
Abstract
Purpose
The proteasome inhibitor bortezomib (PS-341) has displayed significant efficiency against pancreatic cancer cells. However, the underlying mechanisms are not fully understood. Here, we tested if ceramide production was involved in the bortezomib’s effect.
Methods
Two transformed pancreatic cancer cell lines (PANC-1 and Mia) and the primary pancreatic cancer cells were used. Cell death was analyzed by MTT viability assay and trypan blue staining. Cell apoptosis was analyzed by Histone DNA-ELISA assay and Annexin V FACS. Western blots were used to test signal protein changes. The cellular ceramide level after bortezomib treatment was also determined.
Results
In cultured pancreatic cancer cells, bortezomib increased cellular ceramide production to promote cell apoptosis. The ceramide de novo synthase inhibitor fumonisin B1 (F-B1) suppressed bortezomib-induced ceramide production and apoptosis, while exogenously added C6-ceramide facilitated bortezomib-induced pancreatic cancer cell death. Meanwhile, 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP), the inhibitor of glucosylceramide synthetase as well as the sphingosine kinase 1 inhibitors (SKI-II and SKI-IV), facilitated bortezomib-induced ceramide production and subsequent cell apoptosis. Further, bortezomib-induced pro-apoptotic c-Jun N-terminal kinase (JNK) activation was also associated with ceramide production. JNK activation by bortezomib was suppressed by F-B1, but was enhanced by SKI-II and PDMP in pancreatic cancer cells. Finally, C6-ceramide, SKI-II, and PDMP dramatically enhanced bortezomib-induced cytotoxicity in primary cultured pancreatic cancer cells.
Conclusions
We found that bortezomib-induced apoptosis was associated with ceramide production in primary and transformed pancreatic cancer cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pancreatic cancer has one of the worst prognoses among all malignancies [1, 2]. It is typically diagnosed at an advanced stage, and surgery can no longer remove the entire tumor [1]. The current standard therapies for this devastating disease include radiation and gemcitabine [3]. However, pancreatic cancers are among the most intrinsically resistant tumors to both radiation and chemotherapeutic drugs [4]. Hence, the search for novel and efficient agents against pancreatic cancer is necessary and extremely important [5, 6].
Bortezomib (PS-341, Velcade), a potent and selective inhibitor of the proteasome inhibitor, has displayed broad antitumor activities [7, 8]. Bortezomib was approved by the United States Food and Drug Administration (FDA) for the treatment for refractory or relapsed multiple myeloma (MM) [7–9]. Meanwhile, this drug is currently undergoing clinical and preclinical evaluations in the treatment for pancreatic cancers and many other tumors. As a matter of fact, bortezomib has shown significant efficiency against pancreatic cancer cells [10]. However, the underlying mechanisms of such an effect are not fully understood.
The ubiquitin–proteasome system plays an important role in the cellular homeostasis. The inhibition of the 26S proteasome by bortezomib leads to the accumulation of misfolded proteins, resulting in endoplasmic reticulum stress to cause unfolded protein response and cell apoptosis [10–12]. Recent studies have proposed other signaling mechanisms involved in bortezomib-induced cancer cell apoptosis. For example, Podar et al. [13] showed that bortezomib induces myeloid cell leukemia-1 (Mcl-1) down-regulation in MM cells to cause cell apoptosis. In hepatocellular carcinoma cells, Akt inhibition is a major molecular determinant of bortezomib-induced apoptosis [14]. Lauricella et al. [15] showed that bortezomib activates c-Jun N-terminal kinase (JNK)-dependent cell apoptosis. In the current study, we focused on the role of ceramide in bortezomib’s anticancer ability. Ceramide is the well-known apoptosis mediator [16]. A number of anti-cancer agents enhance intracellular ceramide production to promote cell apoptosis [16]. We here discovered that bortezomib-induced apoptosis is also associated with ceramide production in primary and transformed pancreatic cancer cells.
Materials and methods
Chemical and reagents
Bortezomib was purchased from Selleck.com.cn (Shanghai, China). Fumonisin B1 (F-B1) and 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) were purchased from Sigma (Shanghai, China). Sphingosine kinase 1 (SphK1) inhibitors (SKI)-II and SKI-IV were purchased from Calbiochem (Shanghai, China). C6-ceramide was obtained from Avanti (Alabaster, AB). Antibodies used were obtained from the following commercial sources: Anti-JNK1, apoptosis signal-regulating kinase 1 (ASK1), rabbit and mouse horseradish peroxidase (HRP)-conjugated IgG antibodies were purchased from Santa Cruz biotechnology (Santa Cruz, CA). Anti-ceramide synthase 1 and Anti-glucosylceramide synthase (GCS) antibodies were purchased from Abcam (Shanghai, China). All other antibodies used in this study were purchased form Cell Signaling Tech (Denver MA).
Cell culture
The pancreatic cancer PANC-1 and MIA PaCa-2 (MIA) cells were maintained in RPMI-1640 medium (Invitrogen, Shanghai, China), supplemented with a 10 % fetal bovine serum (FBS, Sigma, Shanghai, China), penicillin/streptomycin (1:100, Sigma, Shanghai, China), and 4 mM l-glutamine (Sigma), in a CO2 incubator at 37 °C.
Primary pancreatic adenocarcinoma cells isolation and culture
The primary cultured pancreatic adenocarcinoma cells from a patient with early-stage disease were obtained at the time of surgery. The surgery-isolated tissue was thoroughly washed in phosphate buffer solution (PBS) containing 100 units/ml penicillin–streptomycin and 2 mM DTT (wash buffer), the tissue was then minced by scalpel into small pieces, and was maintained into DMEM containing 100 units/ml penicillin–streptomycin. Pancreatic cancer cell pellets were thoroughly washed and then repelleted at 400 g for 5 min. Single-cell suspensions were achieved by re-suspending cells in 0.15 % (w/v) collagenase dissolved in DMEM and incubating the suspension at 37 °C and 5 % CO2 for 1 h; individual cell was pelleted and rinsed twice with DMEM before re-suspending the cell pellets in cell culture medium (DMEM, 20 % FBS, 2 mM glutamine, 1 mM pyruvate, 10 mM HEPES, 100 units/ml penicillin/streptomycin, 0.1 mg/ml gentamicin, 0.2 units/ml insulin, 0.1 mg/ml hydrocortisone, and 2 g/liter fungizone). The study was approved by the institutional review board of all authors’ institution, and written informed consent was obtained from the patient.
Trypan blue staining of “dead” cells
The number of dead tumor cells (trypan blue positive) after indicated treatment was counted, and the death percentage (%) was calculated by the number of the trypan blue positive cells divided by the total cell number.
Cell viability detection
The pancreatic cancer cell viability was measured by the 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide (MTT) assay [17].
Cell apoptosis quantification by Histone DNA-ELISA
The Cell Apoptosis ELISA Detection Kit (Roche, Palo Alto, CA) was used to detect pancreatic cancer cell apoptosis after the indicated treatment according to the manufacturer’s protocol [17].
Flow cytometry detecting Annexin V positive (“apoptotic”)cells
The pancreatic cancer cell apoptosis was also determined by the Annexin V In Situ Cell Apoptosis Detection Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. Pancreatic cancer cells were also stained with propidium iodide (PI, Molecular Probes). Annexin V+ cells (the apoptotic cells) were recorded through a flow cytometry (BD Bioscience).
Western blotting and data quantification
The cells were washed with ice-cold PBS before lysed with the lysis buffer (Beyotime, Shanghai, China). The lysates were separated by the 10 % SDS-polyacrylamide gel and were electro-transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). The membranes were blocked with 10 % milk in PBS plus Tween-20 (0.5 %) (TBST), incubated overnight at 4 °C with the primary antibody, and then incubated with HRP-conjugated secondary antibody. The detection was performed by Supersignal West Pico Enhanced Chemiluminescent (ECL, Pierce, Rockville, IL). The blot intensity was quantified by Image J software. The intensity of each phosphorylated band was normalized to the intensity of non-phosphorylated kinase band (the loading control).
Immunoprecipitation (IP)
After the indicated treatment, PANC-1 cell lysates (800 μg) in 1 mL IP lysis buffer (Beyotime, Shanghai, China) were precleared with 20 μl of protein A/G-agarose (Sigma) for 30 min. After centrifugation for 10 min at 4 °C in a microcentrifuge, the supernatants were rotated overnight with 2 μg of indicated primary antibody (anti-JNK1, Santa Cruz). Samples were then centrifuged for 5 min at 4 °C in a micro-centrifuge to remove nonspecific aggregates that formed overnight. Protein A/G-agarose (35 μl) was then added to the supernatants for 3 h at 4 °C. Pellets were washed six times with PBS, resuspended in the lysis buffer, and then assayed in Western blots detecting phospho- and total ASK1 and JNK1.
Enzymatic measurement of ceramide levels
The intracellular ceramide level was measured as previously reported [18] and was valued as fmol by nmol of phospholipid. The ceramide level in the treatment group was expressed as the percentage change in the untreated control group. Each measurement was done in triplicate.
Sphingosine kinase activity assay
As previously reported [18], pancreatic cancer cells were collected and lysed by the same lysis buffer for Western blotting. After centrifuging at 13,000 × g for 60 min, proteins (100 μg) in supernatant were then incubated with 25 μM D-erythrosphingosine dissolved in 0.1 % Triton X-100, 2 mM ATP, and [γ−32P] ATP (3.7 × 105 Bq dissolved in 20 mM MgCl2) for 30 min at 37 °C in a final volume of 200 μl. The reaction was stopped by adding 20 μl of HCl (1 N), followed by 800 μl of chloroform/methanol/HCl (100:200:1, v/v). After vigorous vortexing, 250 μl of chloroform and 250 μl of KCl (2 M) were added, and phases were separated by centrifugation. The organic layer was dried and resuspended in chloroform/methanol/HCl (100:100:0.2, v/v). Lipids were resolved on silica TLC plates in 1-butanol/acetic acid/water (3:1:1, v/v). The labeled sphingosine-1-phosphate (S1P) spots were visualized by autoradiography and quantified by scraping and counting in a scintillation counter. The sphingosine kinase activity was valued as pmol/h/g protein and was expressed as the percentage of the untreated control. Each measurement was done in triplicate.
Statistical analyses
The data were expressed as mean ± standard deviation (SD). Data were collected using three set of independent experiments. Statistical differences were analyzed by one-way ANOVA followed by multiple comparisons performed with post hoc Bonferroni test (SPSS version 16). Values of p < 0.05 were considered statistically significant. The significance of any differences between two groups was tested using paired-samples t test when appropriated.
Results
C6-ceramide enhances bortezomib-induced cytotoxicity in cultured pancreatic cancer cells
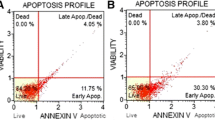
Here, we wanted to know the potential role of ceramide in bortezomib-induced cytotoxicity and to see whether ceramide manipulating could affect pancreatic cancer cells’ response to bortezomib. As shown in Fig. 1, in PANC-1 and MIA pancreatic cancer cells, C6-ceramide and bortezomib alone only induced moderate cell viability loss (Fig. 1a, e), cell death (Fig. 1b), and apoptosis (Fig. 1c, d and f). However, a combination of the two induced the dramatically increased cell death and apoptosis (Fig. 1). For example, bortezomib and C6-ceramide combination induced 57.34 ± 7.56 % of cell apoptosis (Annexin V positive cells) in PANC-1 cells, while bortezomib or C6-ceramide alone only induced 32.65 ± 2.65 or 17.41 ± 1.53 % of cell apoptosis, respectively (Fig. 1c).

C6-ceramide enhances bortezomib-induced cytotoxicity in cultured pancreatic cancer cells. PANC-1 and MIA pancreatic cancer cells were treated with vehicle (V, 0.1 % DMSO), or with bortezomib (100 nM) in the presence or absence of C6-ceramide (10 μg/ml) for 48 h, cell viability was analyzed by MTT assay (a and e), trypan blue was used to stain the “dead” cells (b), cell apoptosis was examined by Annexin V FACS sorting (c) and Histone DNA-ELISA assay (d and f). Experiments in this figure were repeated three times. *p < 0.05 versus the bortezomib treatment group, # p < 0.05 versus the C6-ceramide treatment group
Bortezomib induces ceramide production through de novo synthesis pathway, enhanced by PDMP
Next, we tested whether ceramide was involved in bortezomib-induced cytotoxicity in PANC-1 cells. Results in Fig. 2a showed that bortezomib increased cellular ceramide production in PANC-1 cells. F-B1, the ceramide de novo synthase inhibitor [19], suppressed ceramide production by bortezomib. Subsequently, bortezomib-induced cell viability loss (Fig. 2b, e) and apoptosis (Fig. 2c, e) were inhibited by F-B1, indicating that bortezomib induced pro-apoptotic ceramide production through the de novo synthesis pathway. On the other hand, the GCS inhibitor, PDMP, [20] facilitated bortezomib-induced ceramide production (Fig. 2a). Correspondingly, bortezomib-induced cytotoxicity (Fig. 2b, d) and apoptosis (Fig. 2c, e) were also enhanced by PDMP. Together, these results suggested that bortezomib-induced apoptosis in pancreatic cancer cells might be associated with ceramide production.

Bortezomib induces ceramide production through de novo synthesis pathway, enhanced by PDMP. PANC-1 and MIA cells were treated with vehicle (V, 0.1 % DMSO), or with bortezomib (100 nM) in the presence or absence of fumonisin B1 (F-B1, 20 μM) or 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP, 20 μM), cells were further cultured for indicated time points, cellular ceramide level was analyzed as described (a), cell viability and apoptosis were examined by MTT assay (b and d) and Annexin V FACS sorting (c and e), respectively. Experiments in this figure were repeated three times. *p < 0.05
Bortezomib-induced ceramide production and cytotoxicity in pancreatic cancer cells are enhanced by SphK1 inhibitors
Sphingosine kinase 1 is an oncogenic sphingolipid-metabolizing enzyme that catalyzes the formation of the mitogenic second messenger S1P, while consuming pro-apoptotic ceramide. Thus, blockage of S1P formation provides a pro-apoptotic outcome through preventing ceramide metabolism [21]. We have shown that bortezomib induced ceramide-dependent apoptosis in pancreatic cancer cells. Results in Fig. 3a, b showed that bortezomib alone had no significant effect on SphK1 activity. Interestingly, SKI-II and SKI-V, two SphK1 inhibitors (see Fig. 3a), facilitated bortezomib-induced ceramide production (Fig. 3b) and cytotoxicity (Fig. 3c–e). As compared to pancreatic cancer cells treated with bortezomib or SphK1 inhibitor alone, the cells stimulated with both agents displayed a profound cell viability loss (Fig. 3c, e) and a significantly increased cell apoptosis (Fig. 3d). We also tested whether above agents could affect the response of bortezomib in primary cultured pancreatic cancer cells. Results in Fig. 3f, g demonstrated that bortezomib-induced cell viability loss (Fig. 3f) and cell death (Fig. 3g) were dramatically enhanced by C6-ceramide, the SphK1 inhibitor SKI-II, and PDMP, suggesting that ceramide is also involved in bortezomib-induced cytotoxicity in primary pancreatic cancer cells. Note that the expressions of SphK1, GCS, and ceramide synthase were not affected by bortezomib in both PANC-1 (Fig. 3h) and MIA (Fig. 3i) cells.

Bortezomib-induced ceramide production and cytotoxicity are enhanced by SphK inhibitors. PANC-1 and MIA cells were treated with vehicle (V, 0.1 % DMSO), or with bortezomib (100 nM) in the presence or absence of SphK1 inhibitor SKI-II (5 μM) or SKI-V (5 μM), SphK1 activity (a) and cellular ceramide level (b) were analyzed 6 h after treatment as described. The cell viability (c and e) and Annexin V(D) were also determined 48 h after treatment. Primary cultured pancreatic cancer cells were treated with vehicle (V, 0.1 % DMSO), or with bortezomib (100 nM) in the presence or absence of C6-ceramide (10 μg/ml), PDMP (20 μM), or SKI-II (5 μM), cells were further cultured for 48 h, cell viability was analyzed by MTT assay (f), and cell death was detected by trypan blue staining (g). The expressions of SphK1, glucosylceramide synthase (GCS), and ceramide synthase in PANC-1 (H) or MIA (i) cells treated with vehicle (V, 0.1 % DMSO), or with bortezomib (100 nM) for indicated time. Experiments in this figure were repeated three times. *p < 0.05. # p < 0.05 versus the bortezomib treatment group
Bortezomib-induced JNK activation is associated with ceramide production
The earlier studies have shown that bortezomib-induced cancer cell apoptosis is associated with the ASK1-JNK activation [15, 22]. However, how JNK is activated by bortezomib is not fully addressed. ASK1/JNK is known to play a role in ceramide-mediated cell apoptosis [23, 24]. In PANC-1 cells, we also observed a significant ASK1/JNK activation by bortezomib (Fig. 4a). Immunoprecipitation (IP) results in Fig. 4b showed that bortezomib induced ASK1/JNK complex formation in PANC-1 cells. More importantly, bortezomib-induced JNK activation was significantly inhibited by F-B1, but was enhanced by PDMP and SKI-II (Fig. 4c). These results suggested that bortezomib-induced pro-apoptotic ASK1/JNK activation in pancreatic cancer cells might also be associated with ceramide production. Results in Fig. 4d, e demonstrated that JNK inhibitor SP 600125 significantly reduced bortezomib or bortezomib+C6-ceramide-induced PANC-1 cell viability loss and apoptosis, which again confirmed that JNK activation by bortezomib was pro-apoptotic.

Bortezomib-induced JNK activation is associated with ceramide production. PANC-1 cells were treated with vehicle (V, 0.1 % DMSO) or bortezomib (100 nM) for indicated time points, phospho- and total ASK1 and JNK were tested by western blots, the association between JNK1 and ASK1 was tested by IP (b). PANC-1 cells were treated with vehicle (V, 0.1 % DMSO), or with bortezomib (100 nM) in the presence or absence of PDMP (20 μM), F-B1 (20 μM) or SKI-II (5 μM), cells were further cultured for 6 h, phospho- and total ASK1 and JNK were tested by Western blots (c), the JNK phosphorylation was quantified (d). PANC-1 cells were treated with vehicle (V, 0.1 % DMSO), bortezomib (100 nM) or bortezomib (100 nM) plus C6-ceramide (10 μg/ml) (bortezomib + C6) in the presence or absence of JNK inhibitor SP 600125 (20 μM) for 48 h, cell viability (e) and Annexin V percentage (f) were tested as described. (g) The proposed signaling pathways of this study: in cultured pancreatic cancer cells (both primary and transformed), bortezomib induces pro-apoptotic ceramide production through the de novo synthesis pathway, which is inhibited by ceramide synthase inhibitor fumonisin B1 (F-B1). PDMP, the inhibitor of glucosylceramide synthetase (GCS) as well as the SphK1 inhibitors (SKI-II and SKI-IV), facilitates bortezomib-induced ceramide production and apoptosis. Bortezomib-induced pro-apoptotic ASK1/JNK activation in pancreatic cancer cells is also associated with ceramide production. Experiments in this figure were repeated three times. *p < 0.05, # p < 0.05 versus the bortezomib treatment group
Discussion
In the current study, we found that bortezomib induced pro-apoptotic ceramide production probably through de novo synthesis pathway in cultured pancreatic cancer cells. The ceramide de novo synthase inhibitor F-B1 suppressed bortezomib-induced ceramide production and apoptosis. On the other hand, exogenously added C6-ceramide facilitated bortezomib-induced pancreatic cancer apoptosis. Meanwhile, the GCS inhibitor PDMP and the SphK1 inhibitors facilitated bortezomib-induced ceramide production and cancer cell apoptosis (Fig. 4g). For the mechanism study, we observed that bortezomib-induced ceramide production was important for pro-apoptotic ASK1/JNK activation. ASK1/JNK activation by bortezomib was suppressed by F-B1, but was increased by SKI-II and PDMP in pancreatic cancer cells (Fig. 4g). Finally, in primary cultured pancreatic cancer cells, bortezomib-induced cell death was enhanced by C6-ceramide, SKI-II, and PDMP.
As a potent and selective 26S proteasome inhibitor, bortezomib has displayed significant cytotoxicity in a variety of tumor cells in phase I clinical trials [25], and it recently received FDA approval for the treatment for advanced MMs [26]. Despite the recent research successes of bortezomib as an anti-pancreatic cancer agent [10, 27–29], the underlying mechanisms of such an effect remain to be characterized. Here, we proposed that bortezomib treatment results in the ceramide production, leading to JNK activation and cancer cell apoptosis.
Ceramide can be formed through the de novo synthesis pathway which starts with the condensation of serine and palmitoyl-CoA catalyzed by serine palmitoyl transferase (SPT) to generate 3-keto-dihydrosphingosine [30, 31]; the latter is subsequently reduced to form dihydrosphingosine (sphinganine). Ceramide synthase then acts on dihydrosphingosine (or sphingosine) to form ceramide [30]. In the current study, we found that ceramide synthase inhibitor F-B1 suppressed bortezomib-induced ceramide production, ASK1/JNK activation, and cell apoptosis in pancreatic cancer cells, indicating that bortezomib-induced ceramide production is mediated through de novo synthesis pathway.
Ceramide, once generated, can be consumed in the biosynthetic reactions for the synthesis of sphingomyelin, glucosylceramide, or ceramide 1-phosphate, with the help from sphingomyelin synthase, GCS, or SphK1, respectively [30, 31]. In the current study, our data suggested that bortezomib-induced ceramide was also subjected to metabolism clearance in pancreatic cancer cells. The SphK1 inhibitors (SKI-II and SKI-IV) and the GCS inhibitor (PDMP) enhanced bortezomib-induced ceramide production, JNK activation, and following cell apoptosis. Hence, we suggested that bortezomib-induced cancer cell apoptosis can be strengthened by preventing ceramide metabolism. Interestingly, the expressions of GCS, SphK1, and ceramide synthase were not affected by bortezomib.
Several known anticancer drugs and stress-inducers could increase cellular ceramide production through the de novo synthesis [30, 32, 33]. Ceramide trigger cell apoptosis signaling cascades by regulating multiple signaling molecular. Studies have shown that ceramide-dependent cell apoptosis is associated with JNK activation, ASK1 works as an upstream of JNK and forms a complex with JNK [34, 35].The ASK1-JNK activation and consequent cell apoptosis are seen in a number of stress conditions where the cellular ceramide level is increased [18, 36, 37]. Here, we suggested that bortezomib-induced ceramide production mediates pancreatic cancer cell apoptosis probably through activating JNK. In conclusion, we indicated that bortezomib-induced apoptosis in cultured pancreatic cancer cells is associated with ceramide production.
Abbreviations
- JNK:
-
C-Jun N-terminal kinase
- SphK1:
-
Sphingosine kinase 1
- F-B1:
-
Fumonisin B1
- PDMP:
-
1-Phenyl-2-decanoylamino-3-morpholino-1-propanol
References
Vincent A, Herman J, Schulick R, Hruban RH, Goggins M (2011) Pancreatic cancer. Lancet 378(9791):607–620. doi:10.1016/S0140-6736(10)62307-0
Hidalgo M (2010) Pancreatic cancer. N Engl J Med 362(17):1605–1617. doi:362/17/1605
Colucci G, Labianca R, Di Costanzo F, Gebbia V, Carteni G, Massidda B, Dapretto E, Manzione L, Piazza E, Sannicolo M, Ciaparrone M, Cavanna L, Giuliani F, Maiello E, Testa A, Pederzoli P, Falconi M, Gallo C, Di Maio M, Perrone F (2010) Randomized phase III trial of gemcitabine plus cisplatin compared with single-agent gemcitabine as first-line treatment of patients with advanced pancreatic cancer: the GIP-1 study. J Clin Oncol 28(10):1645–1651. doi:10.1200/JCO.2009.25.4433
Jenks S (2011) AACR highlights: promise for treating pancreatic cancer. J Natl Cancer Inst 103(10):786–787. doi:10.1093/jnci/djr183
Costello E, Neoptolemos JP (2011) Pancreatic cancer in 2010: new insights for early intervention and detection. Nat Rev Gastroenterol Hepatol 8(2):71–73
Wong HH, Lemoine NR (2009) Pancreatic cancer: molecular pathogenesis and new therapeutic targets. Nat Rev Gastroenterol Hepatol 6(7):412–422. doi:10.1038/nrgastro.2009.89
Boccadoro M, Morgan G, Cavenagh J (2005) Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int 5(1):18. doi:1475-2867-5-18
Montagut C, Rovira A, Mellado B, Gascon P, Ross JS, Albanell J (2005) Preclinical and clinical development of the proteasome inhibitor bortezomib in cancer treatment. Drugs Today (Barc) 41(5):299–315. doi:893706
Richardson PG, Mitsiades C, Hideshima T, Anderson KC (2006) Bortezomib: proteasome inhibition as an effective anticancer therapy. Annu Rev Med 57:33–47. doi:10.1146/annurev.med.57.042905.122625
Nawrocki ST, Carew JS, Dunner K Jr, Boise LH, Chiao PJ, Huang P, Abbruzzese JL, McConkey DJ (2005) Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res 65(24):11510–11519. doi:65/24/11510
Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr, Huang P, Abbruzzese JL, McConkey DJ (2005) Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res 65(24):11658–11666. doi:65/24/11658
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569(1–2):29–63. doi:S0027-5107(04)00371-9
Podar K, Gouill SL, Zhang J, Opferman JT, Zorn E, Tai YT, Hideshima T, Amiot M, Chauhan D, Harousseau JL, Anderson KC (2008) A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene 27(6):721–731. doi:1210679
Chen KF, Yeh PY, Yeh KH, Lu YS, Huang SY, Cheng AL (2008) Down-regulation of phospho-Akt is a major molecular determinant of bortezomib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res 68(16):6698–6707. doi:10.1158/0008-5472.CAN-08-0257
Lauricella M, Emanuele S, D’Anneo A, Calvaruso G, Vassallo B, Carlisi D, Portanova P, Vento R, Tesoriere G (2006) JNK and AP-1 mediate apoptosis induced by bortezomib in HepG2 cells via FasL/caspase-8 and mitochondria-dependent pathways. Apoptosis 11(4):607–625. doi:10.1007/s10495-006-4689-y
Dimanche-Boitrel MT, Rebillard A, Gulbins E (2011) Ceramide in chemotherapy of tumors. Recent Pat Anticancer Drug Discov 6(3):284–293. doi:10.2174/157489211796957838
Wu C-H, Cao C, Kim JH, Hsu C-H, Wanebo HJ, Bowen WD, Xu J, Marshall J (2012) Trojan-horse nanotube on-command intracellular drug delivery. Nano Lett 12(11):5475–5480. doi:10.1021/nl301865c
Yao C, Wu S, Li D, Ding H, Wang Z, Yang Y, Yan S, Gu Z (2012) Co-administration phenoxodiol with doxorubicin synergistically inhibit the activity of sphingosine kinase-1 (SphK1), a potential oncogene of osteosarcoma, to suppress osteosarcoma cell growth both in vivo and in vitro. Mol Oncol 6(4):392–404. doi:10.1016/j.molonc.2012.04.002
Merrill AH Jr, van Echten G, Wang E, Sandhoff K (1993) Fumonisin B1 inhibits sphingosine (sphinganine) N-acyltransferase and de novo sphingolipid biosynthesis in cultured neurons in situ. J Biol Chem 268(36):27299–27306
Yao C, Wei JJ, Wang ZY, Ding HM, Li D, Yan SC, Yang YJ, Gu ZP (2012) Perifosine induces cell apoptosis in human osteosarcoma cells: new implication for osteosarcoma therapy? Cell Biochem Biophys. doi:10.1007/s12013-012-9423-5
Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S (2008) Targeting SphK1 as a new strategy against cancer. Curr Drug Targets 9(8):662–673
Yu C, Rahmani M, Dent P, Grant S (2004) The hierarchical relationship between MAPK signaling and ROS generation in human leukemia cells undergoing apoptosis in response to the proteasome inhibitor Bortezomib. Exp Cell Res 295(2):555–566. doi:10.1016/j.yexcr.2004.02.001
Kurinna SM, Tsao CC, Nica AF, Jiffar T, Ruvolo PP (2004) Ceramide promotes apoptosis in lung cancer-derived A549 cells by a mechanism involving c-Jun NH2-terminal kinase. Cancer Res 64(21):7852–7856. doi:64/21/7852
Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN (1996) Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 380(6569):75–79. doi:10.1038/380075a0
Adams J (2002) Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr Opin Chem Biol 6(4):493–500. doi:S1367593102003435
Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC (2003) A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 348(26):2609–2617. doi:10.1056/NEJMoa030288
Bold RJ, Virudachalam S, McConkey DJ (2001) Chemosensitization of pancreatic cancer by inhibition of the 26S proteasome. J Surg Res 100(1):11–17. doi:10.1006/jsre 2001.6194
Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP (2001) 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J Cell Biochem 82(1):110–122. doi:10.1002/jcb.1150
Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ (2004) The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther 3(1):59–70
Mullen TD, Obeid LM (2012) Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents Med Chem 12(4):340–363
Tafesse FG, Ternes P, Holthuis JC (2006) The multigenic sphingomyelin synthase family. J Biol Chem 281(40):29421–29425. doi:R600021200
Andrieu-Abadie N, Gouaze V, Salvayre R, Levade T (2001) Ceramide in apoptosis signaling: relationship with oxidative stress. Free Radic Biol Med 31(6):717–728. doi:S0891584901006554
Hannun YA, Obeid LM (2002) The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem 277(29):25847–25850. doi:10.1074/jbc.R200008200
Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 2(3):389–395
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2(3):222–228
Morad SA, Cabot MC (2013) Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer 13(1):51–65. doi:10.1038/nrc3398
Chen MB, Zhang Y, Wei MX, Shen W, Wu XY, Yao C, Lu PH (2013) Activation of AMP-activated protein kinase (AMPK) mediates plumbagin-induced apoptosis and growth inhibition in cultured human colon cancer cells. Cell Signal. doi:S0898-6568(13)00157-5
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Lei Gong, Bo Yang and Ming Xu have contributed equally to this work.
Rights and permissions
About this article
Cite this article
Gong, L., Yang, B., Xu, M. et al. Bortezomib-induced apoptosis in cultured pancreatic cancer cells is associated with ceramide production. Cancer Chemother Pharmacol 73, 69–77 (2014). https://doi.org/10.1007/s00280-013-2318-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-013-2318-3