
Abstract
Purpose
Despite overall successful application to multiple myeloma patients, clinical efficacy of the proteasome inhibitor bortezomib is typically challenged by primary and secondary resistance of unknown origin. So far, the potential impact of intracellular concentrations on drug efficacy of bortezomib and the influence of drug transporters are unknown.
Methods
We determined cellular bortezomib kinetics in nine myeloma cell lines using ultrahigh-performance liquid chromatography coupled to tandem mass spectrometry. The potential influence of drug transporters on the uptake kinetics observed in these cell lines was investigated by testing substrate characteristics of bortezomib for several transporters in over-expressing model cells. Additionally, transporter mRNA expression was quantified in myeloma cell lines by real-time polymerase chain reaction (RT-PCR).
Results
All myeloma cells revealed an extensive intracellular bortezomib accumulation (47.5–183 ng/ml) exceeding extracellular concentrations (0.04–0.17 ng/ml) by more than factor 1,000. Only organic anion-transporting polypeptide 1B1 facilitated the uptake in over-expressing cells, however, to a negligible extent (factor 1.36). Bortezomib efflux via P-glycoprotein was confirmed by demonstrating reduced sensitivity (IC50 11.6 vs. 2.8 ng/ml) and intracellular concentrations (−56.1 %) in over-expressing cells compared to controls. RT-PCR revealed a varying but overall weak transporter expression in the studied myeloma cells without any correlation to intracellular concentrations. Although principally valid as demonstrated in the P-glycoprotein over-expressing cell model, there was no significant correlation between intracellular concentrations and bortezomib efficacy in myeloma cell lines.
Conclusion
Differences in intracellular concentrations in myeloma cell lines neither result from variable transporter expression nor represent the main factor determining bortezomib efficacy in vitro.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The accumulation of clonal plasma cells in the bone marrow of multiple myeloma patients causes a variety of symptoms that usually result from the displacement of normal hematopoiesis, the formation of osteolytic bone lesions, and the production of monoclonal protein [1, 2]. However, the administration of the reversible proteasome inhibitor bortezomib has significantly improved the outcome in a great number of affected myeloma patients [3, 4].
Although plasma pharmacokinetics showed a strong distribution [5–7], which suggests considerable intracellular enrichment, cellular bortezomib kinetics in targeted cells (e.g., myeloma) has not been studied so far. Moreover, it is unclear whether variability in cellular uptake or enrichment modulates bortezomib efficacy and possibly also resistance. This is particularly interesting because multiple myeloma still cannot be cured in the majority of patients [8]. So far, the molecular principles of both primary and secondary resistance to bortezomib are poorly understood [9, 10].
Among the different proposed resistance mechanisms such as gene mutations of proteasome subunits [11], altered proteasome turnover [12], over-expression of heat shock proteins [13], and formation of stress granules [14], multidrug resistance (MDR) mechanisms also need to be considered because of the generally high predisposition of myeloma cells to altered gene expression [15, 16]. While over-expression of drug transporters has already been observed in myeloma patients [17], the general impact of both influx and efflux transporters on intracellular bortezomib concentrations in myeloma cells has not been thoroughly investigated. In particular, comprehensive investigation of drug efflux mechanisms should clarify the controversial discussion about the contribution of MDR transporters to bortezomib resistance in leukemic and myeloma cells [11, 18–24].
In this study, we therefore determined intracellular concentration–time profiles of bortezomib in nine different myeloma cell lines and investigated whether differences in intracellular concentrations observed in these cell lines might be determined by variable drug transporter expression and to which extent the divergent intracellular concentrations are related to the efficacy of bortezomib.
Materials and methods
Materials
Bortezomib was purchased from Absource Diagnostics (München, Germany) while the internal standard D8-bortezomib was obtained from Toronto Research Chemicals (Toronto, Canada). Olmesartan was purchased from Sequoia Research Products (Pangbourne, UK). Dulbecco’s modified Eagle medium (DMEM), M199, RPMI-1640, glutamine, penicillin, streptomycin sulfate, trypsin, ethylenediaminetetraacetic acid (EDTA), phosphate-buffered saline (PBS), 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazoliumbromide (MTT), probenecid, aprotinin, and anti-β-actin (Clone AC-74) were purchased from Sigma-Aldrich (Taufkirchen, Germany). Fetal calf serum (FCS) and geneticin (G418) were supplied by PAA (Pasching, Austria). Iscove’s modified Dulbecco’s medium (IMDM) and 2-mercaptoethanol (2-ME) were purchased from Invitrogen (Karlsruhe, Germany). DMEM/Ham’s F12 was purchased from Biochrom (Berlin, Germany). Zosuquidar (LY335979) was kindly provided by Eli Lilly (Bad Homburg, Germany), while 5-(3-(2-(7-chloroquinolin-2-yl)ethenyl)phenyl)-8-dimethyl-carbamyl-4,6-dithiaoctanoic acid sodium salt hydrate (MK571) was purchased from Enzo Life Science (Lörrach, Germany). Crystal violet, dimethyl sulfoxide (DMSO), rifampicin, Triton® X-100, sodium dodecyl sulfate (SDS), glycerol, 2-amino-2-(hydroxymethyl)-propan-1,3-diol (TRIS), dithiothreitol (DTT), and Tween®20 were purchased from AppliChem (Darmstadt, Germany). Pefabloc and Collagen R were obtained from Serva (Heidelberg, Germany), and vincristine and the antibody against human P-glycoprotein (P-gp) clone C219 from Calbiochem (Darmstadt, Germany). Bromphenol blue, leupeptin, and pepstatin were from Biomol (Hamburg, Germany). The BCA® Protein Assay Kit and the SuperSignal® West Pico Chemiluminescent Substrate Kit were purchased from Pierce (Rockford, USA), Slim-Fast® from Allpharm (Messel, Germany), and the nitrocellulose membranes (Optitran BA-S 85) from Schleicher & Schuell BioScience (Dassel, Germany). The secondary anti-mouse antibody was obtained from Amersham (Freiburg, Germany), the RNeasy Mini-Kit from Qiagen (Hilden, Germany), and the RevertAid™ H Minus First Strand cDNA Synthesis Kit from Fermentas (St. Leon-Rot, Germany). Methanol, ethanol, 2-propanol, formic acid, acetic acid, sodium hydroxide solution (NaOH), acetonitrile (ACN), and Rotiphorese® gel 30 were obtained from Carl Roth GmbH (Karlsruhe, Germany). Methyl-tert-butylether (MTBE) and hydrochloric acid (HCl) were from Merck KGaA (Darmstadt, Germany). Casy® ton, Casy® clean, and the Cytotoxicity Detection Kit (LDH) were obtained from Roche Diagnostics and Roche Applied Science (Mannheim, Germany). Primers were synthesized by Eurofins MWG Operon (Ebersberg, Germany). The Absolute QPCR SYBR Green Mix was from Abgene (Hamburg, Germany), and the 96-well plates for PCR were obtained from Biozym (Hessisch Oldendorf, Germany). Cell culturing bottles, reaction tubes, and 6-well cell culture plates were purchased from Greiner (Frickenhausen, Germany). Ninety-six-well (300 µl) microtiter plates were supplied by Nunc (Wiesbaden, Germany) and glass vials by VWR International (Darmstadt, Germany).
Cell lines
Myeloma cell lines
Cellular uptake kinetics of bortezomib were investigated in nine myeloma cell lines. Briefly, four non-adherently (Karpas-620, KMS-12-BM, L363, OPM-2) and five partly adherently (EJM, KMM-1, LP-1, RPMI-8226, U266) growing myeloma cell lines were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) and cultured as previously described [25]. The culture medium consisted of IMDM for EJM and LP-1 and RPMI-1640 medium for all other myeloma cell lines, respectively. Each culture medium was supplemented with 10 % FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin sulfate.
HEK-293 cells
In order to investigate human organic anion-transporting polypeptides (OATPs = solute carriers of organic anions (SLCOs)), HEK-293 cells over-expressing SLCO1B1 (HEK-OATP1B1), SLCO1B3 (HEK-OATP1B3), or an empty control vector (HEK-293-VC G418) were used for bortezomib uptake studies. The cell lines were kindly provided by D. Keppler (German Cancer Research Centre, Heidelberg, Germany). Following standard cell culture conditions, cells were cultured in DMEM supplemented with 10 % FCS, 2 mM glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin sulfate, and 800 µg/ml G418.
CHO cells
Similarly, organic anion transporter 1 (OAT1) was tested for bortezomib uptake in over-expressing CHO-hOAT cells and control CHOpIRES cells, transfected only with the empty vector. Both cell lines were cultured in DMEM/Ham’s F12 supplemented with 10 % FCS, 100 U/ml penicillin, 100 µg/ml streptomycin sulfate, and 600 µg/ml G418. The cells were kindly provided by T. Cihlar (Gilead Sciences, Foster City, USA).
LLC cells
Possible transport of bortezomib by P-gp was investigated in LLC-MDR1 (over-expressing P-gp (MDR1/ABCB1)) cells in comparison with the parental cell line LLC-PK1 (available at ATCC, Manassas, USA). The over-expressing cells were kindly provided by A. H. Schinkel (The Netherlands Cancer Institute, Amsterdam, the Netherlands). Both LLC cell lines were cultured in M199 medium supplemented with 10 % FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin sulfate. Vincristine (0.64 µM) was only added to the culture medium of LLC-MDR1 cells to maintain P-gp over-expression. For the experiments, both cell lines were either directly seeded in (see growth inhibition assay) or fed with (see cellular uptake) vincristine-free medium 1 day prior to the respective assay.
MDCKII cells
Potential transport of bortezomib by other common multidrug resistance transporters [26] was investigated in the respective over-expressing MDCKII cell lines. MDCKII-MRP1, MDCKII-MRP2, MDCKII-MRP3 (over-expressing multidrug resistance-associated protein 1 (MRP1/ABCC1), 2 (MRP2/ABCC2), and 3 (MRP3/ABCC3)), and MDCKII-BCRP (over-expressing breast cancer resistance protein (BCRP/ABCG2)) cell lines were kindly provided by P. Borst and A. H. Schinkel (The Netherlands Cancer Institute, Amsterdam, the Netherlands). Following standard cell culture conditions, all MDCKII cells were cultured in DMEM supplemented with 10 % FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin sulfate.
Determination of cellular uptake of bortezomib
Cellular uptake of bortezomib was determined with ultrahigh-performance liquid chromatography coupled to tandem mass spectrometry (UPLC/MS/MS) after liquid–liquid extraction according to our previously described method [27]. Because of its instability in vitro and the potential differences between the used cell lines, it is not sufficient to only determine absolute intracellular bortezomib concentrations [27]. Thus, concentrations in the extracellular compartment (medium) were also quantified, because intracellular bortezomib concentrations can only be reliably interpreted in relation to the actual extracellular exposure. However, since the process of decay did not substantially influence the interpretation of the results (correlations or significances), we limited the discussion to absolute intracellular concentrations and derived areas under the intracellular concentration–time curves (AUCs) to facilitate the comprehensibility of the study results.
Both for incubation in experiments and UPLC/MS/MS calibration, we prepared a single stock solution (1.32 mM) by dissolving bortezomib in acetonitrile/H2O (1/1, v/v +0.01 % formic acid). Uptake kinetics including intracellular and extracellular concentration–time profiles were determined in nine different myeloma cell lines over 48 h of incubation with 0.2, 1, 5, and 25 nM bortezomib (0.08, 0.38, 1.92, and 9.61 ng/ml), which covers a concentration range around determined IC50 values (Table 1). To investigate whether bortezomib is a substrate of OATP1B1 or OATP1B3, intracellular concentrations of bortezomib were quantified after 10 and 60 min of incubation with 1 and 5 nM (0.38 and 1.92 ng/ml) both in parental HEK-293 cells and corresponding cells over-expressing the respective transporter. For control, experiments were performed with or without rifampicin (20 µM), a well-known OATP inhibitor [28]. Similar experiments were conducted in CHO cells over-expressing OAT1 with the use of probenecid (500 µM) or olmesartan (10 µM) as inhibitor. To verify the P-gp substrate characteristics of bortezomib, its uptake was assessed in over-expressing LLC cells and compared to the respective parental cell line after 24 h of incubation with 1 and 5 nM (0.38 and 1.92 ng/ml). For control, experiments were performed with or without the specific P-gp inhibitor LY335979 (2 µM).
Growth inhibition assays
Using growth inhibition assays with crystal violet staining, we screened for possible involvement of efflux transporters (P-gp, BCRP, and MRP1-3) in the disposition of bortezomib; by lowering intracellular concentrations, active efflux would yield higher IC50 values in transporter over-expressing cell lines compared to parental cells [29]. Additionally, the specific inhibitors LY335979 (for P-gp), FTC (for BCRP), and MK571 (for MRP1-3) were used to further confirm involvement of the respective transporter in the observed efflux changes. Moreover, growth inhibition assays using the tetrazolium dye procedure were used to assess the cellular sensitivity to bortezomib in myeloma cell lines and relate it to both transporter expression and cellular uptake.
Both growth inhibition assays were conducted according to previously established methods [30]. For quantification, mean absorption of cell-free wells (background, equal to 0 % proliferation) was first subtracted from each cell-containing well. The final proliferation rate was then calculated from the ratio of absorption in compound-containing wells to drug-free wells (corresponding to 100 % proliferation). Each experiment investigated an octuplet of each concentration and was performed four times. Sigmoid concentration–response curves and IC50 values were calculated by GraphPad Prism version 5.01 (GraphPad Software Inc., La Jolla, USA).
Quantification of transporter mRNA expression by RT-PCR
The mRNA expression of the transporter genes ABCB1 and SLCO1B1 was investigated in all myeloma cell lines to assess their overall expression level and to determine possible differences in their relative expression. Similarly, basal mRNA expression of ABCB1 was compared between LLC-MDR1 cells and the corresponding parental cell line.
RNA was extracted from harvested cells using the RNeasy Mini-Kit. Before storage at −80 °C until analysis, purity and concentration of the isolated RNA were determined spectrophotometrically. According to the manufacturer’s instructions, cDNA was synthesized with the RevertAid™ H Minus First Strand cDNA Synthesis Kit. mRNA expression was quantified by RT-PCR with the LightCycler® 480 (Roche Applied Science, Mannheim, Germany). PCR amplification was carried out in 20 µl reaction volume containing 5 µl 1:10 diluted cDNA and 1 × Absolute QPCR SYBR Green Mix. The utilized primer sequences for both target and housekeeping genes were published previously [29, 31].
Since the comparison of gene expression between different cell lines can be hampered by different expression levels of housekeeping genes among the cell lines, basal mRNA expressions in the myeloma cell lines were normalized to the following three housekeeping genes which were expressed at a similar level in all investigated cell lines: glucose-6-phosphate dehydrogenase (G6PDH), β2-microglobulin (β2 mg), and hypoxanthine-phosphoribosyltransferase 1 (hPRT). As a calibrator, a mixture (in equal parts) of the cDNA of all tested cell lines was used.
Data were evaluated by calibrator-normalized relative quantification with efficiency correction using the LightCycler® 480 software version 1.5 (Roche Applied Science, Mannheim, Germany). This software calculates the relative amount of the target gene and the housekeeping gene based on the crossing points (Cp) and the underlying calibration curve. The results were expressed as the target/reference ratio divided by the target/reference ratio of the calibrator. The results are therefore corrected for sample inhomogeneities and variance caused by detection. All samples were amplified at least in duplicate. Whenever mRNA expression was below the detection limit (Cp value >35), the respective cell line was excluded from analysis and marked (#) in the figure.
Western blot analysis of P-gp
Protein expression of P-gp in LLC-MDR1 cells and the respective parental cell line were compared by SDS-polyacrylamide gel electrophoresis followed by Western blotting as published previously [29]. Blots were semiquantified by ImageJ 1.43u (NIH, USA).
Statistical analysis
Data were analyzed using GraphPad Prism version 5.01 and GraphPad InStat version 3.05 (GraphPad Software Inc., La Jolla, USA). Intracellular concentrations of bortezomib per ml cell volume were calculated by dividing the quantified amount in each cell pellet by the pellet’s mean volume (product of mean individual cell volume and number of cells). AUCs of the intracellular (cell samples) and extracellular (supernatant samples) concentration–time curves were calculated for the entire incubation period of 48 h (AUC0–48 h). Intracellular concentrations of myeloma, HEK-293, and LLC cells were compared and tested using ANOVA with Tukey’s post hoc test. All IC50 values were calculated from sigmoid concentration–response curves (nonlinear regression) and tested using ANOVA with Dunnett’s post hoc test. Correlation analysis of transporter expression with AUC0–48 h of the intracellular concentration–time curves was performed using the Pearson’s correlation coefficient. Spearman’s rank correlation coefficient was utilized for the correlation with cellular sensitivities because of the exceptionally lower IC50 value of EJM cells (Table 1). p ≤ 0.05 was considered significant.
Results
Cellular uptake kinetics of bortezomib in myeloma cells
All intracellular concentration–time curves revealed an almost similar shape after incubation with 0.2 and 1 nM bortezomib over the entire incubation period of 48 h (Fig. 1a, 1 nM). An extensive cellular uptake exceeding the continuously decreasing extracellular concentrations was demonstrated (Fig. 1a). For instance, after only 1 h of incubation with 1 nM bortezomib, bortezomib accumulated 43-fold into myeloma cell lines compared to extracellular concentrations. Steadily increasing intracellular concentrations reached their maximum after 24 h in the majority of myeloma cell lines. However, this maximum concentration was between 47.5 ng/ml (Karpas-620) and 183 ng/ml (EJM) after incubation with 1 nM bortezomib and thus differed significantly (p < 0.001) between the evaluated cell lines. At the respective intracellular peak concentration, median intracellular bortezomib concentrations (21.3, 104, 304, and 495 ng/ml) exceeded the corresponding median extracellular concentrations (0.02, 0.06, 0.58, and 5.58 ng/ml) by far for all exposure concentrations (0.2, 1, 5, and 25 nM). Thus, bortezomib extensively accumulated in myeloma cells resulting in intracellular to extracellular concentration ratios of 1,065, 1,733, 524, and 89 (for exposure concentrations of 0.2, 1, 5, and 25 nM). At higher extracellular exposure concentrations, such as 5 and 25 nM, intracellular peak concentrations were reached earlier.

Cellular uptake of bortezomib in nine myeloma cell lines after up to 48 h of incubation. Time-dependent intracellular and extracellular concentration profiles over 48 h are exemplarily depicted for incubation with 1 nM (a). Each data point represents the mean concentration ± SD of three samples at the respective point in time. Box-and-whisker plots depict the range of the calculated AUC0–48 h of intracellular and extracellular bortezomib concentrations after incubation with 0.2, 1, 5, and 25 nM (b). Given numbers represent the median AUC
The total intracellular bortezomib accumulation (AUC0–48 h) varied considerably between the myeloma cell lines, in particular at low exposure concentrations (Fig. 1b). For example, AUCs differed up to factor 3.3 (from 2,127 (ng/ml) × 48 h (Karpas-620) to 7,010 (ng/ml) × 48 h (EJM)) after incubation with 1 nM bortezomib. Except for extracellular EJM medium (9.70 (ng/ml) × 48 h), the AUCs of the extracellular concentration–time curves ranged only from 3.37 (ng/ml) × 48 h to 5.35 (ng/ml) × 48 h after incubation with 1 nM. Moreover, the total intracellular bortezomib accumulation (AUC0–48 h) generally increased with growing exposure concentrations (0.2–25 nM), but the slope of the accumulation was reduced at higher exposure concentrations (Fig. 1b). In contrast, the AUCs of the extracellular bortezomib concentrations constantly followed a linear trend.
Assessment of bortezomib influx via OATP1B1, OATP1B3, and OAT1
The contribution of OATP influx transporters to the extensive intracellular accumulation of bortezomib was investigated in over-expressing HEK-293 cell lines (Fig. 2). Cells over-expressing OATP1B1 revealed significantly higher intracellular concentrations of bortezomib than the parental cell line after 60 min of incubation (p < 0.001). However, absolute intracellular concentrations only slightly increased from 4.6 ng/ml by +1.7 ng/ml (1 nM incubation) and from 20.4 ng/ml by +7.1 ng/ml (5 nM incubation). The addition of the OATP1B1 inhibitor rifampicin to HEK-OATP1B1 cells revoked the additional uptake (p = 0.63). Incubation for only 10 min revealed very similar findings (+0.75 and +3.84 ng/ml, p < 0.01). The additional uptake in OATP1B1 over-expressing cells was relatively higher (factor 1.58 and 1.62) compared to 60 min (factor 1.36 and 1.35). In contrast, intracellular concentrations of bortezomib did not differ between OATP1B3 over-expressing and parental cells, which indicates that bortezomib is not a substrate of OATP1B3 (Fig. 2). Moreover, no relevant additional uptake via OAT1 could be demonstrated (data not shown).

Comparison of the intracellular concentrations in different HEK-293 cell lines after 60 min of incubation with 1 nM (a) or 5 nM (b) bortezomib. Data represent mean ± SD of a triplicate for each cell line with or without addition of rifampicin (RIF; 20 µM); *p < 0.05; **p < 0.01; ***p < 0.001, ns: p > 0.05
Assessment of bortezomib efflux via P-gp, BCRP, and MRP1-3
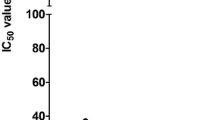
LLC-MDR1 cells were more resistant to bortezomib than the parental cell line (IC50 value increased from 2.8 to 11.6 ng/ml, p < 0.001), which indicates active bortezomib transport by P-gp (Fig. 3a). The addition of the specific P-gp inhibitor LY335979 restored the sensitivity of the over-expressing cells to the level of the parental cell line (IC50: 2.7 ng/ml, p = 0.94). Consistently, intracellular bortezomib concentrations were reduced by 56.1 % (−119 ng/ml) and 72.8 % (−499 ng/ml) in LLC-MDR1 cells compared to the parental LLC-PK1 cell line (p < 0.001) after incubation with 1 and 5 nM, respectively. The addition of LY335979 abolished this effect resulting in intracellular concentrations in LLC-MDR1 cells no longer significantly different to the parental cell line (p = 0.13).

Comparison of concentration-dependent proliferation rates (a), intracellular concentrations after 24 h of incubation with 1 nM bortezomib (b), and P-gp expression in Western blot (c) between P-gp over-expressing (LLC-MDR1) and parental (LLC-PK1) cells. Each data point of the proliferation curves represents the mean ± SD of n = 32 samples. Intracellular concentration data represent the mean ± SD of a triplicate for each cell line with or without addition of LY335979 (2 µM); *p < 0.05; **p < 0.01, ns: p > 0.05. The depicted blot utilizes β-actin for normalization and is representative for a triplicate
In contrast, sensitivities of MDCKII cell lines over-expressing BCRP and MRP1-3 did not differ from the parental control cell line, indicating that bortezomib is not a substrate of these transporters (data not shown).
Comparison of P-gp expression in LLC cells
Both qualitative protein expression (Fig. 3c) and quantitative mRNA expression (factor 24,000) differed considerably between LLC-MDR1 cells and the respective parental cell line.
Comparison of basal transporter mRNA expression in myeloma cells
To evaluate the potential contribution of drug transporters to varying intracellular bortezomib concentrations observed in the studied myeloma cells, the basal mRNA transporter expression of ABCB1 and SLCO1B1 was assessed in the myeloma cell lines. Overall, drug transporters were weakly expressed in most myeloma cells and were below the detection limit in about 50 % of the cell lines investigated. Higher rates of ABCB1 mRNA expression were found in LP-1, KMM-1, and EJM cells (Fig. 4a), whereas five tested cell lines revealed no detectable expression at all (Cp values >35). While there was no traceable SLCO1B1 mRNA expression in four myeloma cell lines either, EJM and KMM-1 cells exhibited the highest relative expression levels of this gene (Fig. 4b).

Basal mRNA expression in different myeloma cell lines in relation to the mean of all tested cell lines. Expression data for ABCB1 (a) and SLCO1B1 (b) are normalized to a set of housekeeping genes (G6PDH, β2 mg, and hPRT) and expressed as mean ± SD for n = 8 (4 biological replicates and 2 PCR replicates for each sample)
Association of basal mRNA expression and intracellular bortezomib accumulation
Despite some identified differences in the expression of transporter genes, baseline mRNA expression and total intracellularly accumulated bortezomib (AUC0–48 h) did not correlate for ABCB1 (p = 0.80) and SLCO1B1 (p = 0.22) expression in the myeloma cell lines. Besides, transporter expression was not significantly related to the bortezomib efficacies determined in the growth inhibition assays (ABCB1: p = 0.93; SLCO1B1: p = 0.09).
Sensitivity of myeloma cells to bortezomib
After 48 h of constant drug exposure, the sensitivity to bortezomib differed up to factor 5.4 between the myeloma cell lines showing a median IC50 value of 6.4 ± 0.1 nM (Table 1). In comparison with the other myeloma cell lines, sensitivity of EJM cells was exceptionally higher (IC50 = 1.6 nM).
Association of total cellular uptake (AUC0–48 h) and sensitivity of myeloma cells to bortezomib
Overall, there was no significant correlation between the sensitivity of the myeloma cell lines (IC50 values) and the total cellular uptake (AUC0–48 h) in myeloma cells (Table 1). In brief, Spearman’s rank correlation for incubation with 0.2, 1, 5, and 25 nM resulted in coefficients of −0.38 (p = 0.31), −0.63 (p = 0.08), −0.45 (p = 0.23), and −0.37 (p = 0.34), respectively. Moreover, additionally calculated ratios of intracellular to extracellular AUC for each cell line (factor 464–1,131) did not reveal any association with cellular sensitivities either (p > 0.39).
Discussion
Despite the successful administration of bortezomib to patients for more than a decade [3, 4], multiple myeloma is still far from curable in the majority of patients because of primary or ultimately emerging secondary resistance [8]. A detailed characterization of bortezomib’s cellular uptake kinetics might help to better comprehend the so far poorly understood mechanisms that lead to different bortezomib sensitivities and thus varying drug efficacy [9, 10, 21]. As described in various other cancer entities [32], drug transporters are an important example of intracellular concentration modulators and related drug efficacy.
Plasma pharmacokinetics of bortezomib has shown extensive distribution into peripheral tissues suggesting intracellular enrichment [5–7]. However, the observed extent of bortezomib accumulation in the myeloma cell lines exceeded this assumption by far. Hence, the observed steep gradient between intracellular and extracellular concentrations raised the question about additional factors promoting this exceptional intracellular accumulation. So far, the process of cellular uptake of bortezomib has been explained exclusively by free diffusion across cell membranes [33]. Indeed, higher exposure concentrations led to higher intracellular concentrations in our experiments reaching saturation only at cytotoxic exposure concentrations, whereas the quantified extracellular concentrations followed a continuously linear trend. Searching for potential influx transporters facilitating bortezomib uptake, we could only identify OATP1B1 as a mediator of additional bortezomib uptake. However, the extent of OATP1B1-mediated influx was marginal despite artificial over-expression in the model cells rapidly diminished within the first hour after incubation, and only accounted for a small proportion of the total intracellular accumulation of bortezomib. Thus, it is very unlikely for influx via OATP1B1 to play a major role even at higher expression levels. Apart from influx transporters, the massive enrichment might rather be explained—at least in part—by the strong binding of bortezomib to proteasomes [34, 35] withdrawing bortezomib from the diffusion equilibrium across the cell membrane and thus maintaining a driving force for the extensive uptake by diffusion. Additionally, this effect might be intensified by still unknown unspecific binding to other intracellular components such as organelles or cytoplasmic proteins.
Although considerable accumulation was observed in all studied cell lines, its extent varied increasingly when reaching the respective maximum of intracellular concentrations. Given the demonstrated outbound gradient, active efflux transporters might considerably contribute to the varying extent of bortezomib accumulation. The screening for efflux transporters of bortezomib via growth inhibition assays excluded common drug transporters like BCRP or MRP1-3, but confirmed P-gp as a transporter of bortezomib [18, 20, 23]. This finding was subsequently confirmed by assessing the intracellular bortezomib concentrations in P-gp over-expressing model cells, which simultaneously showed both reduced intracellular concentrations and reduced bortezomib efficacy. Regarding the controversially discussed relevance of P-gp for bortezomib resistance [18–24], our observation principally confirms the impact of P-gp-mediated efflux for intracellular bortezomib concentration and efficacy. However, since the required level of P-gp over-expression needs to be very high to evoke this effect, applicability, e.g., to myeloma cell lines, greatly depends on their level of P-gp expression. To investigate whether the observed differences in the intracellular bortezomib concentrations of the studied myeloma cell lines are attributable to variable transporter gene expression, we quantified their relative drug transporter mRNA expression levels. Already very low at the mRNA level, transporter expression could not be validated at the protein level by less sensitive Western blotting.
Overall, ABCB1 was only weakly expressed in about half of the cell lines, which is consistent with previous findings [36]. Together with the equally marginal expression of SLCO1B1 in most of the myeloma cell lines, these results obviously question the relevance of these two transporters for the different intracellular bortezomib concentrations observed. Not surprisingly, transporter expression was correlated neither with intracellular concentrations nor with cellular bortezomib sensitivities. Considering that previous studies always demonstrated acquired resistance to bortezomib after long-time exposure to be completely independent of transporter expression [11, 37], their variable expression therefore does not seem to be a main factor determining intracellular bortezomib concentrations in myeloma cells.
Since the detected efflux of bortezomib via P-gp in the respective over-expressing model cells clearly demonstrated the principal relation between reduced intracellular concentrations and weaker efficacy, those myeloma cells with even greater differences in their intracellular concentrations were investigated for a similar association. In addition to the demonstrated irrelevance of drug transporters in these cell lines, the amount of intracellular bortezomib also did not appear to be a crucial determinant of its efficacy. Instead, the uptake of bortezomib in myeloma cells reflected by the AUCs of intracellular concentration–time curves was not significantly correlated with the IC50 values of bortezomib. Taking the in vitro decay of bortezomib into account [27], there was also no correlation with the enrichment ratios (intracellular AUC divided by extracellular AUC). However, altered intracellular concentrations might still be relevant in combination with other factors influencing cellular sensitivities, such as variable expression levels of the pharmacological target (proteasome) [38] or other interacting components [11–14]. Also, quantification of bortezomib in myeloma cells derived from in vivo samples could yield a stronger correlation because bortezomib could distribute within an entire organism resulting in lower intracellular concentrations. This might be the decisive difference because even very low concentrations of bortezomib have been described to be sufficient for a substantial inhibition of cellular proteasome activity [39]. In contrast, the rather high intracellular concentrations observed in our myeloma cells possibly occurred at a plateau of only barely modifiable proteasome inhibition without linear dose–response relationship, thus having no effect on the sensitivity, whereas intracellular concentrations in the model cells were reduced to a relevant and thus effective level. In line with this, similar proteasome inhibition has been demonstrated in several myeloma cell lines after incubation with a similar range of extracellular bortezomib concentrations, although the cells differed in their sensitivity to bortezomib [12]. Despite incubation with clinically observable plasma concentrations in our experiments [40], these considerations might explain the missing significant connection between intracellular bortezomib quantities and efficacy as an artifact that only occurs in vitro.
Conclusion
In conclusion, we described a very extensive but also varying accumulation of bortezomib in myeloma cells. Whereas both OATP1B1 and particularly P-gp were identified as principal transporters of bortezomib, their expression was too weak in the studied myeloma cell lines to influence intracellular concentrations. However, we were able to demonstrate the principal connection between P-gp over-expression, reduced intracellular concentrations, and bortezomib efficacy in model cells. In contrast, intracellular bortezomib concentrations were not correlated with bortezomib efficacy in myeloma cells. Since the differing bortezomib concentrations observed in vitro might have occurred at levels of already saturated proteasome inhibition, re-evaluation of these findings in vivo together with an investigation of additional factors determining intracellular bortezomib concentrations is indicated.
References
Kyle RA, Rajkumar SV (2004) Multiple myeloma. N Engl J Med 351:1860–1873
Barlogie B, Tricot GJ, van Rhee F, Angtuaco E, Walker R, Epstein J, Shaughnessy JD, Jagannath S, Bolejack V, Gurley J, Hoering A, Vesole D, Desikan R, Siegel D, Mehta J, Singhal S, Munshi NC, Dhodapkar M, Jenkins B, Attal M, Harousseau JL, Crowley J (2006) Longterm outcome results of the first tandem autotransplant trial for multiple myeloma. Br J Haematol 135:158–164
Neben K, Lokhorst HM, Jauch A, Bertsch U, Hielscher T, van der Holt B, Salwender H, Blau IW, Weisel K, Pfreundschuh M, Scheid C, Dührsen U, Lindemann W, Schmidt-Wolf IG, Peter N, Teschendorf C, Martin H, Haenel M, Derigs HG, Raab MS, Ho AD, van de Velde H, Hose D, Sonneveld P, Goldschmidt H (2012) Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood 119:940–948
Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348:2609–2617
Reece DE, Sullivan D, Lonial S, Mohrbacher AF, Chatta G, Shustik C, Burris H III, Venkatakrishnan K, Neuwirth R, Riordan WJ, Karol M, von Moltke LL, Acharya M, Zannikos P, Keith Stewart A (2011) Pharmacokinetic and pharmacodynamic study of two doses of bortezomib in patients with relapsed multiple myeloma. Cancer Chemother Pharmacol 67:57–67
Hemeryck A, Geerts R, Monbaliu J, Hassler S, Verhaeghe T, Diels L, Verluyten W, van Beijsterveldt L, Mamidi RN, Janssen C, De Coster R (2007) Tissue distribution and depletion kinetics of bortezomib and bortezomib-related radioactivity in male rats after single and repeated intravenous injection of 14 C-bortezomib. Cancer Chemother Pharmacol 60:777–787
Andriamanana I, Gana I, Duretz B, Hulin A (2013) Simultaneous analysis of anticancer agents bortezomib, imatinib, nilotinib, dasatinib, erlotinib, lapatinib, sorafenib, sunitinib and vandetanib in human plasma using LC/MS/MS. J Chromatogr B Anal Technol Biomed Life Sci 926:83–91
de la Puente P, Azab AK (2013) Contemporary drug therapies for multiple myeloma. Drugs Today (Barc.) 49:563–573
Dispenzieri A, Jacobus S, Vesole DH, Callandar N, Fonseca R, Greipp PR (2010) Primary therapy with single agent bortezomib as induction, maintenance and re-induction in patients with high-risk myeloma: results of the ECOG E2A02 trial. Leukemia 24:1406–1411
Richardson PG, Xie W, Mitsiades C, Chanan-Khan AA, Lonial S, Hassoun H, Avigan DE, Oaklander AL, Kuter DJ, Wen PY, Kesari S, Briemberg HR, Schlossman RL, Munshi NC, Heffner LT, Doss D, Esseltine DL, Weller E, Anderson KC, Amato AA (2009) Single-agent bortezomib in previously untreated multiple myeloma: efficacy, characterization of peripheral neuropathy, and molecular correlations with response and neuropathy. J Clin Oncol 27:3518–3525
Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G (2008) Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 112:2489–2499
Shabaneh TB, Downey SL, Goddard AL, Screen M, Lucas MM, Eastman A, Kisselev AF (2013) Molecular basis of differential sensitivity of myeloma cells to clinically relevant bolus treatment with bortezomib. PLoS ONE 8:e56132
Chauhan D, Li G, Shringarpure R, Podar K, Ohtake Y, Hideshima T, Anderson KC (2003) Blockade of Hsp27 overcomes Bortezomib/proteasome inhibitor PS-341 resistance in lymphoma cells. Cancer Res 63:6174–6177
Gareau C, Fournier MJ, Filion C, Coudert L, Martel D, Labelle Y, Mazroui R (2011) p21 (WAF1/CIP1) upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PLoS ONE 6:e20254
Seckinger A, Meissner T, Moreaux J, Goldschmidt H, Fuhler GM, Benner A, Hundemer M, Rème T, Shaughnessy JD Jr, Barlogie B, Bertsch U, Hillengass J, Ho AD, Pantesco V, Jauch A, De Vos J, Rossi JF, Möhler T, Klein B, Hose D (2009) Bone morphogenic protein 6: a member of a novel class of prognostic factors expressed by normal and malignant plasma cells inhibiting proliferation and angiogenesis. Oncogene 28:3866–3879
Neben K, Jauch A, Hielscher T, Hillengass J, Lehners N, Seckinger A, Granzow M, Raab MS, Ho AD, Goldschmidt H, Hose D (2013) Progression in smoldering myeloma is independently determined by the chromosomal abnormalities del(17p), t(4;14), gain 1q, hyperdiploidy, and tumor load. J Clin Oncol 31:4325–4332
Dalton WS, Grogan TM, Meltzer PS, Scheper RJ, Durie BG, Taylor CW, Miller TP, Salmon SE (1989) Drug-resistance in multiple myeloma and non-Hodgkin’s lymphoma: detection of P-glycoprotein and potential circumvention by addition of verapamil to chemotherapy. J Clin Oncol 7:415–424
Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D, Oerlemans R, Chan ET, Kirk CJ, Peters GJ, van der Heijden JW, de Gruijl TD, Scheper RJ, Jansen G (2012) Inactivating PSMB5 mutations and P-glycoprotein (multidrug resistance-associated protein/ATP-binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 341:174–182
Zheng B, Zhou R, Gong Y, Yang X, Shan Q (2012) Proteasome inhibitor bortezomib overcomes P-gp-mediated multidrug resistance in resistant leukemic cell lines. Int J Lab Hematol 34:237–247
Minderman H, Zhou Y, O’Loughlin KL, Baer MR (2007) Bortezomib activity and in vitro interactions with anthracyclines and cytarabine in acute myeloid leukemia cells are independent of multidrug resistance mechanisms and p53 status. Cancer Chemother Pharmacol 60:245–255
Lü S, Chen Z, Yang J, Chen L, Zhou H, Xu X, Li J, Han F, Wang J (2010) The effects of proteasome inhibitor bortezomib on a P-gp positive leukemia cell line K562/A02. Int J Lab Hematol 32:e123–e131
Rumpold H, Salvador C, Wolf AM, Tilg H, Gastl G, Wolf D (2007) Knockdown of PgP resensitizes leukemic cells to proteasome inhibitors. Biochem Biophys Res Commun 361:549–554
O’Connor R, Ooi MG, Meiller J, Jakubikova J, Klippel S, Delmore J, Richardson P, Anderson K, Clynes M, Mitsiades CS, O’Gorman P (2013) The interaction of bortezomib with multidrug transporters: implications for therapeutic applications in advanced multiple myeloma and other neoplasias. Cancer Chemother Pharmacol 71:1357–1368
Nakamura T, Tanaka K, Matsunobu T, Okada T, Nakatani F, Sakimura R, Hanada M, Iwamoto Y (2007) The mechanism of cross-resistance to proteasome inhibitor bortezomib and overcoming resistance in Ewing’s family tumor cells. Int J Oncol 31:803–811
Seckinger A, Meissner T, Moreaux J, Depeweg D, Hillengass J, Hose K, Rème T, Rösen-Wolff A, Jauch A, Schnettler R, Ewerbeck V, Goldschmidt H, Klein B, Hose D (2012) Clinical and prognostic role of annexin A2 in multiple myeloma. Blood 120:1087–1094
Tiwari AK, Sodani K, Dai CL, Ashby CR Jr, Chen ZS (2011) Revisiting the ABCs of multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol 12:570–594
Clemens J, Longo M, Seckinger A, Hose D, Haefeli WE, Weiss J, Burhenne J (2014) Stability of the proteasome inhibitor bortezomib in cell based assays determined by ultra-high performance liquid chromatography coupled to tandem mass spectrometry. J Chromatogr A 1345:128–138
Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K (2002) Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology 36:164–172
König SK, Herzog M, Theile D, Zembruski N, Haefeli WE, Weiss J (2010) Impact of drug transporters on cellular resistance towards saquinavir and darunavir. J Antimicrob Chemother 65:2319–2328
Peters T, Lindenmaier H, Haefeli WE, Weiss J (2006) Interaction of the mitotic kinesin Eg5 inhibitor monastrol with P-glycoprotein. Naunyn Schmiedebergs Arch Pharmacol 372:291–299
Zembruski NC, Büchel G, Jödicke L, Herzog M, Haefeli WE, Weiss J (2011) Potential of novel antiretrovirals to modulate expression and function of drug transporters in vitro. J Antimicrob Chemother 66:802–812
Gottesman MM (2002) Mechanisms of cancer drug resistance. Annu Rev Med 53:615–627
Pharmacology review part 1 (p. 8), drug approval package of bortezomib, Center of Drug Evaluation and Research, Food and Drug Association (FDA), c2003 [cited 2014 August 5]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2003/21602_Velcade_pharmr_P1.pdf
Kisselev AF, van der Linden WA, Overkleeft HS (2012) Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 19:99–115
Beck P, Dubiella C, Groll M (2012) Covalent and non-covalent reversible proteasome inhibition. Biol Chem 393:1101–1120
Schwarzenbach H (2002) Expression of MDR1/P-glycoprotein, the multidrug resistance protein MRP, and the lung-resistance protein LRP in multiple myeloma. Med Oncol 19:87–104
de Wilt LH, Jansen G, Assaraf YG, van Meerloo J, Cloos J, Schimmer AD, Chan ET, Kirk CJ, Peters GJ, Kruyt FA (2012) Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem Pharmacol 83:207–217
Busse A, Kraus M, Na IK, Rietz A, Scheibenbogen C, Driessen C, Blau IW, Thiel E, Keilholz U (2008) Sensitivity of tumor cells to proteasome inhibitors is associated with expression levels and composition of proteasome subunits. Cancer 112:659–670
Shringarpure R, Catley L, Bhole D, Burger R, Podar K, Tai YT, Kessler B, Galardy P, Ploegh H, Tassone P, Hideshima T, Mitsiades C, Munshi NC, Chauhan D, Anderson KC (2006) Gene expression analysis of B-lymphoma cells resistant and sensitive to bortezomib. Br J Haematol 134:145–156
Moreau P, Karamanesht II, Domnikova N, Kyselyova MY, Vilchevska KV, Doronin VA, Schmidt A, Hulin C, Leleu X, Esseltine DL, Venkatakrishnan K, Skee D, Feng H, Girgis S, Cakana A, van de Velde H, Deraedt W, Facon T (2012) Pharmacokinetic, pharmacodynamic and covariate analysis of subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma. Clin Pharmacokinet 51:823–829
Acknowledgments
This work was supported by Grants from the Deutsche Forschungsgemeinschaft (SFB/TRR79; Bonn, Germany) and the EU 7th framework program “OverMyR.” We thank A. H. Schinkel and P. Borst (The Netherlands Cancer Institute, Amsterdam, the Netherlands) for generously providing the cell lines LLC-MDR1, MDCKII-MDR1, MDCKII-BCRP, MDCKII-MRP1, MDCKII-MRP2, and MDCKII-MRP3. Furthermore, we thank D. Keppler (German Cancer Research Centre, Heidelberg, Germany) for generously providing the cell lines HEK-OATP1B1, HEK-OATP1B3, and HEK-KoG418 and T. Cihlar for providing the cell lines CHO-hOAT and CHOpIRES. We also thank J. Kocher, C. Mueller, S. Rosenzweig, M. Maurer, and A. Deschlmayr for their excellent technical assistance.
Conflict of interest
None to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Clemens, J., Seckinger, A., Hose, D. et al. Cellular uptake kinetics of bortezomib in relation to efficacy in myeloma cells and the influence of drug transporters. Cancer Chemother Pharmacol 75, 281–291 (2015). https://doi.org/10.1007/s00280-014-2643-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-014-2643-1