
Abstract
Objective
To characterize the population pharmacokinetics of plitidepsin (Aplidin®) in cancer patients.
Methods
A total of 283 patients (552 cycles) receiving intravenous plitidepsin as monotherapy at doses ranging from 0.13 to 8.0 mg/m2 and given as 1- or 24-h infusions every week; 3- or 24-h infusion biweekly; or 1-h infusion daily for 5 consecutive days every 21 days were included in the analysis. An open three-compartment pharmacokinetic model and a nonlinear binding to red blood cells model were used to describe the plitidepsin pharmacokinetics in plasma and blood, respectively, using NONMEM V software. The effect of selected covariates on plitidepsin pharmacokinetics was investigated. Model evaluation was performed using goodness-of-fit plots, posterior predictive check and bootstrap.
Results
Plasma clearance and its between subject variability (%) was 13.6 l/h (71). Volume of distribution at steady-state was calculated to be 4791 l (59). The parameters B max and C 50 of the non-linear blood distribution were 471 μg/l (56) and 41.6 μg/l, respectively. Within the range of covariates studied, age, sex, body size variables, aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), total bilirubin, creatinine clearance, albumin, total protein, performance status, co-administration of inhibitors or inducers of CYP3A4 and presence of liver metastases were not statistically related to plitidepsin pharmacokinetic parameters. Bootstrap and posterior predictive check evidenced the model was deemed appropriate to describe the time course of plitidepsin blood and plasma concentrations in cancer patients.
Conclusions
The integration of phase I/II pharmacokinetic data demonstrated plitidepsin linear elimination from plasma, dose-proportionality up to 8.0 mg/m2, and time-independent pharmacokinetics. The distribution to red blood cells can be considered linear at doses lower than 5 mg/m2 administered as 3-h or longer infusion. No clinically relevant covariates were identified as predictors of plitidepsin pharmacokinetics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plitidepsin (Aplidin®) is a cyclic depsipetptide compound isolated in 1988 from a Mediterranean marine tunicate, Aplidium albicans and now, is produced synthetically [1]. Although the exact mechanism of action of plitidepsin has not been completely elucidated, this drug exert its antitumor activity, at least in part, by combining the induction of apoptosis via activation of Jun N-terminal Kinase (JNK) activity and the antiangiogenic activity mediated by reducing the active secretion of the vascular endothelial growth factor (VEGF) [2–10]. Plitidepsin has shown important in vitro antitumour activity against a number of human solid tumor cell lines, including bladder, breast, gastric, lung carcinoid, melanoma, neuroblastoma, prostate, thyroid, leukaemia, myeloma and lymphoma [5, 11–16]. Also, using the hollow fiber model, bladder, gastric and prostate tumors were shown to be susceptible to plitidepsin in vivo. With xenograft models, pharmacological activity was noted against Burkitt’s, gastric, pancreas, thyroid, and renal tumors [17, 18]. The antitumor activity of plitidepsin is currently under clinical investigation with promising results observed in ongoing phase II clinical studies.
Several Phase I dose-escalation studies in subjects with advanced tumors were conducted to determine the maximum tolerated dose of intravenous plitidepsin and characterize its safety and pharmacokinetics as a single agent [19–21]. In these studies, doses ranging from 0.13 to 8.00 mg/m2 were administered as 1- or 24-h infusion weekly, 3- or 24-h infusion biweekly or, 1-h infusion for 5 consecutive days every 3 weeks. Under these dosage regimens, linear and time-independent pharmacokinetics in plasma was observed after several cycles of administration.
These pharmacokinetic studies have shown that after intravenous administration, plitidepsin plasma concentrations decline in a multi-exponential manner, with a terminal half-life ranging from 21 to 44 h [19]. Plitidepsin is widely distributed, with apparent volumes of distribution at steady state (V ss) ranging from 500 to 1,350 l. The Phase I studies found that blood concentrations were fourfold higher than plasma concentrations, suggesting that blood cells were an important distribution compartment. The human plasma protein binding of plitidepsin was 97.3% [22].
Plitidepsin plasma clearance ranged from 22 to 49 l/h. Urinary excretion of unchanged compound is a minor elimination route, with average recovery of about 4% and less than 15% in all patients, over 48 h, and less than 15% in all patients [19]. Preliminary results of metabolism studies indicated that plitidepsin undergoes moderate microsomal-mediated metabolism in mouse, rat, dog, monkey, minipig and human [18, 23].
In the present paper, plasma and blood concentrations obtained from four Phase I and three Phase II clinical studies conducted in cancer patients were pooled to examine the pharmacokinetic behavior of plitidepsin. The objectives of this population pharmacokinetic analysis were three-fold: (1) to model plasma and blood plitidepsin pharmacokinetics after intravenous administration, (2) to obtain estimates of population pharmacokinetic parameters in cancer subjects, and (3) to evaluate the influence of demographic characteristics and other covariates on plitidepsin pharmacokinetics.
Methods
Patient eligibility criteria and study design
Data from four Phase I studies with extensive pharmacokinetic sampling and three Phase II studies with sparse pharmacokinetic sampling from patients with advanced cancer were pooled (Table 1). All studies were conducted in accordance with principles for human experimentation as defined in the Declaration of Helsinki and were approved by the Human Investigational Review Board of each study center. Informed consent was obtained from each subject after being told the potential risks and benefits, as well as the investigational nature of the study.
Patients were eligible if they had histological or cytological confirmation of malignant tumor not amenable to established forms of effective therapy. Other eligibility criteria included a World Health Organization performance status of 0–2, anticipated life expectancy of at least 3 months, and age >18 years. Previous anticancer radiation therapy and/or chemotherapy, if given, had to be discontinued for at least 4 weeks before entry into the study, or 6 weeks in the case of pretreatment with nitrosoureas or mitomycin C. Patients had to have had a negative pregnancy test (only for female patients with reproductive potential), and normal hepatic and renal function, defined as bilirubin ≤1.5 times normal upper limit, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤2.5 times normal upper limit (≤5 times normal upper limit in case of hepatic metastases), and serum creatinine ≤1.5 times normal upper limit. All patients had to have had acceptable bone marrow function, defined as white blood cells >3,500 μl, neutrophil count >1,500 μl, and platelets >100,000 μl. Patients with one or more of the following criteria were not selected: prior extensive radiation therapy (>25% of bone marrow reserve); prior bone marrow transplantation or high dose chemotherapy with marrow or stem cell rescue; concurrent radiation therapy, chemotherapy, hormonal therapy or immunotherapy; participation in a clinical trial involving an investigational drug in the past 30 days or concurrent enrollment in another investigational trial; and, any coexisting medical condition that was likely to interfere with study procedures and/or results. A summary of patient characteristics at baseline is presented in Table 2.
In these studies, patients received intravenous plitidepsin as monotherapy at doses ranging from 0.13 to 8.0 mg/m2 and given as 1- or 24-h infusions weekly; 3- or 24 h biweekly; or 1-h infusion daily for 5 consecutive days every 21 days. Table 1 provides a summary of the study characteristics.
Bioanalytical methods
All venous blood samples were collected in heparinized tubes and transported to the Mario Negri Institute for analysis. Plasma and blood samples were analyzed using a validated high performance liquid chromatography with electrospray ionisation tandem mass spectrometric detection (HPLC/MS method). The lower limit of quantification (LLOQ) was 0.25 ng/ml and the mean overall coefficient of variation was less than 13% across the validated range of concentration, which included up to 250 ng/ml [13].
Pharmacokinetic model development
Software
Nonlinear mixed-effects modeling by extended least squares regression using the first order (FO) approximation method with the POSTHOC option was implemented with the NONMEM V level 1.1 software package (GloboMax, Hanover, MD, USA) [24]. First-order conditional estimation (FOCE) method, which implements first-order expansion about values of the Bayes’ estimates of the interindividual errors rather than zero, was also investigated. Compilations were achieved using DIGITAL Visual Fortran Version 6.0A. Graphical and all other statistical analyses, including evaluation of NONMEM outputs, were performed using S-Plus 6.1 Professional Edition for Windows (Insightful, Seattle, WA, USA).
Structural model selection
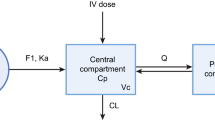
Based on the graphical exploratory analysis, an open, three-compartment model with linear elimination and linear distribution from the central to peripheral compartments was selected to describe the time course of plitidepsin plasma concentrations after intravenous administration. This model was parameterized in terms of clearance (CL), central volume of distribution (V 1), and the intercompartmental exchange flows and volume of distribution for the shallow (Q 2 and V 2) and the deep compartments (Q 3 and V 3), respectively. Further graphical analysis of the predicted blood to plasma concentration ratios suggested that the distribution of plitidepsin into the blood cells might be not linear, as the ratios tended to decrease as concentrations in plasma increase. Therefore, a non-linear distribution to blood cells was modelled, according to the Eq. 1.
where B max corresponds to the maximal plitidepsin concentration bound to blood cells, C 50 is the plitidepsin plasma concentration at which the plitidepsin bound to red blood cells is half-maximal and HCT is the hematocrit of each patient.
The interindividual (IIV, between subject) and interoccasion (IOV, within subject) [25] variabilities in the pharmacokinetic parameters were assumed to follow the lognormal distribution according to the equation:
where P jk is an individual pharmacokinetic parameter for the jth individual and kth occasion, P* is the typical value of the pharmacokinetic parameter, η pj and τ pk are a normally distributed between and within subject random variable with zero-mean and variance \( \omega_{\text{P}}^{2} \) and \( \pi_{\text{P}}^{2} \), respectively. The magnitudes of IIV and IOV were expressed as coefficients of variations (CVs). Three occasions were distinguishable at maximum: two for the full pharmacokinetic profiles and one for the isolated measurements after a drug administration (for instance, trough levels). Residual variability was evaluated using an additive error model after natural logarithmic transformation of the measured concentrations and model predictions, according to the Eq. 2.
where C obs was the observed plasma or blood plitidepsin concentration; C pred was the corresponding model predicted concentration; PB is a dummy variable that takes the value 1 for plasma concentrations and the value 0 for blood concentrations, and ε plasma and ε blood were the residual departure of the natural logarithm of the observed concentration from the predicted concentration in plasma and blood, respectively. Both were assumed to follow an independent Gaussian distribution with zero-mean and variances \( \sigma_{\text{plasma}}^{2} \) and \( \sigma_{\text{blood}}^{2} \).
Covariate analyses
Covariates explored as possible sources of IIV in plitidepsin pharmacokinetics are listed in Table 2. Height was not tested as independent covariate because of its correlation with body weight. Height was only used to calculate the body surface area, which was explored graphically but not formally tested for significance. If after the covariates analysis, body weight was to be identified as a significant covariate, then body surface area was to be evaluated to determine whether this covariate improve the fit relative to the body weight. Hemoglobin was not tested as independent covariate because of its correlation with hematocrit, which is already in the model. Missing values for the quantitative covariates were imputed using the median value in each data set.
Once the structural model was identified, empirical Bayes estimates of the interindividual random effects were computed. The covariate screening was guided by graphical assessment and stepwise linear regression of the relationships between the Bayesian estimates of interindividual random effects and the covariates. Those covariates identified by the screening analysis as having a potential influence on a particular parameter were statistically tested one by one for inclusion in the population pharmacokinetic model (forward inclusion). Categorical covariates were incorporated into the model as index variable, whereas continuous covariates were evaluated using power equations after centering on the median.
Covariates with statistically significant effects on pharmacokinetic parameters were incorporated into the population model simultaneously, and subsequently, the covariate screening process was repeated. A full model was identified when no further covariate additions were possible. Then, the relative contribution of each covariate to the goodness of fit of the full model to the dataset was evaluated one at a time by deleting it from the model (backward elimination) [26]. All non-significant effects on pharmacokinetic parameters were removed from the model and the covariate model was obtained.
The improvement in the fit obtained for each model was assessed in several ways. First, the resulting NONMEM-generated minimum value of the objective function (MVOF) after fitting the models evaluated was used to perform the likelihood ratio test (LRT). This test is based on the change in the minimum value of the objective function (ΔMVOF), which is equal (up to a constant) to minus twice the log-likelihood of the data and is asymptotically distributed like χ 2 with the degrees of freedom (df) equal to the number of parameters added to the model. For hierarchical models, a ΔMVOF of ≥10.83 was required to reach statistical significance (p = 0.001) for the addition of one fixed effect. These stringent statistical criteria were used to avoid the inclusion of weak and clinically non-relevant effects due to the multiple comparisons inherent in the forward inclusion and the backward elimination procedures and the use of FO method [27]. In addition, the improvement in the fit was assessed by the reduction in the IIV, IOV and residual variability, the reduction of the standard errors, and the examination of diagnostic plots such as the scatter plots of observed vs. predicted plitidepsin concentrations, scatter plots of weighted residuals vs. predicted plitidepsin concentrations and time since last dose.
Model refinement
The distribution of the interindividual random effects and the correlation between them were examined graphically to evaluate the normality and the independence assumption, respectively. A model including all the non-diagonal elements of the random effects matrix was fitted to the data. The random effects with the highest correlation were tested by including the corresponding non-diagonal elements in the matrix of random effects. If implementing a correlation significantly improved the fit (ΔMVOF ≥ 10.83), the off-diagonal element of the random effects matrix was kept in the model and the process was repeated until no further improvement of the fit could be achieved.
Pharmacokinetic model qualification and final model development
A non-parametric bootstrap analysis [28] was performed as an internal model evaluation technique, using the package Wings for NONMEM (N. Holford, Version 4.04, June 2003, Auckland, New Zealand). A new replication of the original dataset (a bootstrap sample) was obtained by N random draws of individual data (with replacement) from the dataset including Phase I and Phase II clinical studies. The final population pharmacokinetic model was re-fitted to each new dataset and this process was repeated 1,000 times with different random draws. The stability of the final model was evaluated by comparing the final model parameter estimates to the mean and 95% confidence intervals of the non-parametric bootstrap replicates of the final model. If the parameter estimates fall into the 95% confidence interval obtained from the bootstrap analysis the model was considered unbiased.
In addition, a posterior predictive check was performed using the technique described by Yano et al. [29]. The parameter estimates obtained by fitting the population pharmacokinetic model to the final model were used to simulate the population pharmacokinetic profile of plitidepsin in plasma after the intravenous administration 5 mg/m2 as a 1, 3 and 24-h infusion. A non-parametric 90% prediction interval around the median plasma concentration was constructed to quantify the variability in the model predictions, and to visually compare with the observed dose-normalized plasma concentrations.
Results
Data sets analyzed
A total of 138 patients (179 cycles) receiving intravenous plitidepsin as monotherapy at doses ranging from 0.133 to 7 mg/m2 and given as 1- or 24-h infusion weekly, 3- or 24-h infusion biweekly or, 1-h infusion for 5 consecutive days every 3 weeks, were used to characterize the plitidepsin in plasma and a total of 1,759 plasma concentrations were included in the database. In addition, a total of 152 patients (234 cycles) receiving intravenous plitidepsin as monotherapy at doses ranging from 0.54 to 8 mg/m2 and given as 1- or 24-h infusion weekly, 3- or 24-h infusion biweekly or, 1-h infusion for 5 consecutive days every 3 weeks; were used to characterize the plitidepsin in blood and a total of 2,143 blood concentrations were included in the database.
Structural model selection
An open three compartment disposition model with linear elimination and distribution was used to describe the time course of plasma concentration following plitidepsin administration. The non-linearity in the distribution of plitidepsin to red blood cells improve significantly the model fit (ΔMVOF = −44.22, df = 1, p < 0.001) with respect to the linear model. Substantial further improvement of the fit was achieved by including IOV in V 1, V 2, CL, Q 2, Q 3 (ΔMVOF = −608.015) in the model. The magnitude of the IOV for C 50, B max and V 3 was negligible and, as a consequence, were fixed to 0. At this stage, the interindividual random effect of V 3 turned out to be negligible and, therefore, was excluded. Diagnostic plots of the structural model showed tight random normal scatter around the line of identity and indicated an absence of bias. Histograms of the individual random effects on parameters showed centered distribution around the population typical value (data not shown).
Covariate analyses
The screening analysis showed that serum albumin was related to the CL and B max; body weight was associated with V c, creatinine clearance was related to CL and Q 3, and ALT was correlated with the B max. All these covariates were further explored in NONMEM. Following the forward selection analysis, none of the previous covariates were found to be significant predictors of any of the pharmacokinetic parameters (Table 3) and, consequently, no covariates were incorporated into the model.
Model refinement
Graphical and statistical methods were used to identify correlations between random interindividual effects. The implementation of a full variance-covariance matrix resulted in further improvement in the MVOF (ΔMVOF = −104). However, only the correlation between V 1 with Q 2 (ΔMVOF = −26.2) and V 2 with Q 2 (ΔMVOF = −34.8) were found to be relevant. Further hypothesis testing evidenced that both correlation coefficients were not different from 1, and, therefore, both were fixed to this value (ΔMVOF = −52.2). After implementing these correlations in the model, the rest of the correlations between random effects were not significant, with r 2 < 0.41. Attempts to fit the final model using FOCE method failed.
The final model parameter estimates and their associated precisions, measured as relative standard error (RSE), are presented in Table 4. Fixed effects were estimated with good precision (RSE < 35%), while the precision in the estimate of random effect was reasonable. In addition, diagnostic plots showed tight random normal scatter around the line of identity and indicated an absence of bias (Figs. 1 and 2) while histograms of the individual Bayesian estimates of pharmacokinetic parameters exhibited centered distribution around the population typical value.

Diagnostic plots for the final model fit to the data set for blood samples. Upper panels show plitidepsin concentrations versus population (left) and individual (right) model predictions. Lower panels show the weighted residuals versus time (left) and versus the population model predictions (right)

Diagnostic plots for the final model fit to the data set for plasma samples. Upper panels show plitidepsin concentrations versus population (left) and individual (right) model predictions. Lower panels show the weighted residuals versus time (left) and versus the population model predictions (right)
Model qualification
Nonparametric bootstrap analysis and posterior predictive check were used to qualify the model developed. From the 1,000 bootstrap replicates, 9 failed to minimize successfully and were excluded from the analysis. Eight out of 1,000 replicates (0.8%) had an abnormal termination while the 0.1% of the replicates failed due to rounding errors. The analysis of the bootstrap replicates, which minimized successfully, is given in Table 4. The population estimates for the final model were very similar to the mean of the 991 bootstrap replicates, and were contained within the 95% confidence intervals obtained from the bootstrap analysis, suggesting the absence of bias in the NONMEM parameter estimates. The precision of the NONMEM parameter estimates was also good, because the relative standard error from the bootstrap analysis was lower than 35%.
The results of the posterior predictive check performed on the plitidepsin 5 mg/m2 administered as a 1-, 3- and 24-h intravenous infusion are presented in Fig. 3. In this figure, the 5th, 50th and 95th percentiles of the model-based prediction for plasma concentrations are presented together with the observed plitidepsin plasma concentrations under the same conditions. This figure evidence that the model developed is appropriate to describe the time course of plitidepsin plasma concentrations in cancer patients.

Posterior predictive check performed on the plitidepsin 5 mg/m2 administered as a 1- or 3- or 24-h infusion. Blue lines represents the 5, 50 and 95th percentiles of the model-based predictions, and the red dots represents the observed plitidepsin concentrations
Discussion
A primary goal of this analysis was to develop a population pharmacokinetic model to characterize the concentration-time profile of plitidepsin in plasma and blood following different dosing schedules. An open, 3-compartment disposition model with linear elimination from the central compartment and non-linear distribution to the red blood cells was used to describe the pharmacokinetics of plitidepsin in plasma and blood after intravenous administration to advance cancer patients, respectively.
A single clearance parameter described elimination of plitidepsin by all routes of elimination, including renal and non-renal pathways. In cancer subjects, the typical value of the estimated plitidepsin plasma clearance was 13.6 l/h. There was a moderate to large between subject (71%) and within subject variability (74%), which is consistent with the variability observed for other anticancer agents [30–36].
The typical volume of the central compartment in cancer subjects was estimated to be 117 l with a between and within subject variability of 42 and 47%, respectively. Plitidepsin was found to be widely distributed as the volume of distribution at steady state was estimated to be 4,791 l. Both the central volume of distribution and the steady-state volume of distribution clearly exceeded the total body water, reflecting the large distribution to peripheral tissues consistent with the lipophilicity of the plitidepsin and indicating an extensive tissue binding. This finding is consistent with what has been observed with other highly lipophilic drugs such as trabectedin [37].
The systemic clearance is smaller and steady state volume of distribution is larger than previous estimates obtained from the non-compartmental analysis of the Phase I clinical studies, where the clearance ranged from 45 to 49 l/h, and steady state volume of distribution ranged 1.036 to 1.124 l [19]. In early clinical studies, however, pharmacokinetic samples were collected only during a limited period of time after the end of the infusion, yielding potentially inaccurate estimation of key pharmacokinetic parameters. Thus, the plasma clearance was overestimated, whereas volume of distribution of plitidepsin was underestimated [19]. Therefore, a more accurate estimation of these parameters can be obtained by using population pharmacokinetic methods and pooling the Phase I data with more recent Phase II clinical studies where pharmacokinetic sampling was performed for a multiple cycles. This situation has been recently described for other drugs [31].
Previous studies evidenced blood cells are an important distribution compartment [19, 38]. In fact, concentrations were about fourfold higher in whole blood than in plasma. Non-linear binding to blood cells is evident, given the improvement in the MVOF from the linear distribution model. This non-linear distribution is described by a B max, which was estimated to be 471 μg/l with a moderate between subject variability (55.6%) and a C 50 which was estimated to be 41.6 μg/l. In this model, the blood to plasma ratio is dependent on the hematocrit, with values that ranged from 4.55 to 5.74 for hematocrit of 30–45% respectively at concentrations lower than C 50 (Fig. 4). However, its clinical relevance is limited since about a 90% of the plasma concentrations achieved with the dosing regimens used in Phase II program are below the C 50 value.

Estimated blood to plasma ratio for plitidepsin concentrations as a function of plasma concentrations. Blue lines represents the 5th, 50th and 95th percentiles of the estimated ratio, and the vertical line represents the mean plasma concentration when dose of 7 mg/m2 is given. The graph has been stratified by hematocrit values from 30 to 45%
The evaluated covariates did not influence the pharmacokinetics of plitidepsin to a significant extent. Since generally body weight is related to the amount of extracelular body water, the volume of the central compartment would be expected to be directly proportional to body weight. However, no association between body weight and central volume of distribution was identified, but currently all regimens with plitidepsin contain a dose adjustment to patients body surface area. In theory, with the current dosing practice, plasma and blood concentration at the end of the short intravenous infusion could potentially be associated with body weight. In any case, given the moderate to large variability in the volume to distribution these potential effects are expected to be negligible and with no clinical relevance. The lack of sex and body weight related differences in the population pharmacokinetic parameters suggest that the scientific rationale for dosing plitidepsin according to body surface area is beyond the pharmacokinetic considerations and, as consequence, plitidepsin could be potentially included in a long list of oncology drugs where dosing according to body surface area might not be necessary, unless that efficacy and safety data evidenced otherwise.
Notably, no difference in plitidepsin clearance between the cancer patients older than 65 years relative to younger subjects was observed. In addition, no correlation between plitidepsin clearance and creatinine clearance was identified as expected based on the urinary excretion of unchanged compound, where recoveries below 15% suggested that renal clearance is a minor elimination route for plitidepsin [19], so dose adjustment in renal insufficiency might not be needed.
Concomitant administration of CYP inducers and inhibitors were evaluated as potential factor that may contribute to the between subject variability of plitidepsin pharmacokinetics. The definition of drug inducers and drug inhibitors was based on the list published online [39] and no effect of concomitant co-medication on plitidepsin pharmacokinetics was observed.
In summary, the integration of the Phase I/II pharmacokinetic data demonstrated plitidepsin linear elimination from plasma, dose-proportionality up to 8.0 mg/m2, and time-independent pharmacokinetics. The plitidepsin distribution to red blood cells can be considered linear at doses lower than 5 mg/m2 when administered as 3-h or longer infusion No clinically significant covariates were identified as predictors of plitidepsin pharmacokinetics. The present model manages to well characterize the pharmacokinetics of plitidepsin as it provides a good description of the nonlinearities found, and both population and individual pharmacokinetics are well fitted, making it suitable for use in modelling of plitidepsin pharmacokinetic/pharmacodynamic relationships, simulate pharmacokinetic profiles under different dosing regimens and to perform a model-based optimization of the pharmacokinetic sampling times to optimize the design of future clinical studies.
References
Rinehart KL (2000) Antitumor compounds from tunicates. Med Res Rev 20:1–27
Brandon EF, Sparidans RW, van Ooijen RD et al (2007) In vitro characterization of the human biotransformation pathways of aplidine, a novel marine anti-cancer drug. Invest New Drugs 25:9–19
Adrian TE (2007) Novel marine-derived anti-cancer agents. Curr Pharm Des 13:3417–3426
Broggini M, Marchini SV, Galliera E et al (2003) Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (VEGF) secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human leukemia cells MOLT-4. Leukemia 17:52–59
Cuadrado A, Garcia-Fernandez LF, Gonzalez L et al (2003) Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, Src, JNK, and p38 MAPK. J Biol Chem 278:241–250
Erba E, Bassano L, Di LG et al (2002) Cell cycle phase perturbations and apoptosis in tumour cells induced by aplidine. Br J Cancer 86:1510–1517
Garcia-Fernandez LF, Losada A, Alcaide V et al (2002) Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated JNK and p38 activation and protein kinase C delta. Oncogene 21:7533–7544
Munoz-Alonso MJ, Gonzalez-Santiago L, Zarich N et al (2008) Plitidepsin has a dual effect inhibiting cell cycle and inducing apoptosis via Rac1/c-Jun NH2-terminal kinase activation in human melanoma cells. J Pharmacol Exp Ther 324:1093–1101
Suarez Y, Gonzalez-Santiago L, Zarich N et al (2006) Plitidepsin cellular binding and Rac1/JNK pathway activation depend on membrane cholesterol content. Mol Pharmacol 70:1654–1663
Taraboletti G, Poli M, Dossi R et al (2004) Antiangiogenic activity of aplidine, a new agent of marine origin. Br J Cancer 90:2418–2424
Biscardi M, Caporale R, Balestri F et al (2005) VEGF inhibition and cytotoxic effect of aplidin in leukemia cell lines and cells from acute myeloid leukemia. Ann Oncol 16:1667–1674
Bresters D, Broekhuizen AJ, Kaaijk P et al (2003) In vitro cytotoxicity of aplidin and crossresistance with other cytotoxic drugs in childhood leukemic and normal bone marrow and blood samples: a rational basis for clinical development. Leukemia 17:1338–1343
Celli N, Mariani B, Di CF et al (2004) Determination of aplidin, a marine-derived anticancer drug, in human plasma, whole blood and urine by liquid chromatography with electrospray ionisation tandem mass spectrometric detection. J Pharm Biomed Anal 34:619–630
Depenbrock H, Peter R, Faircloth GT et al (1998) In vitro activity of aplidine, a new marine-derived anti-cancer compound, on freshly explanted clonogenic human tumour cells and haematopoietic precursor cells. Br J Cancer 78:739–744
Gonzalez-Santiago L, Suarez Y, Zarich N et al (2006) Aplidin induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ 13:1968–1981
Lobo C, García-Pozo SG, Núñez de Castro I et al (1997) Effect of dehydrodidemnin B on human colon carcinoma cell lines. Anticancer Res 17:333–336
Urdiales JL, Morata P, Núñez de Castro I et al (1996) Antiproliferative effect of dehydrodidemnin B (DDB), a depsipeptide isolated from mediterranean tunicates. Cancer Lett 102:31–37
Straight AM, Oakley K, Moores R et al (2006) Aplidin reduces growth of anaplastic thyroid cancer xenografts and the expression of several angiogenic genes. Cancer Chemother Pharmacol 57:7–14
Faivre S, Chieze S, Delbaldo C et al (2005) Phase I and pharmacokinetic study of aplidine, a new marine cyclodepsipeptide in patients with advanced malignancies. J Clin Oncol 23:7871–7880
Jimeno JM (2002) A clinical armamentarium of marine-derived anti-cancer compounds. Anticancer Drugs 13(Suppl 1):S15–S19
Maroun JA, Belanger K, Seymour L et al (2006) Phase I study of Aplidine in a dailyx5 one-hour infusion every 3 weeks in patients with solid tumors refractory to standard therapy. A National Cancer Institute of Canada Clinical Trials Group study: NCIC CTG IND 115. Ann Oncol 17:1371–1378
Yin J (2003) Determination of plasma protein binding properties. PUSA 00147. Pharmamar USA Inc, Cambridge
Casals D (2007) Hepatic microsomal metabolism of 14C aplidine-mass identification of three main metabolites of aplidine. S03/012B-AD.2003.KYMOS, Barcelona, Spain
Beal SL, Sheiner LB (1992) NONMEM users guides. GloboMax, LLC, Hanover
Karlsson MO, Sheiner LB (1993) The importance of modeling interoccasion variability in population pharmacokinetic analyses. J Pharmacokinet Biopharm 21:735–750
Wahlby U, Jonsson EN, Karlsson MO (2002) Comparison of stepwise covariate model building strategies in population pharmacokinetic-pharmacodynamic analysis. AAPS Pharm Sci 4:E27
Wahlby U, Matolcsi K, Karlsson MO et al (2004) Evaluation of type I error rates when modeling ordered categorical data in NONMEM. J Pharmacokinet Pharmacodyn 31:61–74
Efron B, Tibshirani R (1993) An Introduction to the bootstrap. Chapman & Hall/CRC Press, London/Boca Raton
Yano Y, Beal SL, Sheiner LB (2001) Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn 28:171–192
Bruno R, Vivler N, Vergniol JC et al (1996) A population pharmacokinetic model for docetaxel (Taxotere): model building and validation. J Pharmacokinet Biopharm 24:153–172
Henningsson A, Sparreboom A, Sandstrom M et al (2003) Population pharmacokinetic modelling of unbound and total plasma concentrations of paclitaxel in cancer patients. Eur J Cancer 39:1105–1114
Nguyen L, Chatelut E, Chevreau C et al (1998) Population pharmacokinetics of total and unbound etoposide. Cancer Chemother Pharmacol 41:125–132
Periclou AP, Avramis VI (1996) NONMEM population pharmacokinetic studies of cytosine arabinoside after high-dose and after loading bolus followed by continuous infusion of the drug in pediatric patients with leukemias. Cancer Chemother Pharmacol 39:42–50
Schleyer E, Kuhn S, Ruhrs H et al (1997) Oral idarubicin pharmacokinetics–correlation of trough level with idarubicin area under curve. Leukemia 11(Suppl 5):S15–S21
Urien S, Rezai K, Lokiec F (2005) Pharmacokinetic modelling of 5-FU production from capecitabine–a population study in 40 adult patients with metastatic cancer. J Pharmacokinet Pharmacodyn 32:817–833
Xie R, Mathijssen RH, Sparreboom A et al (2002) Clinical pharmacokinetics of irinotecan and its metabolites: a population analysis. J Clin Oncol 20:3293–3301
Perez-Ruixo JJ, Zannikos P, Hirankarn S et al (2007) Population pharmacokinetic meta-analysis of trabectedin (ET-743, Yondelis) in cancer patients. Clin Pharmacokinet 46:867–884
Maroun JA, Goel R, Stewart DJ et al (2001) Phase I study of Aplidin in a 5 day bolus q 3 weeks in patients with solid tumors and lymphomas. In: Proceedings of the American society of clinical oncology, 2001, Abstract #2082. http://www.asco.org
Indiana University School of Medicine Department of Medicine (2008) http://medicine.iupui.edu/flockhart/table.htm
Acknowledgments
The authors would like to thank the patients, investigators and their medical, nursing and laboratory staff who participated in the clinical trials included in the present study. In particular, we recognize the effort from the laboratory staff of the Mario Negri Institute, who participated in the bioanalytical analysis of plitidepsin samples.
Conflict of interest
The authors disclosure none conflict of interest other than that Arturo Soto-Matos and Bernardo Miguel-Lillo are employees of Pharma Mar SA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nalda-Molina, R., Valenzuela, B., Ramon-Lopez, A. et al. Population pharmacokinetics meta-analysis of plitidepsin (Aplidin®) in cancer subjects. Cancer Chemother Pharmacol 64, 97–108 (2009). https://doi.org/10.1007/s00280-008-0841-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-008-0841-4