
Abstract
Background
In this phase II clinical trial, we evaluated the efficacy and safety of S-1 monotherapy in patients with previously treated advanced non-small-cell lung cancer (NSCLC). We also measured plasma concentrations of 5-fluorouracil (5-FU) and 5-chloro-2,4-dihydroxypyridine components of S-1 and examined correlation with effectiveness and toxicity.
Methods
S-1 was given orally at a dose of 80 mg/m2/day for 14 consecutive days, followed by a 7-day rest period. This treatment course was repeated until disease progression or intolerable toxicity.
Results
We enrolled 30 patients. The response rate was 26.7% (8/30), and the disease control rate was 70% (21/30). Median progression-free survival (PFS) was 3.1 months, and median overall survival (OS) was 11.2 months. Mutations in the epidermal growth factor receptor (EGFR) gene were analyzed in 27 patients. The response rate was higher in patients with mutant EGFR (50.0%) than in those with wild-type EGFR (11.8%, P = 0.0288). Median PFS was 4.8 and 2.5 months (P = 0.038), and median OS was 22.4 and 8.4 months (P = 0.071). There was no grade 4 toxicity in this study. Five patients had grade 3 non-hematologic toxicity, and there was a trend toward higher plasma concentrations of 5-FU in those patients than in another patients.
Conclusions
S-1 monotherapy is effective and well-tolerated treatment for previously treated advanced NSCLC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, primary lung cancer has become the leading cause of cancer-related death worldwide. Lung cancer can be classified into two types: small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC). NSCLC accounts for 80% of all lung cancers, and many cases are advanced and not indicated for surgery at the time of detection. Further progress in systemic chemotherapy is thus awaited.
In the 1990s, regimens combining platinum compounds with new anticancer agents were demonstrated to be effective for NSCLC [7, 10]. Such regimens produced higher response rates than those obtained with conventional first-line chemotherapy. However, outcomes were poor in patients who had recurrence after first-line treatment.
Second-line chemotherapy with docetaxel has been reported to significantly prolong survival in patients with NSCLC, as compared with best supportive care [11]. Pemetrexed has been reported to be as effective as docetaxel in patients with recurrent NSCLC [2] and is considered one strategic option for second-line treatment. Phase III studies of patients with advanced NSCLC who had previously received 1 or 2 regimens of chemotherapy showed that erlotinib, an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), significantly prolonged survival compared with a placebo [12]. On the basis of the results of these phase III studies, docetaxel, pemetrexed, and erlotinib are currently recommended for second-line chemotherapy of advanced NSCLC. However, median survival is only 6–8 months, and toxicity precludes the use of these drugs in some patients.
S-1, an oral fluoropyrimidine anticancer agent, was approved for the treatment of NSCLC in 2004. S-1 combines tegafur, a prodrug of 5-fluorouracil (5-FU), with gimeracil (5-chloro-2,4-dihydroxypyridine, CDHP; a reversible antagonist of the rate-limiting enzyme for the degradation of 5-FU) and oteracil potassium (a reversible inhibitor of phosphorylating enzymes of 5-FU). This formulation was designed to enhance antitumor activity by increasing plasma concentrations of 5-FU, while reducing attendant increases in gastrointestinal toxicity [13, 14, 16, 18, 21].
A late phase II clinical study of S-1 monotherapy reported a response rate of 22% and a median survival time of 10.2 months in patients with previously untreated NSCLC. As for safety in that study, 1 patient (1/56) had grade 4 neutropenia. However, there was no grade 4 non-hematologic toxicity [5]. In patients with previously treated NSCLC, phase II studies reported a response rate of 7.1–20.0% and a median survival time 7.3–16.4 months. Treatment-related grade 4 toxicities rarely occurred [1, 3, 15, 19].
The results of these clinical trials of S-1 were comparable to those obtained with currently available new anticancer agents in patients with advanced NSCLC. In addition, S-1 is well tolerated. S-1 is thus expected to be an effective treatment strategy even in patients with previously treated disease. This study was designed to evaluate the effectiveness and safety of S-1 monotherapy in patients with previously treated advanced NSCLC.
Patients and methods
Patient eligibility
Patients who met the following criteria were eligible for the enrollment in the study: (1) a histologically or cytologically confirmed diagnosis of NSCLC; (2) a history of first-line chemotherapy with a platinum compound plus a new anticancer agent (however, patients who had received palliative radiotherapy to treat lesions other than the primary tumor and assessable lesions were eligible if at least 2 weeks had elapsed since the completion of radiotherapy); (3) an age of 20 years or older; (4) an Eastern Cooperative Oncology Group performance status of 0 to 2; (5) the presence of lesions able to be measured according to the Response Evaluation Criteria in Solid Tumors (RECIST) guidelines, version 1.0; (6) no severe dysfunction of major organs, as defined by the following laboratory findings: neutrophil count ≥1,500/μl, platelet count ≥100 × 103/μl, hemoglobin concentration ≥9.0 g/dl, total bilirubin concentration ≤1.5 mg/dl, aspartate aminotransferase and alanine aminotransferase levels ≤100 IU/l, serum creatinine concentration ≤1.3 mg/dl, creatinine clearance ≥40 ml/min, percutaneous oxygen saturation (SpO2) ≥90% or partial oxygen pressure in arterial blood (PaO2) ≥60 torr; (7) oral intake was possible; and (8) written informed consent was obtained directly from every patient.
Patients were excluded from the study if they had any of the following conditions: serious infections or other serious complications; active double cancers; the presence of massive pleural effusion, ascites, or pericardial effusion interfering with the administration of chemotherapy; clear evidence of interstitial pneumonia or pulmonary fibrosis on plain chest X-ray; brain metastasis associated with central nervous symptoms (patients were eligible if symptoms were controlled by steroids or other treatments); poorly controlled diabetes mellitus; a history of bone transplantation; a history of peripheral blood stem cell transplantation; a distinct history of drug allergies; or lactating or pregnant women. This study was approved by the Institutional Review Board at Kitasato University School of Medicine.
Treatments
S-1 is orally administered at 80 mg/m2/day, divided into two doses given after breakfast and dinner. The dose of S-1 was assigned on the basis of the patient body surface area (BSA) as follows: BSA less than 1.25 m2, 80 mg/day; BSA from 1.25 m2 to less than 1.5 m2, 100 mg/day; and BSA 1.5 m2 or higher, 120 mg/day. S-1 was administered for 2 weeks, followed by a 7-day rest period. This 3-week cycle was repeated.
During the study, treatment with S-1 was discontinued in the event of any of the following abnormalities: neutrophil count <1,000/μl, platelet count <75 × 103/μl, aspartate aminotransferase and alanine aminotransferase levels >100 IU/l, total bilirubin concentration >1.5 mg/dl, any grade 3 or higher non-hematologic toxicity, or infection accompanied by fever. Treatment was resumed after the abnormality regressed. If patients had grade 4 neutropenia, grade 3 or higher neutropenia with a fever of 38°C or higher, grade 4 thrombocytopenia, platelet transfusion, or grade 3 or higher non-hematologic toxicity, and treatment was not resumed because the criteria for starting treatment were not met criteria for at least 2 weeks after the date scheduled for the next course to begin, or if treatment could not be resumed even after 1 week after discontinuation of S-1 within a given course, the dose of S-1 was reduced in decrements of 20 mg/day. If the dose of S-1 was 80 mg/day, the dose was reduced to 50 mg/day for the next course. The dose could be lowered by up to 2 levels. If dose reduction beyond 2 levels was necessary or if adverse events occurred at a dose of 50 mg/day, treatment was discontinued.
Response and toxicity evaluation
Before the start of treatment, the following examinations were performed: hematologic examinations and serum chemical analysis, urinalysis, measurement of tumor marker levels, measurement of percutaneous oxygen saturation, and electrocardiography. Lesions were evaluated by plain chest radiography; computed tomography (CT) or magnetic resonance imaging (MRI) of the chest, abdomen, and cranium; and bone scintigraphy. After initiating treatment, hematologic examinations and serum chemical analysis were performed at 1- to 2-week intervals. To evaluate tumor lesions, chest radiography was performed at 1- to 2-week intervals, CT and MRI at 1-month intervals, and bone scintigraphy at 3-month intervals.
Tumor shrinkage was assessed in accordance with the RECIST guidelines, version 1.0. All evaluations of response were confirmed by an independent evaluator. Adverse reactions were graded according to the Common Terminology Criteria for Adverse Events, version 3.0. Therapy after discontinuation of the study treatment was not specified in the protocol.
Statistical analysis
The primary endpoint of this study was the response rate. Secondary endpoints were the disease control rate, progression-free survival (PFS), overall survival (OS), and safety. Given an expected response rate of 20% and a threshold response rate of 5% with an alpha error of 0.05 and a beta error of 0.20, we estimated that 27 patients would be required to assess tumor response. Fisher’s exact test was used to analyze response rates. All data were analyzed at a cut-off date in May 2011. PFS was defined as the time interval between the date of enrollment and disease progression or patient death. OS was defined as the time interval between the date of enrollment and patient death. PFS and OS were expressed as median and range. Statistical analyses were performed with the Kaplan–Meier method and log-rank test. P values less than 0.05 were considered to indicate a statistically significant difference.
Measurement of plasma concentrations of 5-FU and CDHP
In this study, we measured plasma concentrations of 5-FU and CDHP and examined the correlation with the effectiveness and adverse events of S-1. Blood samples were obtained 12 h after oral administration of S-1 on day 7 of the first course, because at least 2 days would be required to reach the steady state for 5-FU and CDHP [4]. At the assigned time points, 5 ml samples of blood were collected. Each sample was placed in vacuumized blood collection tubes containing heparin, and the tube was gently rotated to mix the contents. The tube was then immediately placed on ice or in a refrigerator. The blood samples were centrifuged for 5–10 min at 3,000 revolutions per minute and a temperature of 4°C. The plasma portion was stored in a freezer at a temperature below −20°C until analysis.
Plasma drug concentrations were measured by the method of Matsushima et al. [6] 5-FU and CDHP were assayed by gas chromatography-mass spectrometry.
Multivariate logistic regression analysis (JMP 7, SAS Institute Inc., Cary, NC, USA) was used to assess correlations of plasma drug concentrations with effectiveness and adverse events.
Results
Patient characteristics
From March 2007 through March 2009, a total of 30 eligible patients (22 men and 8 women) were enrolled. All patients were included in efficacy and safety analyses. Table 1 shows the patient characteristics. Their median age was 65 years (range, 49–73). The clinical stage of disease was stage IV in 29 patients. One patient had stage IIIA disease associated with secondary lung cancer, which developed after left lower lobectomy. Radical radiotherapy was precluded by pulmonary dysfunction. The histologic type was adenocarcinoma in 26 patients, squamous-cell carcinoma in 3, and others (non-small-cell cancer) in 1. The performance status was 0 in 18 patients and 1 in 12. Twenty-five patients had a history of smoking, and 5 had never smoked. All patients had previously received 1–5 regimens of chemotherapy. The number of previously administered regimens was 1 in 10 patients, 2 in 13, 3 in 3, 4 in 3, and 5 in 1. Five patients had received prior radiotherapy. Four patients had previously received EGFR-TKI such as gefitinib and erlotinib, and in total, 14 patients (all ten patients with EGFR-positive tumors, three of 17 patients with EGFR-negative tumors, and one patient with unknown EGFR mutation status) received EGFR-TKI at anytime of their treatment. EGFR mutations were examined in 27 patients by the polymerase chain reaction clamping method (Mitsubishi Chemical Medience Corporation, Tokyo, Japan). EGFR mutations were positive in 10 tumors and negative in 17. The following EGFR mutations were detected in the 10 positive tumors: exon 19 deletion in 6 tumors, leucine-to-arginine substitution in codon 858 (L858R) of exon 21 in 3 tumors, and leucine-to-glutamine substitution in codon 861 (L861Q) of exon 21 in 2 tumors (accompanied by exon 19 deletion in 1).
Drug administration
The 30 subjects received a total of 226 treatment cycles. The median number of cycles per patient was 4 (range, 1–45). Twenty-seven patients (90%) received at least 3 consecutive cycles of treatment. Two patients discontinued treatment after 2 cycles, because of tumor progression due to bone metastasis in 1 patient and grade 3 interstitial pneumonia in the other. The dose of S-1 was reduced in 3 patients. The reasons for dose reduction were grade 3 anorexia (course 2) in 1 patient, grade 3 diarrhea in 1 (course 2), and elevated total bilirubin concentration in 1. In the third patient, the dose was reduced because the total bilirubin concentration at the start of course 2 did not meet the criteria of ≤1.5 mg/dl. The actually delivery dose/projected dose intensity was 94%.
Efficacy
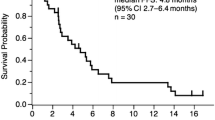
Among the 30 patients, the response to treatment was partial response (PR) in 8, stable disease (SD) in 13, and progressive disease (PD) in 9. The response rate was 26.7% (8/30; 95% confidence interval [CI], 12.3–45.9), and the disease control rate (complete response + PR + SD) was 70.0% (21/30; 95% CI, 50.6–85.3) (Table 2). As of May 26, 2011, 5 of the 30 patients are alive. The median PFS was 3.1 months (95% CI, 2.4–4.7), the median OS was 11.2 months (95% CI, 6.1–17.5), and the 1-year survival rate was 43.3% (Fig. 1).

Progression-free survival and overall survival. PFS progression-free survival, OS overall survival
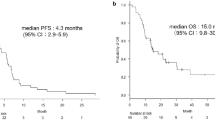
In the 27 patients in whom EGFR mutations were assessed, the response rate was significantly higher in patients with EGFR mutation-positive tumors (50.0% [5/10]; 95% CI, 18.7–81.3) than in those with EGFR mutation-negative tumors (11.8% [2/17]; 95% CI, 1.5–36.4; P = 0.029) (Table 2). There was a significant difference in median PFS between patients with EGFR mutation-positive tumors (4.8 months; 95% CI, 1.2–7.5) and those with EGFR mutation-negative tumors (2.5 months; 95% CI, 1.9–3.4; P = 0.038) (Fig. 2). The median OS was tended to be longer in patients with EGFR mutation-positive tumors (22.4 months; 95% CI, 3.7–51.4) than in those with EGFR mutation-negative tumors (8.4 months; 95% CI, 5.2–17.5; P = 0.071).

Progression-free survival according to EGFR mutation status. The P value for patients with EGFR mutant tumors (solid line) versus those with wild-type tumors (dashed line) is 0.0384
Toxicity
Table 3 shows the main toxic effects. No patient had grade 3 or higher hematologic toxicity. Hematologic toxicity included leukopenia, decreased hemoglobin level, neutropenia, and thrombocytopenia, but all reactions were grade 2 or lower. As for non-hematologic toxicity, 5 patients had grade 3 reactions: anorexia in 1, diarrhea in 1, interstitial pneumonia in 1, stomatitis in 1, and dizziness in 1. There was no treatment-related mortality. In the patient with grade 3 interstitial pneumonia, treatment was discontinued because of toxicity. In the two patients with grade 3 dizziness and stomatitis, treatment was discontinued at the patients’ request. In the other patients with grade 3 toxicity (anorexia, diarrhea, and stomatitis), treatment could be continued after a rest period or a decrease in the dose of S-1.
Relation of plasma drug concentrations to response and toxicity
In this study, we examined the relations of the plasma concentrations of 5-FU and CDHP to effectiveness and adverse events. The plasma concentration of 5-FU significantly correlated with the plasma concentration of CDHP (P < 0.001) (Fig. 3). Neither the plasma concentration of 5-FU nor that of CDHP significantly correlated with the response to S-1. The plasma 5-FU concentration in patients with grade 3 non-hematologic toxicity was significantly higher than that in patients with grade 2 or lower non-hematologic toxicity (P = 0.007). Plasma 5-FU concentrations were particularly high in 1 patient with grade 3 diarrhea and the 1 with grade 3 anorexia.

Plasma concentrations of 5-FU and CDHP. Y = 1.70 X + 18.35. R = 0.86. 5-FU fluorouracil, CDHP gimeracil
Discussion
The recent advent of new anticancer agents, molecular targeted agents, and improvements in supportive care have contributed to progress in chemotherapy for advanced NSCLC. However, patients with recurrence after first-line chemotherapy continue to have poor outcomes [2, 11, 12].
A late phase II clinical trial of S-1 monotherapy in patients with previously untreated NSCLC reported a response rate of 22%, a median survival time of 10.2 months, and good tolerability, with a low incidence of serious adverse events [5]. In the present study, we evaluated the effectiveness and safety of S-1 monotherapy in previously treated patients. The response rate was 26.7%, with a disease control rate of 70.0%, a median PFS of 3.1 months, and a median OS of 11.2 months. These results showed that S-1 monotherapy was as effective as other anticancer agents for second-line or subsequent chemotherapy.
In this study, all 8 patients with a PR received S-1 as third-line or subsequent chemotherapy. This finding suggests that S-1 monotherapy can be adequately effective even in heavily pretreated patients. Although none of the patients who received S-1 as second-line treatment had a PR, the disease control rate was good (70%, 7/10). One of the reasons for the lack of a PR in this subgroup of patients might have been the relatively low number of patients who received S-1 as second-line chemotherapy.
When response rates were analyzed according to EGFR mutation status in this study, we interestingly found that the response rate was significantly higher in patients with EGFR mutation-positive tumors (50.0%) than in those with EGFR mutation-negative tumors (11.8%, P = 0.028). The median PFS was significantly longer in patients with EGFR mutation-positive tumors (4.8 months) than in those with EGFR mutation-negative tumors (2.5 months, P = 0.038), and the median OS was tended to be longer in patients with EGFR mutation-positive tumors (22.4 months) (8.4 months, P = 0.071). EGFR mutations are an important predictive factor indicating the response to EGFR-TKIs. To our knowledge, no study has demonstrated a direct relation between EGFR mutations and the response to S-1, but several reports have discussed the relation between EGFR-TKIs and S-1 [8, 9]. S-1 suppresses tumor growth by inhibiting the target enzyme thymidylate synthase in tumor cells. Okabe et al. [9] reported that the EGFR-TKI gefitinib inhibits thymidylate synthase expression. S-1 and gefitinib are thus expected to act synergistically. Okabe et al. [8] also reported that the addition of S-1 to EGFR-TKIs might be able to overcome EGFR-TKI resistance caused by MET amplification.
The effect of EGFR mutations on the response to UFT (tegafur plus uracil), an oral anticancer agent belonging to the same category as S-1, was studied by Suehisa et al. [17] in patients with adenocarcinoma of the lung. Postoperative adjuvant chemotherapy with UFT was associated with longer survival in patients with EGFR wild-type tumors than in those with EGFR mutant tumors [17]. However, unlike our study, the investigation of Suehisa et al. was a retrospective analysis of patients who received postoperative chemotherapy, making it difficult to draw firm conclusions. The same study reported that cell lines with L858R in exon 21 had low sensitivity to 5-FU in vitro, whereas cell lines that concurrently had resistant genes with threonine-to-methionine substitution in codon 790 (T790 M) were more sensitive to 5-FU than were cell lines with only L858R. The present study did not examine genes associated with EGFR-TKI resistance; the impact of such genes on the sensitivity to S-1 thus remains unclear.
In this study, no patient had grade 3 or higher hematologic toxicity. Grade 3 non-hematologic toxicity occurred in 5 patients: anorexia in 1, diarrhea in 1,interstitial pneumonia in 1, stomatitis in 1, and inner ear disorder in 1. In the patient with interstitial pneumonia, the results of a whole-blood drug-induced lymphocyte stimulation test were positive for S-1, suggesting drug-induced pneumonia. At the same time, pneumocystis pneumonia was also suspected. The patient responded to steroids and a combination of sulfamethoxazole and trimethoprim. During treatment, the dose of S-1 had to be reduced in only 3 patients; the actually delivery dose/projected dose intensity was good (94%). In a late phase II clinical study of S-1 monotherapy in patients with previously untreated NSCLC, S-1 was given for 4 weeks, followed by a 2-week rest period. In our study, S-1 was given for 2 weeks, followed by 1 week of rest, resulting in high dose intensity. Tsukuda et al. [20] studied treatment schedules of S-1 in patients with advanced head and neck cancer and found that S-1 given for 2 weeks with a 1-week rest was associated with a lower incidence of adverse events than was S-1 given for 4 weeks with a 2-week rest. The former schedule allowed a longer duration of treatment, resulting in more prolonged antitumor effectiveness [20]. Because hematologic and non-hematologic toxicities were mild in our study, all patients could receive the second and subsequent courses on an outpatient basis. Seven patients received 10 or more courses of treatment with S-1. The patient who was given the maximum number of 45 courses is still receiving S-1 and continues to have SD. Our results suggest that the ability to continuously administer S-1 monotherapy to patients with previously treated NSCLC is an important factor.
In conclusion, our study confirmed that S-1 monotherapy has effects and is well tolerated in patients with previously treated, advanced NSCLC. Further clinical studies of larger numbers of patients are necessary to confirm our findings.
References
Govindan R, Morgenzstern D, Kommor MD, Herbst RS, Schaefer P, Gandhi J, Saito K, Zergebel C, Schiller J (2011) Phase II trial of S-1 as second-line therapy in patients with advanced non-small cell lung cancer. J Thorac Oncol 6:790–795
Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, Gatzemeier U, Tsao TC, Pless M, Muller T, Lim HL, Desch C, Szondy K, Gervais R, Shaharyar, Manegold C, Paul S, Paoletti P, Einhorn L, Bunn PA Jr (2004) Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol 22:1589–1597
Hashizume T, Nakada Y (2009) S-1 monotherapy in patients with pretreated advanced non-small cell lung cancer. Gan To Kagaku Ryoho 36:963–967
Hirata K, Horikoshi N, Aiba K, Okazaki M, Denno R, Sasaki K, Nakano Y, Ishizuka H, Yamada Y, Uno S, Taguchi T, Shirasaka T (1999) Pharmacokinetic study of S-1, a novel oral fluorouracil antitumor drug. Clin Cancer Res 5:2000–2005
Kawahara M, Furuse K, Segawa Y, Yoshimori K, Matsui K, Kudoh S, Hasegawa K, Niitani H (2001) Phase II study of S-1, a novel oral fluorouracil, in advanced non-small-cell lung cancer. Br J Cancer 85:939–943
Matsushima E, Yoshida K, Kitamura R (1997) Determination of S-1 (combined drug of tegafur, 5-chloro-2, 4-dihydroxypyridine and potassium oxonate) and 5-fluorouracil in human plasma and urine using high-performance liquid chromatography and gas chromatography-negative ion chemical ionization mass spectrometry. J Chromatogr B Biomed Sci Appl 691:95–104
Ohe Y, Ohashi Y, Kubota K, Tamura T, Nakagawa K, Negoro S, Nishiwaki Y, Saijo N, Ariyoshi Y, Fukuoka M (2007) Randomized phase III study of cisplatin plus irinotecan versus carboplatin plus paclitaxel, cisplatin plus gemcitabine, and cisplatin plus vinorelbine for advanced non-small-cell lung cancer: four-arm cooperative study in Japan. Ann Oncol 18:317–323
Okabe T, Okamoto I, Tsukioka S, Uchida J, Hatashita E, Yamada Y, Yoshida T, Nishio K, Fukuoka M, Janne PA, Nakagawa K (2009) Addition of S-1 to the epidermal growth factor receptor inhibitor gefitinib overcomes gefitinib resistance in non-small cell lung cancer cell lines with met amplification. Clin Cancer Res 15:907–913
Okabe T, Okamoto I, Tsukioka S, Uchida J, Iwasa T, Yoshida T, Hatashita E, Yamada Y, Satoh T, Tamura K, Fukuoka M, Nakagawa K (2008) Synergistic antitumor effect of S-1 and the epidermal growth factor receptor inhibitor gefitinib in non-small cell lung cancer cell lines: role of gefitinib-induced down-regulation of thymidylate synthase. Mol Cancer Ther 7:599–606
Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH (2002) Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med 346:92–98
Shepherd FA, Dancey J, Ramlau R, Mattson K, Gralla R, O’rourke M, Levitan N, Gressot L, Vincent M, Burkes R, Coughlin S, Kim Y, Berille J (2000) Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol 18:2095–2103
Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353:123–132
Shirasaka T, Shimamato Y, Ohshimo H, Yamaguchi M, Kato T, Yonekura K, Fukushima M (1996) Development of a novel form of an oral 5-fluorouracil derivative (S-1) directed to the potentiation of the tumor selective cytotoxicity of 5-fluorouracil by two biochemical modulators. Anticancer Drugs 7:548–557
Shirasaka T, Shimamoto Y, Fukushima M (1993) Inhibition by oxonic acid of gastrointestinal toxicity of 5-fluorouracil without loss of its antitumor activity in rats. Cancer Res 53:4004–4009
Shiroyama T, Komuta K, Imamura F, Hirashima T, Kijima T, Tachibana I, Kawase I (2011) Phase II study of S-1 monotherapy in platinum-refractory, advanced non-small cell lung cancer. Lung Cancer 74:85–88
Spears CP, Gustavsson BG, Mitchell MS, Spicer D, Berne M, Bernstein L, Danenberg PV (1984) Thymidylate synthetase inhibition in malignant tumors and normal liver of patients given intravenous 5-fluorouracil. Cancer Res 44:4144–4150
Suehisa H, Toyooka S, Hotta K, Uchida A, Soh J, Fujiwara Y, Matsuo K, Ouchida M, Takata M, Kiura K, Date H (2007) Epidermal growth factor receptor mutation status and adjuvant chemotherapy with uracil-tegafur for adenocarcinoma of the lung. J Clin Oncol 25:3952–3957
Tatsumi K, Fukushima M, Shirasaka T, Fujii S (1987) Inhibitory effects of pyrimidine, barbituric acid and pyridine derivatives on 5-fluorouracil degradation in rat liver extracts. Jpn J Cancer Res 78:748–755
Totani Y, Saito Y, Hayashi M, Tada T, Kohashi Y, Mieno Y, Kato A, Imizu H, Yoneda Y, Hoshino T, Uchiyama Y, Takeuchi Y, Okazawa M, Sakakibara H (2009) A phase II study of S-1 monotherapy as second-line treatment for advanced non-small cell lung cancer. Cancer Chemother Pharmacol 64:1181–1185
Tsukuda M, Kida A, Fujii M, Kono N, Yoshihara T, Hasegawa Y, Sugita M (2005) Randomized scheduling feasibility study of S-1 for adjuvant chemotherapy in advanced head and neck cancer. Br J Cancer 93:884–889
Wilkinson DS, Tlsty TD, Hanas RJ (1975) The inhibition of ribosomal rna synthesis and maturation in novikoff hepatoma cells by 5-fluorouridine. Cancer Res 35:3014–3020
Acknowledgments
We are indebted to Drs. Toshiyuki Sawa and Kaoru Matsui for evaluating response in this study. We are also indebted to Prof. J. Patrick Barron of the Department of International Medical Communications of Tokyo Medical University for his review of this manuscript.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wada, M., Yamamoto, M., Ryuge, S. et al. Phase II study of S-1 monotherapy in patients with previously treated, advanced non-small-cell lung cancer. Cancer Chemother Pharmacol 69, 1005–1011 (2012). https://doi.org/10.1007/s00280-011-1795-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-011-1795-5