. Thus, the ratio of pro- and anti-apoptotic members of the Bcl-2 family determines the response of mitochondria to apoptotic stimulation. When the balance of pro- and anti-apoptotic members of the Bcl-2 family is damaged, mitochondrial integrity could be disrupted, leading to the release of pro-apoptotic factors like Cyto C [33]. Then, pro-apoptotic factors would activate caspases, a group of intracellular cysteine proteases that execute apoptosis by cleaving their substrates [12]. The previous literature has shown that apoptosis plays an important role in cisplatin-induced ototoxicity [1, 6, 10, 27]. In the present study, we evaluated the effect of GB on cisplatin-induced apoptosis in inner ear hair cells. We found that GB could concentration-dependently inhibit apoptosis and associated mitochondrial molecular events induced by cisplatin, indicating that the inhibition of mitochondrial apoptosis may be involved in the protective effect of GB against cisplatin-induced ototoxicity.
In a body, redox balance is controlled by ROS generation and antioxidant defense systems. Once excessive ROS is generated or antioxidant defense is destroyed, oxidative stress would occur, resulting in DNA, lipid, and protein damage. It is usually believed that oxidative stress is closely associated with apoptosis through mitochondrial pathway [5, 20]. Mitochondria and NADPH oxidases are major sources of ROS generation in a cell [23, 24]. Nrf2 is a well-known transcription factor, which controls redox balance through regulating a variety of antioxidant enzymes [22, 35], including HO-1. HO-1 is reported to possess antioxidant, anti-apoptotic, and anti-inflammatory properties via its products bilirubin/biliverdin and carbon monoxide [26]. In the context of cisplatin-induced ototoxicity, oxidative stress is a primary determinant of cell survival or death [29, 30, 37]. In the current study, we examined the effect of GB on redox balance in cisplatin-treated cells. We found that GB inhibited cellular and mitochondrial ROS generation and enhanced Nrf2-regulated HO-1 antioxidant defense in cells exposed to cisplatin. GB exerted inhibitory effects on NOX2 expression induced by cisplatin, indicating that inhibition of NOX-derived ROS may be involved in the antioxidant effect of GB. Moreover, interference of Nrf2 and inhibition of HO-1 significantly blocked the cytotoxic effect of GB, implicating that enhancement of Nrf2–HO-1 pathway was responsible for GB-induced protective effect against cisplatin-induced ototoxicity.
Under several conditions, Akt signaling is usually involved in Nrf2 activation exerted by intracellular stimuli and natural products [25, 39]. In this study, we also tested the possibility of the involvement of Akt in GB-exhibited regulation of antioxidant and anti-apoptotic effect. The results showed that GB inhibited cisplatin-induced decrease in Akt phosphorylation and that inhibition of Akt significantly blocked GB-enhanced Nrf2/HO-1 expression and GB-inhibited apoptosis in cisplatin-exposed cells. The results indicated that Akt signaling mediated GB-exerted regulation of Nrf2–HO-1 pathway and anti-apoptotic activity.
Finally, we tested the hypothesis in rats exposed to cisplatin injection. It was noted that GB could protect animals against cisplatin-induced hearing loss and vestibular dysfunction. Inhibition of Akt and HO-1 significantly suppressed the protective effect of GB on hearing loss and vestibular dysfunction exerted by cisplatin, indicating that Akt-mediated regulation of Nrf2–HO-1 pathway also plays a role in the otoprotective effect of GB in vivo.
In conclusion, the main and novel finding of this study is that GB decreases ROS generation through reducing NOX2 expression and enhancing Nrf2–HO-1 antioxidant pathway via activation of Akt signaling, resulting in inhibition of mitochondrial apoptosis and final reduction of cisplatin-induced ototoxicity in vitro and in vivo (Fig. 7). Our findings have appointed a new path toward the understanding of pharmacological activities of GB and gained an insight into the molecular mechanism underlying the protective effect of GB against cisplatin-induced ototoxicity.

Schematic summary of the obtained results and hypotheses. Cisplatin induced ototoxicity through generation of ROS by NOX2 and mitochondria and subsequent mitochondrial apoptosis. GB decreases ROS generation through reducing NOX2 expression and enhancing Nrf2–HO-1 antioxidant pathway via activation of Akt signaling, resulting in inhibition of mitochondrial apoptosis and final reduction of cisplatin-induced ototoxicity
Abbreviations
- CHIP:
-
Chromatin immunoprecipitations
- Cyto C:
-
Cytochrome c
- GB:
-
Ginkgolide B
- HEI-OC1:
-
House Ear Institute-Organ of Corti 1
- HO-1:
-
Heme oxygenase-1
- NOX:
-
Nicotinamide adenine dinucleotide phosphate oxidase
- MMP:
-
Mitochondrial membrane potential
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- PARP:
-
Poly (ADP-ribose) polymerase
- PBS:
-
Phosphate-buffered saline
- qRT-PCR:
-
Real-time polymerase chain reaction
- Rho123:
-
Rhodamine 123
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling
- ZnPP:
-
Zinc protoporphyrin IX
References
Altun Z, Olgun Y, Ercetin P, Aktas S, Kirkim G, Serbetcioglu B, Olgun N, Guneri EA (2014) Protective effect of acetyl-l-carnitine against cisplatin ototoxicity: role of apoptosis-related genes and pro-inflammatory cytokines. Cell Prolif 47:72–80
Black FO, Myers EN, Schramm VL, Johnson J, Sigler B, Thearle PB, Burns DS (1982) Cisplatin vestibular ototoxicity: preliminary report. Laryngoscope 92:1363–1368
Burschka MA, Hassan HA, Reineke T, van Bebber L, Caird DM, Mosges R (2001) Effect of treatment with Ginkgo biloba extract EGb 761 (oral) on unilateral idiopathic sudden hearing loss in a prospective randomized double-blind study of 106 outpatients. Eur Arch Otorhinolaryngol 258:213–219
Chan WH, Hsuuw YD (2007) Dosage effects of ginkgolide B on ethanol-induced cell death in human hepatoma G2 cells. Ann N Y Acad Sci 1095:388–398
Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV (2014) Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int 2014:238463
Cho SI, Lee JH, Park JH, Do NY (2014) Protective effect of (-)-epigallocatechin-3-gallate against cisplatin-induced ototoxicity. J Laryngol Otol 128:1–6
Choi SJ, Kim SW, Lee JB, Lim HJ, Kim YJ, Tian C, So HS, Park R, Choung YH (2013) Gingko biloba extracts protect auditory hair cells from cisplatin-induced ototoxicity by inhibiting perturbation of gap junctional intercellular communication. Neuroscience 244:49–61
Choung YH, Kim SW, Tian C, Min JY, Lee HK, Park SN, Lee JB, Park K (2011) Korean red ginseng prevents gentamicin-induced hearing loss in rats. Laryngoscope 121:1294–1302
Devarajan P, Savoca M, Castaneda MP, Park MS, Esteban-Cruciani N, Kalinec G, Kalinec F (2002) Cisplatin-induced apoptosis in auditory cells: role of death receptor and mitochondrial pathways. Hear Res 174:45–54
Goncalves MS, Silveira AF, Teixeira AR, Hyppolito MA (2013) Mechanisms of cisplatin ototoxicity: theoretical review. J Laryngol Otol 127:536–541
Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13:1899–1911
Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, Yeh WC, Ohashi P, Wang X, Mak TW (2005) Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell 121:579–591
Hinduja S, Kraus KS, Manohar S, Salvi RJ (2014) d-methionine protects against cisplatin-induced neurotoxicity in the hippocampus of the adult rat. Neurotox Res [Epub ahead of print]
Huang X, Whitworth CA, Rybak LP (2007) Ginkgo biloba extract (EGb 761) protects against cisplatin-induced ototoxicity in rats. Otol Neurotol 28:828–833
Kose K, Dogan P (1995) Lipoperoxidation induced by hydrogen peroxide in human erythrocyte membranes. 2. Comparison of the antioxidant effect of Ginkgo biloba extract (EGb 761) with those of water-soluble and lipid-soluble antioxidants. J Int Med Res 23:9–18
Li P, Nijhawan D, Wang X (2004) Mitochondrial activation of apoptosis. Cell 116: S57-9, 2 p following S59
Liu J, Zhang H, Zhang Y, Li N, Wen Y, Cao F, Ai H, Xue X (2014) Homing and restorative effects of bone marrow-derived mesenchymal stem cells on Cisplatin injured ovaries in rats. Mol Cells 37:865–872
Ma L, Liu X, Zhao Y, Chen B, Li X, Qi R (2013) Ginkgolide B reduces LOX-1 expression by inhibiting Akt phosphorylation and increasing Sirt1 expression in oxidized LDL-stimulated human umbilical vein endothelial cells. PLoS One 8:e74769
Maclennan KM, Darlington CL, Smith PF (2002) The CNS effects of Ginkgo biloba extracts and ginkgolide B. Prog Neurobiol 67:235–257
Martinez-Reyes I, Cuezva JM (2014) The H(+)-ATP synthase: a gate to ROS-mediated cell death or cell survival. Biochim Biophys Acta 1837:1099–1112
Martinou JC, Green DR (2001) Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol 2:63–67
Na HK, Surh YJ (2014) Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic Biol Med 67:353–365
Nemoto S, Takeda K, Yu ZX, Ferrans VJ, Finkel T (2000) Role for mitochondrial oxidants as regulators of cellular metabolism. Mol Cell Biol 20:7311–7318
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Niture SK, Khatri R, Jaiswal AK (2014) Regulation of Nrf2-an update. Free Radic Biol Med 66:36–44
Ollinger R, Yamashita K, Bilban M, Erat A, Kogler P, Thomas M, Csizmadia E, Usheva A, Margreiter R, Bach FH (2007) Bilirubin and biliverdin treatment of atherosclerotic diseases. Cell Cycle 6:39–43
Orzaez M, Sancho M, Marchan S, Mondragon L, Montava R, Valero JG, Landeta O, Basanez G, Carbajo RJ, Pineda-Lucena A, Bujons J, Moure A, Messeguer A, Lagunas C, Herrero C, Perez-Paya E (2014) Apaf-1 inhibitors protect from unwanted cell death in in vivo models of kidney ischemia and chemotherapy induced ototoxicity. PLoS One 9:e110979
Paksoy M, Ayduran E, Sanli A, Eken M, Aydin S, Oktay ZA (2011) The protective effects of intratympanic dexamethasone and vitamin E on cisplatin-induced ototoxicity are demonstrated in rats. Med Oncol 28:615–621
Poirrier AL, Pincemail J, Van Den Ackerveken P, Lefebvre PP, Malgrange B (2010) Oxidative stress in the cochlea: an update. Curr Med Chem 17:3591–3604
Rybak LP, Whitworth CA, Mukherjea D, Ramkumar V (2007) Mechanisms of cisplatin-induced ototoxicity and prevention. Hear Res 226:157–167
Sakamoto M, Kaga K, Kamio T (2000) Extended high-frequency ototoxicity induced by the first administration of cisplatin. Otolaryngol Head Neck Surg 122:828–833
Schacht J, Talaska AE, Rybak LP (2012) Cisplatin and aminoglycoside antibiotics: hearing loss and its prevention. Anat Rec 295:1837–1850
Shore GC, Nguyen M (2008) Bcl-2 proteins and apoptosis: choose your partner. Cell 135:1004–1006
Tian CJ, Kim YJ, Kim SW, Lim HJ, Kim YS, Choung YH (2013) A combination of cilostazol and Ginkgo biloba extract protects against cisplatin-induced Cochleo-vestibular dysfunction by inhibiting the mitochondrial apoptotic and ERK pathways. Cell Death Dis 4:e509
Vriend J, Reiter RJ (2014) The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome. Mol Cell Endocrinol 401C:213–220
Waissbluth S, Daniel SJ (2013) Cisplatin-induced ototoxicity: transporters playing a role in cisplatin toxicity. Hear Res 299:37–45
Waissbluth S, Pitaro J, Daniel SJ (2012) Gene therapy for cisplatin-induced ototoxicity: a systematic review of in vitro and experimental animal studies. Otol Neurotol 33:302–310
Waissbluth S, Salehi P, He X, Daniel SJ (2013) Systemic dexamethasone for the prevention of cisplatin-induced ototoxicity. Eur Arch Otorhinolaryngol 270:1597–1605
Wang X, Wu H, Chen H, Liu R, Liu J, Zhang T, Yu W, Hai C (2012) Does insulin bolster antioxidant defenses via the extracellular signal-regulated kinases-protein kinase B-nuclear factor erythroid 2 p45-related factor 2 pathway? Antioxid Redox Signal 16:1061–1070
Wood JW, Bas E, Gupta C, Selman Y, Eshraghi A, Telischi FF, Van De Water TR (2014) Otoprotective properties of mannitol against gentamicin induced hair cell loss. Otol Neurotol 35:e187–e194
Yang TH, Young YH, Liu SH (2011) EGb 761 (Ginkgo biloba) protects cochlear hair cells against ototoxicity induced by gentamicin via reducing reactive oxygen species and nitric oxide-related apoptosis. J Nutr Biochem 22:886–894
Yang YI, Ahn JH, Choi YS, Choi JH (2014) Brown algae phlorotannins enhance the tumoricidal effect of cisplatin and ameliorate cisplatin nephrotoxicity. Gynecol Oncol 136:355–364
Yoshikawa T, Naito Y, Kondo M (1999) Ginkgo biloba leaf extract: review of biological actions and clinical applications. Antioxid Redox Signal 1:469–480
Zhang C, Tian X, Luo Y, Meng X (2011) Ginkgolide B attenuates ethanol-induced neurotoxicity through regulating NADPH oxidases. Toxicology 287:124–130
Zhang S, Chen B, Wu W, Bao L, Qi R (2011) Ginkgolide B reduces inflammatory protein expression in oxidized low-density lipoprotein-stimulated human vascular endothelial cells. J Cardiovasc Pharmacol 57:721–727
Zhong Q, Gao W, Du F, Wang X (2005) Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121:1085–1095
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ma, W., Hu, J., Cheng, Y. et al. Ginkgolide B protects against cisplatin-induced ototoxicity: enhancement of Akt–Nrf2–HO-1 signaling and reduction of NADPH oxidase. Cancer Chemother Pharmacol 75, 949–959 (2015). https://doi.org/10.1007/s00280-015-2716-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-015-2716-9