
Abstract
Purpose
UGT1A1 genotypes are important when considering treatment with irinotecan-containing regimens. In this study, we determined the dose, efficacy, and tolerability of irinotecan according to UGT1A1 genotypes when combined with capecitabine in patients with metastatic colorectal cancer.
Methods
Patients with histologically confirmed metastatic adenocarcinoma of the colon or rectum were enrolled into a UGT1A1 genotype-directed dose-escalation trial of irinotecan plus fixed-dose capecitabine (2,000 mg/m2/day). The starting dose of irinotecan was different for each genotype group and ranged from 200 to 280 mg/m2. Pharmacokinetic concentrations of irinotecan and metabolites were determined by LC/MS/MS.
Results
Fifty patients were genotyped for UGT1A1 *28 and *6, and grouped according to the numbers of defective alleles (DA): 0, 1, and 2. Plasma concentrations of irinotecan, SN-38, and SN-38G were measured. The maximum tolerated dose of irinotecan was 350 mg/m2 for the 0 and 1 DA groups, and 200 mg/m2 for the 2 DA group. For the 0, 1, and 2 DA groups, mean AUClast ratios of SN-38G to SN-38 were 7.72, 5.71, and 2.72 (P = 0.0023) and relative dose intensities at recommended dose were 85, 83, and 97 %.
Conclusion
Irinotecan dosing based on UGT1A1*28 and *6 is feasible, and higher doses of irinotecan can be safely administered in patients with 0 or 1 DA, compared to those with 2 DA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Irinotecan, one of the pivotal agents in the first-line treatment of metastatic colorectal cancer (MCRC) [5], is a prodrug activated to SN-38 by carboxylesterase. After exertion of its cytotoxic effects, SN-38 is inactivated via glucuronidation [32]. This step is mediated by UDP-glucuronosyltransferase (UGT) 1A enzymes, with UGT1A1 playing a major role in the detoxification of SN-38 [20].
Irinotecan is well-known for its significant interindividual pharmacokinetic variability. Recent pharmacogenetic studies have shown that polymorphisms of the UGT1A1 gene, especially UGT1A1*28, underlie irinotecan-related toxicity in the Caucasian population [30]. Based on these findings, US FDA amended the label of irinotecan to include homozygosity for the UGT1A1*28 genotype as one of the risk factors for severe neutropenia in 2004. Despite these efforts, complete individualization of irinotecan dosing is yet to be established. Several earlier reports have shown that high-dose irinotecan can be safely administered in selected patients [36, 37]. These findings suggest that patients without UGT1A1*28 alleles tolerate higher doses of irinotecan. While UGT1A1*28 is problematic in Western ethnicities, the UGT1A1*6 genotype has been exclusively identified in Asian populations, occurring with a frequency of about 20 % [1, 12]. The UGT1A1*6 genotype is associated with higher exposure to SN-38 and lower relative glucuronidation rates, and thus suggested as a biomarker for irinotecan-induced severe neutropenia in Asian cancer patients [3, 14, 21, 27].
Schemes based on UGT1A1*28 polymorphisms have been applied for individualizing the dose of irinotecan [9, 15, 35]. These studies have verified the concept that patients with genotypes associated with decreased glucuronidation cannot tolerate higher doses of irinotecan, compared to the wild-type population. However, the influence of UGT1A1*6, which is prevalent and problematic in Asian populations, has not been evaluated as yet. At present, data on the optimal dose and clinical outcomes of irinotecan-based regimens regarding UGT1A1*28 and *6 polymorphisms are limited [8, 26].
The main aim of the current genotype-directed phase I study was to determine maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs) of the capecitabine plus irinotecan (XELIRI) regimens according to UGT1A1 genotypes in MCRC patients in Korea. In addition, we investigated the relationship between these genotypes and pharmacokinetics of irinotecan and its metabolites, and further evaluated the safety profiles and antitumor activities.
Patients and methods
Patient selection
Patients with histologically confirmed metastatic adenocarcinoma of the colon or rectum were eligible, provided they met the following criteria: age 18–65 years, genotyped for UGT1A1*28 and *6, Eastern Cooperative Oncology Group (ECOG) performance status of ≤2, life expectancy of longer than 3 months, at least one measurable or evaluable non-measurable disease, no prior systemic chemotherapy for metastatic disease (any previous adjuvant chemotherapy or radiotherapy must have been completed at least 6 months before study inclusion), absolute neutrophil counts (ANCs) ≥1,500/μL, platelets ≥100,000/μL, serum creatinine levels less than 1.5 mg/dL, AST and ALT <3 times the upper limit of normal (ULN), total serum bilirubin level <1.5 times that of ULN. Patients were excluded in cases of central nervous system metastasis, gastrointestinal obstruction, bleeding, current diarrhea greater than grade 2 according to the National Cancer Institute of Common Toxicity Criteria (NCI CTC) version 3.0, and known bleeding diathesis or coagulopathy. The protocol and written informed consent were approved by the institutional review board of Asan Medical Center. All of the patients were enrolled after signing this consent form.
Study design and treatments
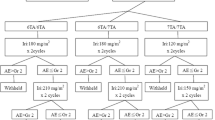
This phase I trial was designed to include up to seven levels using the 3+3 dose-escalation design for each group. The starting dose of irinotecan was different for each group: 0 DA, 280 mg/m2 (20 % above the standard dose of XELIRI); 1 DA, 240 mg/m2 (standard dose); 2 DA, 240 mg/m2 (standard dose), and capecitabine was administered at a fixed dose of 2,000 mg/m2/day at all levels in each group.
Irinotecan was administered intravenously over 90 min in 200 ml of 5 % dextrose water on day 1 every 3 weeks. Fixed-dose capecitabine was administered orally from days 2 to 15 every 3 weeks. All patients were premedicated with antiemetic drugs (e.g., 5-HT3 antagonists). In addition, atropine (0.5 mg) and loperamide (4 mg every 2 h, as necessary) were administered for hypercholinergic syndrome and delayed diarrhea, respectively. Treatment was repeated every 21 days and continued for a maximum of 9 cycles in the absence of progressive disease, development of unacceptable toxicity, or patient refusal.
Dose-limiting toxicity (DLT) and recommended dose (RD)
Toxicity was graded using the National Cancer Institute Common Toxicity Criteria (NCI CTC) version 3.0. Any of the following toxicities occurring during the first cycle of chemotherapy were grouped as DLT: grade 4 neutropenia lasting for more than 5 days, febrile neutropenia, grade 4 thrombocytopenia or any other grade 3 or 4 non-hematological toxicity (including nausea, vomiting, excluding alopecia) that did not improve to at least grade 1 within 2 days after the institution of appropriate therapy, any other toxicities that prevented completion of the prescribed dose of capecitabine during the first cycle, and treatment-related adverse events causing a delay of more than 2 weeks following administration of the second cycle. Dose escalation was continued until DLTs were observed in the first cycle of treatment in two or more of six patients, defined as the maximum tolerated dose (MTD). After determination of MTD, two to six more patients were treated at the dose level below this cutoff, which was defined as the recommended dose (RD). MTD and RD were determined separately for each genotype. Intrapatient dose escalation was not permitted. Dose intensity was defined as the body surface area-normalized irinotecan dose per day during the cycle treatment period (i.e., from the date of irinotecan administration to that before subsequent treatment with irinotecan).
Patient and tumor evaluation
Patients were evaluated at baseline on a weekly basis during the first treatment cycle and at 3-weekly intervals thereafter. Evaluation included complete blood counts with differential, blood chemistry analysis, complete clinical examination, and recording of all adverse events, including severity and outcome.
Responses were evaluated via CT scan every 2 cycles until tumor progression, using the same imaging technique for each assessment as that at baseline. Tumor responses were classified according to the response evaluation criteria in solid tumor (RECIST 1.0) guidelines [34]. Patients with complete response (CR) or partial response (PR) required a confirmatory disease assessment after at least 4 weeks. Patients with no confirmed tumor response were not regarded as responders. PFS was recorded from the start of chemotherapy until documented disease progression or death, and OS determined from the start of chemotherapy until death.
UGT1A1*6 and *28 genotyping
Patients were genotyped for eligibility using a previously described assay [38].
Pharmacokinetics
Serial blood samples were collected at cycle 1 prior to irinotecan infusion and at 0, 0.5, 1.0, 2.0, 3.0, 6.0, 24, and 48 h after infusion. Total plasma concentrations of irinotecan, SN-38, and SN-38G were measured with the HPLC assay. Briefly, proteins were precipitated from plasma (100 μl) after the addition of methanol. After solvent evaporation, samples will be reconstituted in 30 % acetonitrile and 70 % 0.1 M potassium dihydrogen phosphate containing 3 mM heptane sulfonic acid at pH 4 and injected into an HPLC system with a μBondapak C18 column. Standard curves were constructed for irinotecan and SN-38, SN-38G concentration after hydrolysis with 1,000 units of β-glucuronidase (Escherichia Coli type IX-A, Sigma-Aldrich Co., St. Louis, MO). Pharmacokinetic parameters for irinotecan, SN-38, and SN-38G were calculated using non-compartmental analyses with WinNonlin (Professional Network Version 5.2; Pharsight Corporation, Mountain View, CA). Dose-normalized pharmacokinetic parameters were calculated by dividing parameters, including area under the concentration curve (AUClast) and maximum concentration (Cmax), by dose. Pharmacokinetic parameters reflecting the metabolism and disposition of irinotecan were calculated. The biliary index (AUCIrinotecan × AUCSN-38/AUCSN-38G), relative extent of conversion (REC; AUCSN-38/AUCIrinotecan), and relative extent of glucuronidation (REG; AUCSN-38G/AUCSN-38) were calculated to measure the metabolism and disposition of irinotecan [17].
Safety assessment
Toxicity was graded according to National Cancer Institute Common Toxicity Criteria version 3.0. Patients were treated with full doses of irinotecan at ANC counts ≥1,500/μL, platelet counts ≥100,000/μL, and in cases where treatment-related diarrhea was fully resolved. A treatment delay of up to 1 week was permitted without dose reduction.
Statistical analysis
Exploratory analyses were performed to analyze the effect of UGT1A1 genotypes on pharmacokinetic parameters of irinotecan, which were log-transformed and compared using mixed model analysis of variance (ANOVA). Progression-free survival (PFS) was calculated from the commencement date of chemotherapy to disease progression or death from any cause, and overall survival (OS) from the starting date of chemotherapy to death from any cause. Survival analyses were performed using the Kaplan–Meier method with the log-rank test. Data analyses were performed using the Statistical Software Package for Social Sciences (SPSS version 14.0; Chicago, IL).
Results
Patient characteristics and disposition
Between April 2006 and 2009, 50 patients were screened for UGT1A1 genotypes (*28 and *6), and patients stratified into one of 3 groups according to the number of defective alleles (DA): 0 (wild type: *1/*1), 1 (containing only one of the *28 or *6 allele: *1/*28 or *1/*6), and 2 (*28/*28, *6/*6, or double heterozygous for *1/*28 and *1/*6) (Table 1). In the 1 DA group (n = 20), 14 and 6 patients were heterozygous for UGT1A1*6 and *28, respectively. In the 2 DA group (n = 7), two patients were simultaneously heterozygous for UGT1A1*6 and *28, and three and two patients were homozygous for UGT1A1*6 and *28, respectively. Genotypes did not deviate from Hardy–Weinberg equilibrium based on Chi-square test (P = 0.44). Patient characteristics are summarized in Table 2.
Dose escalation, safety, and tolerability
The MTD was 380 mg/m2 for the 0 DA group, 380 mg/m2 for the 1 DA group, and 240 mg/m2 for the 2 DA group. In the 0 DA group, no DLT was evident at levels II and III. Two of the six patients at level V (380 mg/m2) experienced two DLT, and two DLTs were observed at level IV (350 mg/m2), which was expanded to 11 patients. Within the 1 DA group, based on the finding that two DLTs occurred in two patients at level V (380 mg/m2), level IV was expanded to eight patients displaying one DLT. One patient was misallocated to level I (240 mg/m2), and consequently, level I included 4 patients, despite no DLT. In the 2 DA group, two DLTs were observed in three patients classified as level I (240 mg/m2), and thus the dose was lowered to level −1 (200 mg/m2) at which no DLTs were observed (Table 1). RD was designated the dose level below the MTD for each genotype group. The most common DLTs among the treated patients included diarrhea (n = 3), neutropenia (n = 3), and asthenia (n = 3). No treatment-related deaths were evident. At the RD level, relative dose intensities (mean ± SD, mg/m2/day) were 85 % (14.2 ± 2.98), 83 % (13.8 ± 3.36), and 97 % (9.21 ± 0.51) for the 0 DA, 1 DA, and 2 DA groups, respectively. While the cumulative toxicities of patients in each genotype group did not differ significantly at the RD level, neutropenia was often observed in the 1 DA group (Table 3).
Pharmacokinetics
Full pharmacokinetic data were analyzed in all 50 patients. Mean dose-normalized plasma concentration versus time profiles of SN-38 are shown in Fig. 1a. Dose-normalized AUClast values of irinotecan and SN-38G were not significantly different among the genotype groups. However, dose-normalized AUClast of SN-38 revealed significant differences among the genotype groups. The metabolic parameters including biliary index, REC, and REG revealed higher values with higher numbers of DA (P = 0.384, <0.0001 and 0.0023).

Comparison according to the number of UGT1A1 defective alleles for a mean dose-normalized concentration–time profile of SN-38 and b relative extent of glucuronidation (REG)
The genotype group with 2 DA displayed significantly lower values of REG, compared to the 0 and 1 DA groups (P = 0.0017, P = 0.0068; Fig. 1b). At the RD level, patients with 2 DA displayed similar SN-38 exposure (mean ± SD, 381.78 ± 133.11 μg*h/L) as those with 0 and 1 DA (341.22 ± 282.67 μg*hr/L).
Efficacy assessment
Among the 50 patients, 40 were evaluated for response. The ten remaining patients were not assessable, since they did not have measurable disease. The overall response rate in the intention-to-treat population was 65.0 % (95 % confidence interval [CI] 50.2–79.8; Table 4). Patients with 2 DA of the UGT1A1 genotype showed a lower response rate than those classified as 0 or 1 DA. At a median follow-up duration of 22.2 months (range 2.3–45.0 months), median PFS and OS for all patients were 9.1 months (95 % CI 7.8–10.4 months) and 29.3 months (95 % CI 24.9–33.8 months), respectively (Fig. 2). DA number (0, 1 vs. 2) and exposure of SN-38 (≥180 vs. <180 μg*h/L) did not display significant differences in terms of PFS. Seventeen patients (34 %) displayed metastatic lesions limited to the liver. The response rate of these patients was 58 %, and 5 (29.4 %) received metastasectomy.

Kaplan-Meier curves of progression-free survival (PFS) and overall survival (OS) for 40 evaluable patients. Median PFS and OS were 9.1 months (95 % CI 7.8–10.4 months) and 29.3 months (95 % CI 24.9–33.8 months), respectively
Discussion
Conventionally, XELIRI therapy is composed of irinotecan 240 mg/m2 and capecitabine 2,000 mg/m2/day. In our dose-ascending study of irinotecan for XELIRI, the RD of irinotecan for wild type and 1 DA patients was 350 and 200 mg/m2 for patients with 2 DA. The overall response rate was 65.0 %. Median PFS and OS were 9.1 and 29.3 months, respectively. In 2007, the pivotal BICC-C trial has described a median PFS and OS for CapeIRI of 5.8 and 18.9 months [11]. In 2009, Sukumaran et al. reported a median time-to-progression and OS of 7.9 and 15.6 months based on a pooled analysis of 30 non-randomized phase II trials (n = 1,380) along with 6 randomized phase II and 3 phase III trials. In this analysis, however, the daily dose of capecitabine ranged from 1,800 to 2,500 mg/m2 for 7 to 14 days per cycle and the dose of irinotecan 180–350 mg/m2, over a 3-week period per cycle [33]. Our results suggest that irinotecan and capecitabine for XELIRI should not be fixed at the conventional dose, but adjusted on the basis of number of defective alleles related to the UGT1A1 genotype.
The pharmacokinetic-pharmacodynamic changes associated with defective alleles of the UGT1A1 genotype have been described during the past decade [6]. Innocenti et al. [18] predicted severe neutropenia based on UGT1A1 genotypes and pretreatment serum total bilirubin levels. Furthermore, Toffoli and coworkers performed a UGT1A1 genotype-oriented dose-escalating trial of FOLFIRI and reported that DLTs of irinotecan occur at >300 mg/m2, which is approximately twice the conventional dose [35]. While most studies have focused on the defective allele, UGT1A1*28, differences in the UGT1A1 allele frequencies may require consideration for worldwide application of these results. The frequencies of UGT1A1 *28 homozygotes are reported as 10 and 20 % in Caucasians and Africans, while Asians show less than 5 % prevalence. On the other hand, UGT1A1*6 homozygotes have been identified at a frequency of about 5 % exclusively in the Asian population. Moreover, combined heterozygosity of *28 and *6 has been observed in another 5 % of the population [2]. Our results suggest that the dose for irinotecan used in major phase III trials may have been too high. We have observed two DLTs out of three patients with susceptible genotypes which are *28 and *6 homozygotes, compound heterozygotes. These genotypes account for about 10 % of the whole population regardless of ethnicity [29]. Considering the fact that this regimen has been avoided by clinicians due to safety issues, our data suggest that the tolerability of this regimen may have to be re-evaluated. Our study also expands the UGT1A1 genotype-directed approach by including UGT1A1*6, and suggests that irinotecan can be elevated to a dose of 350 mg/m2 for patients with wild-type UGT1A1 or one defective *6 or *28 allele, compared to 240 mg/m2, which is the standard XELIRI regimen dose used for major phase III trials. To our knowledge, this is the first prospective genotype-based approach with irinotecan in the Asian population taking into account both UGT1A1*28 and *6 alleles. One Japanese group conducted a dose-escalation study of irinotecan plus doxifluridine based on UGT1A1*28 [15]. However, they did not include UGT1A1*6, which is more important in the Asian population than UGT1A1*28 and did not escalate the irinotecan dose according to the occurrence of DLT. Alternatively, Ishida et al. [19] identified metastatic colorectal patients susceptible to irinotecan toxicity via studies including both UGT1A1*6 and *28 genotypes, and initiated treatment with oxaliplatin-based chemotherapy, rather than an irinotecan-based regimen.
While diarrhea and neutropenia are well-known toxicities of irinotecan, direct comparisons of the pharmacogenetic-pharmacokinetic-toxicity relationship between trials are difficult owing to differences in the manner of irinotecan administration. A number of studies have shown that the UGT1A1*28/*28 genotype is effective in predicting grade 3/4 neutropenia, but not diarrhea, while others have reported that the genotype predicts grade 3/4 diarrhea, but not hematologic toxicity [18, 23]. Moreover, Hoskins and coworkers suggested that susceptibility of UGT1A1*28 homozygote patients to neutropenia does not occur at low doses of irinotecan (100–125 mg/m2), while Hu et al. consistently reported neutropenia in UGT1A1*28 homozygotes at low doses of irinotecan, but no diarrhea [16, 17]. In addition to UGT1A1, various genes including ABCC2 and SLCO1B1 involved in the metabolism and disposition of irinotecan have been investigated. But due to the limited number of patients in these studies, it is hard to validate the predictive value of comprehensive pharmacogenetic analysis for toxicity, efficacy, and optimal dose [7, 25].
In the present study, we hypothesized that heterozygotes of UGT1A1*28 or *6 have similar effects. Minami and coworkers reported that patients with either UGT1A1*6 or *28 show similar degrees of reduced area under concentration curve ratios [26]. UGT1A1*28 and *6 homozygotes, compound heterozygotes have also shown similar pharmacokinetic profiles and susceptibility to neutropenia induced by irinotecan administered at a dose of 150 mg/m2 with the FOLFIRI regimen [28]. However, despite the dominant effects of UGT1A1 genotypes on irinotecan metabolism, pharmacokinetics remains considerably variable. These results suggest that genetic polymorphisms (carboxylesterase, cytochrome P450 3A, or ABC transporters) or environmental factors other than UGT1A1 additionally contribute to the metabolism of irinotecan [6]. Based on the risks of irinotecan-associated toxicities, the US Food and Drug Administration has recommended the use of reduced dosage of irinotecan in individuals with UGT1A1*28. In Japan, 150 mg/m2 is the approved dose for the FOLFIRI regimen owing to the prevalence of UGT1A1*6 or *28 [28].
While the earlier investigations were mainly based on concerns related to genotype-associated toxicity, we believe that genotypes may also be used for dose escalation in patients without defective alleles. Metastatic colorectal patients treated with FOLFIRI containing higher irinotecan doses (≥260 mg/m2) have been reported to show higher response rates and time-to-progression [24, 35]. We are concerned that there is a possibility of “underdosage” of irinotecan in many UGT1A1 wild-type patients, owing to the lack of genotype-driven dose finding studies.
This study has several limitations. At present, continuous intravenous 5-FU is still preferred when administered in combination with irinotecan (FOLFIRI), compared to XELIRI in metastatic colorectal cancer [11, 22]. However, XELIRI is still under evaluation in various indications [10, 13, 31]. Another drawback was the limited number of patients with two defective alleles. Due to a lack of patients, the study was stopped before the protocol-defined number of patients was enrolled, which limited the defining of RD for 2 DA genotypes. While our study focused on the pharmacogenetics of irinotecan, 5-FU is also subject to individual toxicity related with various genes including dihydropyrimidine dehydrogenase (DPYD) polymorphisms. However, as our study used a fixed dose of capecitabine, we did not consider this in our analysis. Furthermore, DPYD polymorphisms associated with 5-FU toxicity are rarely described in the Korean population [4].
Our results support the feasibility of irinotecan dosing stratified by the UGT1A1*28 and *6 genotype in Korean patients with metastatic colorectal cancer in combination with capecitabine and safe administration of higher doses of irinotecan in patients without 2 defective alleles. We suggest that in the future, the optimal dosage of irinotecan-containing agents can be evaluated based on the present genotype information to maximize their efficacy and minimize toxicity. In addition, prospective analysis of “genotype-targeted dosing” in a standard method of exploratory-confirmatory trials is necessary so that the cost-benefit issues of widespread UGT1A1 genotyping can be settled. At present, a large-scale Asian trial is ongoing to address this issue of UGT1A1 genotyping with FOLFIRI (ClinicalTrials.gov Identifier NCT01271582).
References
Akaba K, Kimura T, Sasaki A, Tanabe S, Ikegami T, Hashimoto M, Umeda H, Yoshida H, Umetsu K, Chiba H, Yuasa I, Hayasaka K (1998) Neonatal hyperbilirubinemia and mutation of the bilirubin uridine diphosphate-glucuronosyltransferase gene: a common missense mutation among Japanese, Koreans and Chinese. Biochem Mol Biol Int 46:21–26
Akiyama Y, Fujita K, Nagashima F, Yamamoto W, Endo H, Sunakawa Y, Yamashita K, Ishida H, Mizuno K, Araki K, Ichikawa W, Miya T, Narabayashi M, Kawara K, Sugiyama M, Hirose T, Ando Y, Sasaki Y (2008) Genetic testing for UGT1A1*28 and *6 in Japanese patients who receive irinotecan chemotherapy. Ann Oncol 19:2089–2090
Araki K, Fujita K, Ando Y, Nagashima F, Yamamoto W, Endo H, Miya T, Kodama K, Narabayashi M, Sasaki Y (2006) Pharmacogenetic impact of polymorphisms in the coding region of the UGT1A1 gene on SN-38 glucuronidation in Japanese patients with cancer. Cancer Sci 97:1255–1259
Cho HJ, Park YS, Kang WK, Kim JW, Lee SY (2007) Thymidylate synthase (TYMS) and dihydropyrimidine dehydrogenase (DPYD) polymorphisms in the Korean population for prediction of 5-fluorouracil-associated toxicity. Ther Drug Monit 29:190–196
Davies JM, Goldberg RM (2008) First-line therapeutic strategies in metastatic colorectal cancer. Oncology (Williston Park) 22:1470–1479
de Jong FA, de Jonge MJ, Verweij J, Mathijssen RH (2006) Role of pharmacogenetics in irinotecan therapy. Cancer Lett 234:90–106
Di Paolo A, Bocci G, Polillo M, Del Re M, Di Desidero T, Lastella M, Danesi R (2011) Pharmacokinetic and pharmacogenetic predictive markers of irinotecan activity and toxicity. Curr Drug Metab 12:932–943
Esaki T, Satoh T, Ura T, Tsujinaka T, Sasaki Y, Yamazaki K, Yamada Y, Ishizuka N, Hyodo I, Sakata Y (2009) A prospective PGx and PK/PD dose-finding study of irinotecan based on UGT1A1*6 and *28 genotyping (UGT0601). J Clin Oncol 27:abstr e14560
Falandry C, You B, Milano G, Chatelut E, Rebischung C, Glehen O, Mille D, Delord J, Lledo G, Trillet-Lenoir V, Freyer G (2007) Individual genotyping to optimize chemotherapy in metastatic colorectal cancer (MCRC): the COLOGEN trial. J Clin Oncol 25(18S):2510
Farhat FS, Kattan J, Ghosn MG (2010) Role of capecitabine and irinotecan combination therapy in advanced or metastatic gastric cancer. Expert Rev Anticancer Ther 10:541–548
Fuchs CS, Marshall J, Mitchell E, Wierzbicki R, Ganju V, Jeffery M, Schulz J, Richards D, Soufi-Mahjoubi R, Wang B, Barrueco J (2007) Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: results from the BICC-C Study. J Clin Oncol 25:4779–4786
Fujita K, Ando Y, Nagashima F, Yamamoto W, Eodo H, Araki K, Kodama K, Miya T, Narabayashi M, Sasaki Y (2007) Genetic linkage of UGT1A7 and UGT1A9 polymorphisms to UGT1A1*6 is associated with reduced activity for SN-38 in Japanese patients with cancer. Cancer Chemother Pharmacol 60:515–522
Garcia-Alfonso P, Munoz-Martin A, Mendez-Urena M, Quiben-Pereira R, Gonzalez-Flores E, Perez-Manga G (2009) Capecitabine in combination with irinotecan (XELIRI), administered as a 2-weekly schedule, as first-line chemotherapy for patients with metastatic colorectal cancer: a phase II study of the Spanish GOTI group. Br J Cancer 101:1039–1043
Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Jang IJ, Lee DH, Lee JS (2006) Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol 24:2237–2244
Hazama S, Nagashima A, Kondo H, Yoshida S, Shimizu R, Araki A, Yoshino S, Okayama N, Hinoda Y, Oka M (2010) Phase I study of irinotecan and doxifluridine for metastatic colorectal cancer focusing on the UGT1A1*28 polymorphism. Cancer Sci 101:722–727
Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL (2007) UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 99:1290–1295
Hu ZY, Yu Q, Pei Q, Guo C (2010) Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: low doses also increase risk. Clin Cancer Res 16:3832–3842
Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramirez J, Rudin CM, Vokes EE, Ratain MJ (2004) Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol 22:1382–1388
Ishida H, Fujita K, Akiyama Y, Sunakawa Y, Yamashita K, Mizuno K, Miwa K, Kawara K, Ichikawa W, Ando Y, Saji S, Sasaki Y (2011) Regimen selection for first-line FOLFIRI and FOLFOX based on UGT1A1 genotype and physical background is feasible in Japanese patients with advanced colorectal cancer. Jpn J Clin Oncol 41:617–623
Iyer L, King CD, Whitington PF, Green MD, Roy SK, Tephly TR, Coffman BL, Ratain MJ (1998) Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest 101:847–854
Jada SR, Lim R, Wong CI, Shu X, Lee SC, Zhou Q, Goh BC, Chowbay B (2007) Role of UGT1A1*6, UGT1A1*28 and ABCG2 c.421C > A polymorphisms in irinotecan-induced neutropenia in Asian cancer patients. Cancer Sci 98:1461–1467
Kohne CH, De Greve J, Hartmann JT, Lang I, Vergauwe P, Becker K, Braumann D, Joosens E, Muller L, Janssens J, Bokemeyer C, Reimer P, Link H, Spath-Schwalbe E, Wilke HJ, Bleiberg H, Van Den Brande J, Debois M, Bethe U, Van Cutsem E (2008) Irinotecan combined with infusional 5-fluorouracil/folinic acid or capecitabine plus celecoxib or placebo in the first-line treatment of patients with metastatic colorectal cancer. EORTC study 40015. Ann Oncol 19:920–926
Marcuello E, Altes A, Menoyo A, Del Rio E, Gomez-Pardo M, Baiget M (2004) UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br J Cancer 91:678–682
Marcuello E, Paez D, Pare L, Salazar J, Sebio A, del Rio E, Baiget M (2011) A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br J Cancer 105:53–57
Marsh S, Hoskins JM (2010) Irinotecan pharmacogenomics. Pharmacogenomics 11:1003–1010
Minami H, Sai K, Saeki M, Saito Y, Ozawa S, Suzuki K, Kaniwa N, Sawada J, Hamaguchi T, Yamamoto N, Shirao K, Yamada Y, Ohmatsu H, Kubota K, Yoshida T, Ohtsu A, Saijo N (2007) Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: roles of UGT1A1*6 and *28. Pharmacogenet Genomics 17:497–504
Sai K, Saeki M, Saito Y, Ozawa S, Katori N, Jinno H, Hasegawa R, Kaniwa N, Sawada J, Komamura K, Ueno K, Kamakura S, Kitakaze M, Kitamura Y, Kamatani N, Minami H, Ohtsu A, Shirao K, Yoshida T, Saijo N (2004) UGT1A1 haplotypes associated with reduced glucuronidation and increased serum bilirubin in irinotecan-administered Japanese patients with cancer. Clin Pharmacol Ther 75:501–515
Satoh T, Ura T, Yamada Y, Yamazaki K, Tsujinaka T, Munakata M, Nishina T, Okamura S, Esaki T, Sasaki Y, Koizumi W, Kakeji Y, Ishizuka N, Hyodo I, Sakata Y (2011) Genotype-directed, dose-finding study of irinotecan in cancer patients with UGT1A1*28 and/or UGT1A1*6 polymorphisms. Cancer Sci 102:1868–1873
Shimoyama S (2010) Pharmacogenetics of irinotecan: an ethnicity-based prediction of irinotecan adverse events. World J Gastrointest Surg 2:14–21
Shulman K, Cohen I, Barnett-Griness O, Kuten A, Gruber SB, Lejbkowicz F, Rennert G (2011) Clinical implications of UGT1A1*28 genotype testing in colorectal cancer patients. Cancer 117:3156–3162
Skof E, Rebersek M, Hlebanja Z, Ocvirk J (2009) Capecitabine plus Irinotecan (XELIRI regimen) compared to 5-FU/LV plus Irinotecan (FOLFIRI regimen) as neoadjuvant treatment for patients with unresectable liver-only metastases of metastatic colorectal cancer: a randomised prospective phase II trial. BMC Cancer 9:120
Slatter JG, Schaaf LJ, Sams JP, Feenstra KL, Johnson MG, Bombardt PA, Cathcart KS, Verburg MT, Pearson LK, Compton LD, Miller LL, Baker DS, Pesheck CV, Lord RS 3rd (2000) Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos 28:423–433
Sukumaran S, Pavlakis N, Pittman KB, Patterson K, Price TJ (2009) Capecitabine and irinotecan (XELIRI) in first-line treatment of metastatic colorectal cancer (mCRC): a systematic review of controlled clinical trials. J Clin Oncol 27:e15100
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Toffoli G, Cecchin E, Gasparini G, D’Andrea M, Azzarello G, Basso U, Mini E, Pessa S, De Mattia E, Lo Re G, Buonadonna A, Nobili S, De Paoli P, Innocenti F (2010) Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J Clin Oncol 28:866–871
Van Cutsem E, Dirix L, Van Laethem JL, Van Belle S, Borner M, Gonzalez Baron M, Roth A, Morant R, Joosens E, Gruia G, Sibaud D, Bleiberg H (2005) Optimisation of irinotecan dose in the treatment of patients with metastatic colorectal cancer after 5-FU failure: results from a multinational, randomised phase II study. Br J Cancer 92:1055–1062
Ychou M, Raoul JL, Desseigne F, Borel C, Caroli-Bosc FX, Jacob JH, Seitz JF, Kramar A, Hua A, Lefebvre P, Couteau C, Merrouche Y (2002) High-dose, single-agent irinotecan as first-line therapy in the treatment of metastatic colorectal cancer. Cancer Chemother Pharmacol 50:383–391
Yea SS, Lee SS, Kim WY, Liu KH, Kim H, Shon JH, Cha IJ, Shin JG (2008) Genetic variations and haplotypes of UDP-glucuronosyltransferase 1A locus in a Korean population. Ther Drug Monit 30:23–34
Acknowledgments
This manuscript was presented in part at the 2009 Annual Meeting of the American Society of Clinical Oncology, Orlando, May 29–June 2, 2009. This study was supported by grants from the National Project for Personalized Genomic Medicine, Korea Health 21 R&D Project (A111218-11-PG02) and Korean Health Technology R&D Project (A102059-HI10C2014), Ministry for Health and Welfare, Republic of Korea, and the Asan Institute for Life Sciences (Seoul, Republic of Korea, 2006–231). Irinotecan was provided by Pfizer Korea and CJ Pharma Korea.
Conflict of interest
All authors have no financial disclosures.
Author information
Authors and Affiliations
Corresponding author
Additional information
Kyu-pyo Kim and Ho-Sook Kim contributed equally as first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Kim, Kp., Kim, HS., Sym, S.J. et al. A UGT1A1*28 and *6 genotype-directed phase I dose-escalation trial of irinotecan with fixed-dose capecitabine in Korean patients with metastatic colorectal cancer. Cancer Chemother Pharmacol 71, 1609–1617 (2013). https://doi.org/10.1007/s00280-013-2161-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-013-2161-6