
Abstract
Purpose
To develop a population pharmacokinetics (PPK) model for rucaparib, an oral poly(ADP-ribose) polymerase inhibitor.
Methods
The PPK analysis used PK data from patients in Study 1014 (NCT01009190, n = 35), Study 10 (NCT01482715, n = 123), and ARIEL2 (NCT01891344, n = 300), including intensive intravenous data (12–40 mg), intensive and sparse oral data (12–360 mg single-dose, 40–500 mg once daily, and 240–840 mg twice daily [BID]), and intensive single-dose oral data under fasted conditions and after a high-fat meal (40, 300, and 600 mg).
Results
Rucaparib PK was well described by a two-compartment model with sequential zero-order release and first-order absorption and first-order elimination. A high-fat meal slightly increased bioavailability at 600 mg but not at lower doses; this is not considered clinically significant, and rucaparib can be taken with or without food. Covariate effects of baseline creatinine clearance and albumin on rucaparib clearance were identified. Despite numerical increases in exposure with renal impairment, no dose adjustment is recommended for patients with mild or moderate renal impairment. No statistically significant relationships were detected for demographics, hepatic function (normal versus mild impairment), CYP1A2 and CYP2D6 phenotypes, or strong CYP1A2 or CYP2D6 inhibitors. Concomitant proton pump inhibitors showed no clinically significant effect on absorption. External validation of the model with data from ARIEL3 (NCT01968213) and TRITON2 (NCT02952534) studies showed no clinically meaningful PK differences across indications or sex.
Conclusion
The PPK model adequately described rucaparib PK, and none of the covariates evaluated had a clinically relevant effect.
ClinicalTrials.gov
Study 1014 (NCT01009190), Study 10 (NCT01482715), ARIEL2 (NCT01891344), ARIEL3 (NCT01968213), and TRITON2 (NCT02952534).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rucaparib is an oral, small molecule inhibitor of poly(ADP-ribose) polymerase (PARP) enzymes, including PARP1, PARP2, and PARP3 [1,2,3]. Rucaparib has been shown to induce cytotoxicity in tumor cells with homologous recombination deficiency (HRD), including those with deleterious mutations in BRCA1 or BRCA2 (BRCA) [3,4,5]. Defects in the homologous recombination repair pathway combine with PARP inhibition in a mechanism known as synthetic lethality, resulting in the accumulation of DNA damage and cell death [3,4,5].
The pharmacokinetics (PK), efficacy, and safety of rucaparib have been evaluated in several studies including Study 1014 (NCT01009190), Study 10 (NCT01482715), ARIEL2 (NCT01891344), ARIEL3 (NCT01968213), and TRITON2 (NCT02952534) [6,7,8,9,10,11,12,13]. Rucaparib is approved as monotherapy in the United States and Europe for maintenance treatment of women with recurrent ovarian cancer who have responded to platinum-based chemotherapy, and as monotherapy treatment for patients with BRCA-mutated, relapsed ovarian cancer who have received ≥ 2 prior lines of platinum-based chemotherapy (in Europe, all patients must have platinum-sensitive disease and, for monotherapy treatment, be unable to tolerate further platinum-based chemotherapy) [14, 15]. Rucaparib is approved in the United States for the treatment of men with BRCA mutation-associated metastatic castration-resistant prostate cancer (mCRPC) after androgen receptor-directed therapy and a taxane-based chemotherapy [15]. The approved starting dose of rucaparib is 600 mg twice daily (BID).
Three clinical trials established the formulation and route of administration, recommended starting dose, and evaluated the efficacy of rucaparib. Study 1014 was a phase 1 dose-escalation study evaluating the safety, PK, and pharmacodynamics (PD) of intravenous (IV) and/or oral rucaparib in combination with several chemotherapeutic regimens in adult patients with advanced solid tumors [6]. Study 10 was a phase 1/2, open-label, safety, PK, and preliminary efficacy study of oral rucaparib in patients with germline BRCA-mutated ovarian cancer or other solid tumors that evaluated a range of oral rucaparib doses to establish the recommended starting dose [7, 8]. ARIEL2 was a two-part phase 2, open-label study of rucaparib (starting dose, 600 mg BID) in patients with relapsed, high-grade epithelial ovarian, fallopian tube, or primary peritoneal cancer [9, 10]. The primary endpoints in ARIEL2 were progression-free survival in patients with relapsed platinum-sensitive ovarian cancer (Part 1) and objective response rate in heavily pretreated patients with relapsed ovarian cancer (Part 2).
In vitro, the solubility of rucaparib camsylate is reasonably high (1.4 nmol/L) due to the relatively high acid dissociation constant of 9.6 [2]. However, in the presence of chloride ions, rucaparib camsylate could precipitate as a HCl salt. Rucaparib showed 70% plasma protein binding and moderate permeability [15, 16]. Clinically, rucaparib showed linear PK and mean absolute oral bioavailability of 36% [6, 17]. Steady state was observed following 1 week BID dosing with a 1.47- to 5.44-fold area under the concentration–time curve (AUC) accumulation ratio at tested oral dose levels [8]. In a 14C-rucaparib (600 mg, 140 μCi) mass balance study in patients with advanced solid tumors, the most abundant metabolite was M324, an inactive carboxylic acid metabolite of rucaparib based on in vitro PARP binding and cellular assay results [16]. Rucaparib showed a mean half-life of 25.9 h [16]. The intact rucaparib and M324 accounted for 64.0% and 18.6% of total radioactivity in plasma, respectively [16]. Urinary and fecal recovery accounted for 17.4% (44.9% rucaparib, 50.0% M324) and 71.9% (94.9% rucaparib, 5.1% M324), respectively, of the administered dose [16].
For the current analysis, PK data from patients in Study 1014, Study 10, and ARIEL2 were used to develop the population PK (PPK) model for IV and oral rucaparib in patients with solid tumors. Clinical factors of interest were tested in the model, and their effects on PK were determined. PK data from ARIEL3 (NCT01968213) and TRITON2 (NCT02952534) were used for external model validation.
Materials and methods
Study populations and data
Data from three studies were pooled for the development of the PPK model. Two additional studies were used for external model validation. The design, study populations, and PK sampling protocols for each of the five studies are summarized in Supplementary Table 1.
Studies used for model development
Study 1014 was a parallel arm, phase 1 safety, PK, and PD study of rucaparib in combination with several chemotherapeutic regimens in adult patients with advanced solid tumors [6]. The study included a lead-in IV rucaparib dose on day −10 and a lead-in oral rucaparib dose on day −5 without concomitant chemotherapies. The IV and oral PK data from the lead-in period were included in the PPK analysis. PK data after cycle 1 day 1 were not included in the PPK analysis due to concomitant chemotherapies. The IV dose was administered as rucaparib phosphate, and the oral dose was given as rucaparib camsylate tablets. In total, 35 patients from Study 1014 with both intensive IV and oral PK data were included in estimation of the final PPK model.
Study 10 was a phase 1/2, open-label, safety, PK, and preliminary efficacy study of oral rucaparib in patients with germline BRCA-mutated ovarian cancer or other solid tumor [7, 8]. In Study 10 Part 1 (n = 56), rucaparib doses ranged from 40 to 500 mg once daily (QD) and from 240 to 840 mg BID, with intensive PK data collected following single-dose administration and at steady state. Sparse trough PK data were also collected in selected subsequent cycles. In addition, a preliminary assessment of the effect of a high-fat meal on rucaparib PK was evaluated in patients who received 40 or 300 mg QD. In Part 2A (n = 42), patients received 600 mg BID rucaparib and sparse trough PK data were collected. In Part 3 (n = 26), intensive PK was collected to assess the PK of a single 600 mg dose of rucaparib under fasted conditions as compared to with a high-fat meal. Intensive PK was also collected at steady state following 600 mg rucaparib BID. Additional sparse trough PK data were collected in selected subsequent treatment cycles.
ARIEL2 is a phase 2, open-label study of rucaparib in patients with relapsed, high-grade ovarian cancer [9, 10]. The starting dose of rucaparib was 600 mg BID in both Part 1 (n = 196) and Part 2 (n = 104). Sparse trough PK data were collected for all patients on cycle 1 day 15 and on day 1 of selected subsequent treatment cycles.
All available PK data from Study 10 and ARIEL2 were combined with the IV and oral data from Study 1014 for PPK model development.
External validation
After finalization of the PPK model, study data became available for ARIEL3 and TRITON2. ARIEL3 is a multicenter, randomized, double-blind, placebo-controlled phase 3 study of rucaparib as maintenance following platinum-based chemotherapy in patients with platinum-sensitive, high-grade serous or endometrioid epithelial ovarian, primary peritoneal, or fallopian tube ovarian cancer [11]. Patients were randomized (2:1) to receive either 600 mg BID rucaparib (n = 375) or placebo (n = 189). TRITON2 is an open-label, phase 2 study evaluating rucaparib as monotherapy for treatment of mCRPC associated with HRD who have progressed on prior androgen receptor-directed therapy and a taxane-based chemotherapy in the castration-resistant setting [12, 13]. Patients (n = 199) initiated treatment with 600 mg BID rucaparib. Data from ARIEL3 and TRITON2 were used for external validation of the PPK model that was developed using data from Study 1014, Study 10, and ARIEL2.
Data handling
For PPK model development, all data from patients who received at least one dose of rucaparib followed by at least one nonexcluded quantifiable concentration were included. The dataset included actual sample collection date and times and dosing date and times when available. For nine subjects in Study 1014, actual sampling dates and times were used. For the remaining 26 of 35 patients, nominal date and sampling times were used. In Study 10, actual times were not available for 641 (of 25,382) dosing records. In these cases, the dosing time was inferred based on protocol design, available dosing records, and PK sampling time as appropriate. In ARIEL2, 14 PK sampling records lacked dates; hence, dates were assigned according to the nominal treatment dates and visit information. Missing PK sampling times were assigned the typical clock time observed in the dataset, and dosing times were derived as 12 h prior to sampling. Prior to data exclusions, the initial full model development dataset included 4176 observations from 453 potentially PK-evaluable patients. After the inclusion of additional oral data from Study 1014, the full dataset used to estimate the final PPK model included 458 patients and 4518 observations prior to exclusions.
Pharmacokinetic sample analysis
Plasma concentrations of rucaparib were determined by Q Squared Solutions BioSciences (Ithaca, NY; formerly Advion Bioanalytical Laboratories) using validated liquid chromatography and tandem mass spectrometric methods as previously reported [8]. The assay has a lower quantification limit of 5 ng/mL and a calibration range of 5–10,000 ng/mL.
PPK model development
The PPK model was developed as described below. The first-order conditional estimation with interaction (FOCE-I) method in NONMEM® (version 7.3 or higher) [18] was used for implementation of all model development (ICON Development Solutions, Ellicott City, MD). Perl-speaks-NONMEM (PsN; version 4.2.0 or higher) [19, 20] was used for model assessment graphics, stepwise covariate modeling, and visual predictive checks (VPCs). R (version 3.2 or higher) was used for dataset construction, exploratory analyses, and postprocessing of PsN results.
Structural model
First, intensively sampled data from Study 10 Part 1 and Part 3 (oral administration) and Study 1014 (oral and IV administration) were used to identify a structural model providing an adequate description of the PK data. On the basis of graphical evaluation of rucaparib plasma concentration versus time profiles, an initial structural model was selected and tested with various modifications. Specification of the structural model included the number of compartments, preliminary random effect structures, and the preliminary residual error models. Sparsely sampled data from all studies were added after identification of the structural model.
Covariate search
PK parameter–covariate relationships were evaluated using a staged process. First, graphical analyses and a univariate search were used to identify statistically significant (p < 0.05) relationships. Next, relationships identified in the univariate search were tested in a stepwise forward–backward search using stepwise covariate modeling [21] in PsN. The likelihood ratio test was used to evaluate the significance of incorporating or removing fixed effects in the PPK model. For forward selection and backward deletion, significance levels of p < 0.01 and p < 0.001 were employed, respectively. All continuous and categorical covariates were initially incorporated into the PPK model as follows:
Continuous covariates:
or
Categorical covariates:
where P is the population estimate of a parameter, Xi is the covariate of subject i for the parameter P, M(Xi) is the median of covariate X for the patient population, θk is the typical value of the parameter P, and θj is a coefficient that reflects the covariate’s effect on the parameter. For categorical covariates, Xi is a binary variable with value 0 for the base case (i.e., without the effect) and value 1 for the test case (i.e., with the effect). When a categorical variable with multiple levels was tested, e.g., food, the effect was parameterized as a series of effects so that one θj is estimated for each category that was not in the base category.
Model finalization and evaluation
The model selection process was driven by the data, prior information regarding rucaparib PK, and various goodness-of-fit indicators, including standard diagnostic plots, precision of parameter estimates, and changes in the objective function value (OFV). Since one of the primary goals for this analysis was to use the final model for prediction of PK exposures for the exposure response analysis, model development was approached to optimize the model for that purpose. Hence, in the structural model development and model optimizations stages, a model with a higher OFV might be selected based on improvement in model diagnostic plots over a model with a lower OFV. The extent of shrinkage derived from the base and final models were assessed for each interindividual variability term (η) [22].
Bootstrap resampling techniques were used as appropriate to evaluate the stability of the final model and to estimate nonparametric confidence intervals (CIs) for the model parameters. This analysis was conducted by repeatedly fitting the final model to bootstrap replicates of the analysis data set. The 95% CI (2.5th–97.5th percentiles) around the parameter estimates of the final model were generated based on nonparametric bootstrap analysis (N ≈ 500 or 1000 bootstrap datasets). The number of runs that converged and minimized successfully was recorded. Prediction-corrected VPCs (pcVPCs) were used to evaluate the predictive ability of the final model and its suitability for use in simulations. PcVPC was performed using the VPC algorithm in PsN. At least 500 replicates (i.e., datasets) were simulated using the relevant models assessed. Within each replicate, the simulated data were summarized by computing the median, 5th and 95th percentiles by population, study phase, and nominal time. Model performance was evaluated based upon the extent to which the observed median values fell within the associated 90% prediction interval (PI).
External validation
PK data from ARIEL3 and TRITON2 were used for an external validation of the final PPK model. PcVPCs were used to evaluate the ability of the final model to describe the data from ARIEL3 and TRITON2. For ARIEL3, sampling times and the date and time of the prior dose were available; hence, pcVPCs were evaluated both as a function of time after dose and time after first dose. For TRITON2, the clock time of the prior dose was not available, thus precluding the ability to calculate time after dose. For TRITON2, pcVPCs were limited to evaluation as a function of time after first dose. Dose times and dates were inferred based on a nominal dosing schedule (one dose every 12 h). If the dose record indicated otherwise (e.g., QD dosing or dose interruptions), the inferred times and dates were adjusted accordingly.
Results
Analysis population
Table 1 provides a summary of the baseline characteristics of the patients included in the model development dataset. The full development dataset (n = 453) comprised 30 patients from Study 1014, 123 patients from Study 10, and 300 patients from ARIEL2 (Supplementary Table 2). Dose-normalized rucaparib concentration–time profiles in Study 10 and ARIEL2 stratified by dose and dosing frequency and in Study 1014 stratified by administration route are shown in Supplementary Fig. 1.
The PPK analysis population was 96.0% (n = 435) female and 81.9% (n = 371) White, with a median age of 61 years and a median weight of 69 kg. Approximately 40% of patients (n = 183) had a deleterious BRCA mutation (109 and 74 patients had a deleterious mutation of BRCA1 and BRCA2, respectively). Patients’ CYP1A2 and CYP2D6 phenotypes and concomitant use of proton pump inhibitors (PPIs) are summarized in Table 1. About 4% of available PK observations were below the limit of quantification and, thus, were excluded from the analysis. In addition to these exclusions, samples that were hemolyzed (n = 58), mislabeled (n = 62), or had potential data errors (n = 74) were excluded. Given the approximately 25.9-h half-life of rucaparib, based on noncompartmental analysis (NCA) analysis [16], samples collected more than 150 h postdose (n = 59) were excluded. Ultimately, 442 PK-evaluable patients with 3730 concentration observations remained in the analysis dataset.
Structural model development
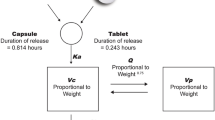
Rucaparib PK data were adequately described by a two-compartment model with sequential zero-order release and first-order absorption, and first-order elimination (Fig. 1). Given the IV and oral data from Study 1014, absolute oral bioavailability (F1) was estimated in the PPK model. Between-subject variability (BSV) was included on clearance (CL), absorption rate constant (Ka), and the duration of the zero-order release (D1).

PPK model diagram for rucaparib. CL clearance, Cp plasma concentration, D1 duration of the zero-order absorption, F1 absolute oral bioavailability, IV intravenous, Ka absorption rate constant, PPK population pharmacokinetics, Q intercompartmental clearance, Vc volume of central compartment, Vp volume of peripheral compartment
Estimates of CL and F1 from the NCA of data from Study 1014 were compared to estimates from the PPK structural model based on the pooled dataset. After a 30-min IV infusion, the mean total plasma CL ranged from 14 to 18 L/h, and steady-state volume of distribution ranged from 113 to 262 L. In the NCA of Study 1014, F1 ranged from 30.1 to 45.3%, with a mean of 36% [6]. Based on the PPK structural model (prior to addition of any covariate effects), F1 of rucaparib was 32.3% (95% CI 26.4–38.7%), the typical estimate of CL was 8.45 L/h (95% CI 6.8–10.1 L/h), and steady-state volume of distribution was 143 L (109–177 L).
Table 2 summarizes the available data by study, food, tablet strength, dose, and sampling intensity. Previous NCA of Study 10 [8] and Study 1014 [6] revealed mild and dose-dependent food effect on oral rucaparib PK. There was no apparent effect of a high-fat meal for 40 or 300 mg rucaparib, but a mild increase in exposure occurred for 600 mg rucaparib. Graphical analyses of post hoc PK parameters suggested an increase in Ka for fasted dosing and decreasing Ka with increasing dose. Additionally, graphical analyses suggested reduced F1 for fasted dosing, relative to a high-fat meal. Given the differences in food effects observed across different dose ranges as well as the dose levels tested in clinical trials, three categorical effects were tested: fasted and dose ≤ 480 mg, fasted and dose > 480 mg, and high-fat meal and dose > 480 mg. The reference case was a patient-selected meal (i.e., no meal restriction) and a dose > 480 mg. The dose category of > 480 mg included 600 mg BID (n = 395) and 840 mg BID (n = 3, Study 10 Part 1). Ultimately, the model included effects of fasted dosing on F1 (≤ 480 mg dose separate from > 480 mg dose), administration following a high-fat meal at dose > 480 mg, effect of dose (continuous) on Ka, effect of fasted dosing on Ka (any dose), and effect of high-fat meal on Ka (any dose).
During the structural model development, several absorption models were evaluated including transit compartment models, parallel first-order absorption process, parallel zero- and first-order processes. The sequential zero-order release and first-order absorption model provided the best description of the limited data collected during the absorption phase. In a separate analysis (not reported), additional semimechanistic and complex empirical absorption models, including saturable absorption mechanisms, were evaluated to provide a more mechanistic description of the effect of dose on Ka. Although single-dose data were better described by a more complex model including parallel saturable and transit-compartment processes, the model became over-parameterized when multiple-dose data, food effects, and other covariates were incorporated. Model-predicted steady-state exposure estimates were similar for the two models; hence, the simpler sequential zero-order release and first-order absorption model was retained.
Covariate effects and model finalization
Starting with the base model including the effects noted above, the remaining parameter-covariate relationships were evaluated in a forward–backward search. Ultimately, effects of baseline CLcr on CL and baseline albumin on CL were added to the PPK model.
A final update to the PPK dataset was made to include additional oral data from patients in Study 1014. The optimized PPK model was re-estimated based on the final dataset. Diagnostic plots, provided in the Supplementary Information (Supplementary Fig. 2), show that the data were adequately described by the model. Model parameter estimates are provided in Table 3. The typical estimate of CL was 10.3 L/h. Intercompartmental clearance (Q) was 17.4 L/h, and the central volume of distribution (Vc) and peripheral volume (Vp) were 16.9 L and 166 L, respectively. Absolute F1 was 37.2%, and the typical estimates of Ka and D1 were 0.0718 h−1 and 0.619 h, respectively. BSV on CL, estimated for all patients was 48.8%. BSV on Ka and D1, limited to patients with intensive sampling data in Study 10 (Part 1 and Part 3) and Study 1014, were 63.5% and 111%, respectively. Estimates of shrinkage for all three BSV parameters were low (< 15%), and epsilon shrinkage was low (5.8%).
The parameter-covariate effects in the final model are shown in Eq. 1 (see Supplemental Information). The primary effects on rucaparib PK were dose and food on F1, dose and food on Ka, CLcr on CL, and albumin on CL. Most data in the PPK dataset (i.e., the reference case) were for 600 mg BID rucaparib with patient-selected food intake (i.e., fed or fasted). At doses approaching the clinical starting dose (i.e., 600 mg), F1 decreased 12.3% under fasted conditions and increased 39% following a high-fat meal. The results suggest a mild variability in F1 when patients took rucaparib without meal restrictions. For doses ≤ 480 mg and under fasted or high-fat conditions, F1 was slightly lower (22%) than under patient-selected food intake. Fasted dosing increased Ka by 40%. Over the full range of doses evaluated (40–840 mg), the effect of the dose on Ka was significant; however, at the clinical dose levels of 300–600 mg BID evaluated in ARIEL2, the model estimated Ka was in a narrow range of 0.0836–0.0667 h−1, suggesting that the effect on Ka is not clinically relevant. Data summarizing the effects of dose and food on F1 and Ka are provided in the Supplementary Information (Supplementary Fig. 3).
The effect of CLcr on CL met the criteria for clinical significance. At the 10th percentile of CLcr observed in the PPK dataset (53 L/min), CL was 8.93 L/h, 13% lower than the typical estimates (10.3 L/h). At the 90th percentile (124 L/min), CL was 11.7 L/h, 13.6% higher than the typical estimate. The effect of albumin on CL was smaller, with an ≈ 18% difference in CL from the 10th to the 90th percentiles of observed albumin. The underlying mechanism for higher CL values at higher albumin levels is not clear, and the effect of albumin may not be clinically significant, especially given rucaparib’s relatively low plasma protein binding (70%). No plausible covariate effects were identified for Vc and D1.
Following continuous 600 mg BID rucaparib dosing, the model-estimated steady-state AUC for patients with mild (GFR 60–89 mL/min, n = 149) and moderate (GFR 30–59 mL/min, n = 76) renal impairment was 15% and 33% higher than that of patients with normal renal function (GFR ≥ 90 mL/min, n = 147), respectively (Supplementary Fig. 4). Polymorphisms of CYP1A2 (normal metabolizers [n = 28], and hyperinducers [n = 136]) and CYP2D6 (slow [n = 9], intermediate [n = 71], normal [n = 76], and ultrarapid [n = 4] metabolizers) did not impact rucaparib PK at 600 mg BID (Supplementary Fig. 5). In an analysis of concomitant medications as time-varying covariates in patients at all starting doses (n = 22) and in patients at the 600 mg BID starting dose (n = 19), dose-normalized steady-state trough concentrations were similar when rucaparib was dosed alone or concomitantly with a PPI (Supplementary Fig. 6).
Model assessment
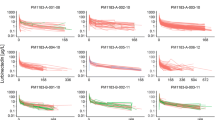
The pcVPC for the final model is shown in Fig. 2, stratified by sampling intensity and food. Overall, the model adequately described the observed PK data. The pcVPC shows greater variability around the steady-state trough concentration than the observed data. Additionally, there is some underprediction of the median and variability of the absorption profile following a high-fat meal. However, the patient population of interest is unlikely to regularly consume high-fat meals, and the model adequately describes the data following a patient-selected meal.

Prediction-corrected visual predictive checks (pcVPCs) for rucaparib. Solid red line represents the median of the observed data. Solid blue lines represent the 5th and 95th percentiles of the observed data. Shaded regions encompass 90% of the simulated values (n = 500) of the predicted medians (pink), 5th (blue), and 95th (blue) percentiles. Data points (Xs) represent the individual observed data. PI prediction interval
In addition to the pcVPC, a bootstrap analysis was also completed. Of the 500 resampled runs, 436 (87.2%) minimized successfully, with 64 failed runs due to rounding or other errors. Parametric and bootstrap estimates were largely similar (Table 3).
External model validation
An external model validation was conducted to validate the final PPK model in a population of women with ovarian cancer receiving maintenance rucaparib (ARIEL3) and in a population of men with mCRPC (TRITON2) taking rucaparib in the treatment setting. For ARIEL3, the pcVPC (Fig. 3) shows that the data are well described by the final rucaparib PPK model and that the observed data are mostly consistent with the 90% PIs. At all timepoints where sufficient observed data are available, the line representing the median of the observed data falls within the median PI. For TRITON2, the pcVPC (Supplementary Fig. 7A) is displayed as a function of time since first dose because actual dose times were not recorded. The median line falls within the median PI, suggesting that the model describes the central tendency of the data reasonably well and that rucaparib PK is similar between ovarian cancer patients (females) and prostate cancer patients (males). There was a slight tendency toward overprediction of the median trough concentration. Additionally, the 5th and 95th percentiles fall outside or barely within the PIs, suggesting the model may not adequately describe the variance of the mCRPC data. Indeed, the OFV was reduced 79 points when the proportional and additive error components of the model were re-estimated (with all other parameters fixed). Estimates of proportional and additive error were reduced from 38.2% and 379 ng/mL to 26.0% and 122 ng/mL, respectively. The pcVPC for the model with updated residual error components is provided in the Supplementary Information (Supplementary Fig. 7B).

External validation of the rucaparib population PK model in ARIEL3. Data points represent the individual observed data. The solid red line represents the median of the observed data. The solid blue lines represent the 5th and 95th percentiles of the observed data. The shaded regions encompass 90% of the simulated values (n = 1000) of the predicted medians (orange), 5th (blue), and 95th (blue) percentiles. PI prediction interval, PK pharmacokinetics
Discussion
Rucaparib PPK was adequately described by a two-compartment PK model with sequential zero-order release and first-order absorption and first-order elimination. Intrinsic (e.g., genetic polymorphisms or hepatic function) and extrinsic covariates (e.g., food intake) of clinical interest [23] were tested. The mild effect of a high-fat meal on rucaparib PK is not considered clinically significant based on collective clinical data, thus patients have been allowed to take rucaparib with no meal restriction (i.e., patient-selected meal) in all subsequent rucaparib safety and efficacy trials. The 200/300 mg strength tablets showed comparable PK with lower-strength tablets, and PPK analyses supported pooling of clinical efficacy data of all tablet strengths.
The effect of baseline CLcr on CL was statistically significant. Despite numerical increases in rucaparib exposure with renal impairment, no clinically meaningful effect on safety has been observed, and no dose adjustment is recommended for patients with mild (CLcr 60–89 mL/min) to moderate (CLcr 30–59 mL/min) renal impairment. In the previously published exposure–response analyses for rucaparib in patients with recurrent ovarian carcinoma from Study 10 and ARIEL2 [24], a significant correlation was observed between exposure and independent radiology review-assessed objective response rate, supporting the 600 mg BID starting dose. Dose reductions are allowed in patients with impaired renal function based on individual patients’ safety data following rucaparib treatment. It is worth mentioning that rucaparib is an inhibitor of renal transporters MATE1, MATE2-K, and OCT2 [16], which are involved in renal excretion of creatinine, and mild creatinine increases have been reported clinically [7, 9, 11,12,13]. As a result, posttreatment CLcr should not be used as a surrogate for renal function. Liver enzymes showed no statistically significant impact on rucaparib PK, and no difference was observed between patients with normal and mildly impaired hepatic function per National Cancer Institute (NCI) guidelines (Supplementary Fig. 4). While the effect of albumin appeared statistically significant, the impact on exposures was minimal and of no clinical significance given rucaparib’s low plasma protein binding.
Polymorphisms of CYP1A2 and CYP2D6 did not impact rucaparib PK at 600 mg BID (Supplementary Fig. 5). Concomitant PPI use showed no apparent effect on rucaparib PK (Supplementary Fig. 6). No difference in rucaparib PK was observed between patients with different BRCA gene mutation (i.e., BRCA1 versus BRCA2) or BRCA status (i.e., germline versus somatic). Insufficient data were available to evaluate the impacts of concomitant CYP or transporter perpetrator drugs, the effect of current smoking, and baseline alpha-1-acid-glycoprotein on rucaparib PK.
Rucaparib absorption was modeled as a sequential zero-order release plus first-order absorption process. In the original model development, the absorption model was selected based on goodness-of-fit plots and a large decrease in OFV (259 units) compared with a simpler first-order absorption model. Addition of covariates to the model resulted in limited reduction in estimates of BSV (< 4%). The relatively high variability on absorption model parameters coupled with the effect of dose on Ka could be due to precipitation of rucaparib in the stomach in the presence of chloride ion, and supported exploration of nonlinear and semimechanistic absorption models. In an analysis of a more limited dataset (not reported), the addition of interoccasion variability on Ka and D1 resulted in dramatic reductions in BSV estimates and large interoccasion variability estimates, suggesting that the elevated BSV on Ka and D1 in the final model might be due to unrecorded variability in dosing conditions for rucaparib (e.g., nonspecified food intake). Attempts to describe the nonlinear absorption and food effects on absorption resulted in overparameterized models, indicating the available data are likely insufficient to evaluate these effects. While a more complex model might better describe single-dose absorption, any improvement is of limited clinical relevance given the relatively flat rucaparib absorption profile at steady state.
In conclusion, the two-compartment PPK model with sequential zero-order release and first-order absorption and first-order elimination adequately described the PK of rucaparib in advanced ovarian cancer patients and other solid tumors. External validation using data from an ovarian cancer maintenance population and an mCRPC population suggest satisfactory model performance and lack of PK difference across indications and between sexes. While the effect of baseline CLcr was statistically significant and numerical increases in rucaparib exposure were observed, no dose adjustment is recommended for patients with mild-to-moderate renal impairment. No clinically relevant effects were observed for mild hepatic impairment, CYP1A2 and CYP2D6 polymorphisms, concomitant PPIs, BRCA mutation, BRCA status, age, or weight.
References
Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell A, Ekblad T, Oncu D, Kull B, Robertson GM, Pellicciari R, Schuler H, Weigelt J (2012) Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol 30(3):283–288. https://doi.org/10.1038/nbt.2121
Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, Maegley KA, Newell DR, Skalitzky D, Wang LZ, Webber SE, Curtin NJ (2007) Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther 6(3):945–956. https://doi.org/10.1158/1535-7163.MCT-06-0552
Robillard L, Nguyen M, Harding TC, Simmons AD (2017) In vitro and in vivo assessment of the mechanism of action of the PARP inhibitor rucaparib. Cancer Res 77(13 suppl):abstract 2475
Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, Mukhopadhyay A, Los G, Hostomsky Z, Plummer ER, Edmondson RJ, Curtin NJ (2011) Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst 103(4):334–346. https://doi.org/10.1093/jnci/djq509
Nguyen M, Simmons AD, Harding TC (2017) Preclinical assessment of the PARP inhibitor rucaparib in homologous recombination deficient prostate cancer models. Cancer Res 77(13 suppl):abstract 2476
Wilson RH, Evans TRJ, Middleton MR, Molife LR, Spicer J, Dieras V, Roxburgh P, Giordano H, Jaw-Tsai S, Goble S, Plummer R (2017) A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br J Cancer 116(7):884–892. https://doi.org/10.1038/bjc.2017.36
Kristeleit R, Shapiro GI, Burris HA, Oza AM, LoRusso P, Patel MR, Domchek SM, Balmana J, Drew Y, Chen LM, Safra T, Montes A, Giordano H, Maloney L, Goble S, Isaacson J, Xiao J, Borrow J, Rolfe L, Shapira-Frommer R (2017) A phase I-II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res 23(15):4095–4106. https://doi.org/10.1158/1078-0432.CCR-16-2796
Shapiro GI, Kristeleit R, Burris HA, LoRusso P, Patel MR, Drew Y, Giordano H, Maloney L, Watkins S, Goble S, Jaw-Tsai S, Xiao J (2019) Pharmacokinetic study of rucaparib in patients with advanced solid tumors. Clin Pharmacol Drug Dev 8(1):107–118. https://doi.org/10.1002/cpdd.575
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, Konecny GE, Coleman RL, Tinker AV, O’Malley DM, Kristeleit RS, Ma L, Bell-McGuinn KM, Brenton JD, Cragun JM, Oaknin A, Ray-Coquard I, Harrell MI, Mann E, Kaufmann SH, Floquet A, Leary A, Harding TC, Goble S, Maloney L, Isaacson J, Allen AR, Rolfe L, Yelensky R, Raponi M, McNeish IA (2017) Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol 18(1):75–87. https://doi.org/10.1016/S1470-2045(16)30559-9
Swisher EM, Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, Coleman RL, Aghajanian C, Konecny GE, O’Malley DM, Leary A, Provencher D, Welch S, Chen LM, Wahner Hendrickson AE, Ma L, Ghatage P, Kristeleit RS, Dorigo O, Musafer A, Kaufmann SH, Elvin JA, Lin DI, Chambers SK, Dominy E, Vo LT, Goble S, Maloney L, Giordano H, Harding T, Dobrovic A, Scott CL, Lin KK, McNeish IA (2021) Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat Commun 12(1):2487. https://doi.org/10.1038/s41467-021-22582-6
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, Leary A, Holloway RW, Gancedo MA, Fong PC, Goh JC, O’Malley DM, Armstrong DK, Garcia-Donas J, Swisher EM, Floquet A, Konecny GE, McNeish IA, Scott CL, Cameron T, Maloney L, Isaacson J, Goble S, Grace C, Harding TC, Raponi M, Sun J, Lin KK, Giordano H, Ledermann JA (2017) Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390(10106):1949–1961. https://doi.org/10.1016/s0140-6736(17)32440-6
Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, Voog EG, Bryce AH, McDermott R, Ricci F, Rowe J, Zhang J, Piulats JM, Fizazi K, Merseburger AS, Higano CS, Krieger LE, Ryan CJ, Feng FY, Simmons AD, Loehr A, Despain D, Dowson M, Green F, Watkins SP, Golsorkhi T, Chowdhury S (2020) Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase 2 TRITON2 study. Clin Cancer Res 26(11):2487–2496. https://doi.org/10.1158/1078-0432.CCR-20-0394
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, Sautois B, Vogelzang NJ, Bambury RM, Voog E, Zhang J, Piulats JM, Ryan CJ, Merseburger AS, Daugaard G, Heidenreich A, Fizazi K, Higano CS, Krieger LE, Sternberg CN, Watkins SP, Despain D, Simmons AD, Loehr A, Dowson M, Golsorkhi T, Chowdhury S, TRITON investigators, (2020) Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol 38(32):3763–3772. https://doi.org/10.1200/JCO.20.01035
Rubraca (rucaparib) tablets [summary of product characteristics] (2021). Clovis Oncology Ireland Ltd., Swords, Ireland
Rubraca (rucaparib) tablets [prescribing information] (2021). Clovis Oncology, Inc., Boulder, CO
Liao M, Watkins S, Nash E, Isaacson J, Etter J, Beltman J, Fan R, Shen L, Mutlib A, Kemeny V, Papai Z, van Tilburg P, Xiao JJ (2020) Evaluation of absorption, distribution, metabolism, and excretion of [14C]-rucaparib, a poly(ADP-ribose) polymerase inhibitor, in patients with advanced solid tumors. Invest New Drugs 38(3):765–775. https://doi.org/10.1007/s10637-019-00815-2
Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, Curtin N, Boddy A, McHugh P, Newell D, Harris A, Johnson P, Steinfeldt H, Dewji R, Wang D, Robson L, Calvert H (2008) Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 14(23):7917–7923. https://doi.org/10.1158/1078-0432.CCR-08-1223
Beal S, Sheiner L, Boeckmann A, Bauer R (2009) NONMEM 7.4 Users Guides. (1989–2017). Ellicott City, MD: Icon Development Solutions. https://nonmem.iconplc.com/nonmem744/guides. Accessed 23 Nov 2021
Lindbom L, Ribbing J, Jonsson EN (2004) Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed 75(2):85–94. https://doi.org/10.1016/j.cmpb.2003.11.003
Lindbom L, Pihlgren P, Jonsson EN (2005) PsN-Toolkit–a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79(3):241–257. https://doi.org/10.1016/j.cmpb.2005.04.005
Khandelwal A, Harling K, Jonsson EN, Hooker AC, Karlsson MO (2011) A fast method for testing covariates in population PK/PD models. AAPS J 13(3):464–472. https://doi.org/10.1208/s12248-011-9289-2
Savic RM, Karlsson MO (2009) Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J 11(3):558–569. https://doi.org/10.1208/s12248-009-9133-0
US Department of Health and Human Services Food and Drug Administration (1999) Guidance for industry—population pharmacokinetics. https://www.fda.gov/drugs/guidances-drugs/all-guidances-drugs. Accessed 4 June 2019
Konecny GE, Oza AM, Tinker AV, Oaknin A, Shapira-Frommer R, Ray-Coquard I, Aghajanian C, Coleman RL, O’Malley DM, Leary A, Chen LM, Provencher D, Ma L, Brenton JD, Castro C, Green M, Simmons AD, Beltman J, Harding T, Lin KK, Goble S, Maloney L, Kristeleit RS, McNeish IA, Swisher EM, Xiao JJ (2021) Population exposure-efficacy and exposure-safety analyses for rucaparib in patients with recurrent ovarian carcinoma from Study 10 and ARIEL2. Gynecol Oncol 161(3):668–675. https://doi.org/10.1016/j.ygyno.2021.03.015
Acknowledgements
Medical writing and editorial support funded by Clovis Oncology, Inc., were provided by Nathan Yardley and Stephen Bublitz of Ashfield MedComms, an Ashfield Health company.
Funding
The current analyses were funded by Clovis Oncology, Inc., and were designed in collaboration with the sponsor.
Author information
Authors and Affiliations
Contributions
MLG and JJX designed the population PK analyses. MLG and SCM conducted the analyses. All authors contributed to data analysis and interpretation. SG, HG, LM, ADS, JB, and TCH were involved in the design of the clinical studies. All authors contributed to drafting and critical revision of the manuscript for important intellectual content, approved the final submission draft, and take responsibility for the integrity of the data.
Corresponding author
Ethics declarations
Conflict of interest
MLG and SCM were employees of Certara and were paid contractors to Clovis Oncology in connection with the analysis and development of this manuscript. SG, HG, LM, ADS, JB, TCH, and JJX are employees of Clovis Oncology and may own stock or have stock options in that company.
Consent for publication
All authors gave consent for publication.
Ethics approval and consent to participate
For the studies that are included in this analysis, the institutional review board or ethics committee at each site approved the study protocols and patients provided written informed consent per the Declaration of Helsinki and Good Clinical Practice.
Availability of data and material
Requests for deidentified datasets for the results reported in this publication will be made available to qualified researchers following submission of a methodologically sound proposal to medinfo@clovisoncology.com. Data will be made available for such requests following online publication of this article and for 1 year thereafter in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data will be provided by Clovis Oncology. Clovis Oncology does not share identified participant data or a data dictionary.
Code availability
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Date of registration: Study 1014 (November 6, 2009), Study 10 (November 30, 2011), ARIEL2 (July 3, 2013), ARIEL3 (October 23, 2013), and TRITON2 (November 2, 2016).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Green, M.L., Ma, S.C., Goble, S. et al. Population pharmacokinetics of rucaparib in patients with advanced ovarian cancer or other solid tumors. Cancer Chemother Pharmacol 89, 671–682 (2022). https://doi.org/10.1007/s00280-022-04413-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-022-04413-7