
Abstract
Purpose
We compared the safety, toxicity, biocompatibility and anti-tumour efficacy of a novel chitosan-egg phosphatidylcholine (ePC) implantable drug delivery system that provides controlled and sustained release of paclitaxel (PTXePC) versus commercial paclitaxel formulated in Cremophor EL (PTXCrEL).
Methods
Toxicity studies were conducted in healthy CD-1 female mice, whereas efficacy studies were performed in the SKOV-3 xenograft model of ovarian cancer. Treatments consisted of intraperitoneal (IP) implantation of drug-free or PTXePC formulations, IP bolus PTXCrEL, or Cremophor EL (CrEL) vehicle. Toxicity was assessed as number of deaths, weight loss, serum hepatic enzyme levels and histopathological changes.
Results
Mice implanted with drug-free or PTXePC formulations did not exhibit observable toxicities, local inflammation or fibrous encapsulation of the implant. In contrast, mice receiving PTXCrEL or CrEL encountered significant toxicity, lethality, abnormal peritoneal organ morphology and hepatic inflammation. The maximum tolerable dose (MTD) of PTXCrEL was 20 mg/kg/week, whereas PTX doses of up to 280 mg/kg/week were well tolerated when administered as PTXePC. Enhanced anti-tumour efficacy was achieved with PTXePC in contrast to PTXCrEL with the same total dose of 60 mg/kg PTX.
Conclusions
The novel PTXePC formulation is a safer and better tolerated method for PTX administration, with significant increase in MTD and enhanced anti-tumour efficacy, suggesting improved therapeutic index with possible clinical implications in the treatment of ovarian tumours.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer is the second most frequent gynecological malignancy and the fifth leading cause of cancer-related deaths among women. Although initial chemotherapy results in a favourable response, the disease generally relapses within a short period of time. Because ovarian cancer is predominantly confined to the peritoneal cavity, investigators have probed the role of chemotherapy administration directly into the peritoneal cavity. This concept has been explored most extensively for over the past two decades, however only recently has it been clinically implicated and favoured [1].
Paclitaxel (PTX) is one of the gold standard chemotherapeutics in the treatment of ovarian cancer. Its main action is to inhibit cell proliferation by inducing a sustained mitotic block at the metaphase/anaphase boundary of the cell cycle. In this context, we hypothesized that localized and sustained intraperitoneal (IP) administration of PTX may result in improved ovarian tumour responsiveness. Recently, a novel chitosan-egg phosphatidylcholine (chitosan-ePC) implantable formulation has been developed and evaluated for localized and sustained delivery of PTX [4]. The implant is composed of poly (lactide) (PLA) nanoparticles loaded with PTX dispersed throughout a chitosan egg-phospatidylcholine (ePC) matrix and provides sustained release of PTX both in vitro and in vivo [4, 5, 17]. Therefore, the focus of our study was to compare the in vivo safety, toxicity, biocompatibility and efficacy of the novel chitosan-ePC implantable delivery system (PTXePC) versus the commercial Taxol ® formulation (PTXCrEL).
Materials and methods
Preparation of PTX chitosan-ePC delivery system
The chitosan-ePC implants were prepared as previously described [4]. Briefly, chitosan flakes were dissolved in a 1% (v/v) acetic acid solution. The lipid, ePC, was mixed with chitosan such that the chitosan : lipid ratio was 1:0.8 (wt/wt). For the drug-free films, the chitosan-lipid solution was poured into a teflon-coated dish and dried over five days. For PTX films, PLA nanoparticles containing 100 mg of PTX, purchased from Bristol-Myers Squibb (Montreal, QC, Canada) and 5 μCi of 14C-PTX from Moravek Biochemicals (Brea, CA, USA) were first prepared via a modified emulsification-diffusion method. To obtain a dry powder, the nanoparticles were dialyzed against distilled water for 24 h and then lyophilized in FreeZone® 6 Liter Freeze Dry System, Labconco Corporation (Kansas City, MO, USA). The nanoparticles were then resuspended in distilled water, mixed with the chitosan-ePC solution and dried in a teflon dish as described above. The final drug to material ratio for the PTXePC implant was 1:8 (wt/wt).
Animals
Healthy female CD-1 mice (4–6 weeks old, 25–30 g) were utilized for the conduction of the release, toxicity and biocompatibility studies and female CD-1 nude mice (4–6 week old, 18–20 g) were utilized for the anti-tumour efficacy studies, all purchased from Charles River (St. Constant, QC, Canada). All animal studies were conducted in accordance with the guidelines of the Canadian Animal Care Council. Animals were fed standard chow diet with water ad libitum and maintained on an automatic 12-h light cycle at 22–24°C.
In vivo PTX release studies from chitosan-ePC delivery system
In vivo release was assessed as previously described [5]. Briefly, PTXePC formulations were surgically implanted intra-peritoneally (IP) in healthy mice (n = 8), which were anesthetized under Isoflurane from Abbott Laboratories Ltd (Saint-Laurent, QC, Canada). Animals were housed in metabolic cages for 24-h periods over the course of 2 weeks. Feces and urine were collected at the end of each 24-h time period for PTX analysis. At the end of the study period, animals were sacrificed and the remainder of the implant system was weighed and solubilized for the determination of 14C-PTX by scintillation counting. In mice, approximately 50% of PTX is excreted in feces [15], therefore the total daily amount of PTX released from the implant was estimated based on the amount excreted in feces as well as on the remainder of PTX in the implant which was quantified at the end of the study period following euthanization.
Evaluation of chitosan-ePC delivery system
Mice (n = 4–8 per group) were anesthetized as above and surgically implanted IP under sterile conditions with PTXePC implants (20, 50, 100 and 200 mg, providing 35, 70, 140, 280 mg/kg/week of PTX, respectively) and with drug-free implants of similar sizes as controls. Drug-free implants remained in animals for 2, 3, 4, 12 weeks, whereas PTXePC implants remained for a period of 2, 3, 4 weeks.
Evaluation of commercial PTX formulation
The commercially available formulation of PTX, Taxol® (PTXCrEL), 6 mg/ml from Bristol-Myers Squibb (St-Laurent, QC, Canada) was diluted in sterile 0.9% sodium chloride solution for IP administration in a total volume of 300 μl. IP administration was chosen since this route is more relevant to the method of PTX delivery from the implant. Initially, different bolus PTXCrEL doses (15, 20, 25, 30 mg/kg) were administered IP to animals every other day (n = 3 per group) since this dosage has been previously reported as tolerable [7, 10, 12, 14]. However, this dosing regimen was not well tolerated in our experimental animals. Therefore, in order to mimic more clinically relevant dosing and minimize toxicity, bolus PTXCrEL was administered IP on a q7d × 3 schedule (15, 20, 25 and 30 mg/kg, n = 6–8 per group). CrEL and dehydrated ethanol (1:1, v/v) vehicle from Sigma Chemical Co. (St. Louis, MO, USA) was also administered as control in the same fashion (25–50 μl CrEL and 25–50 μl dehydrated ethanol (1:1, v/v) were diluted in 250–275 μl of 0.9% sodium chloride solution for a total of 300 μl IP bolus injection).
Toxicity assessment
Toxicity was assessed as number of deaths, weight loss, general appearance, serum hepatic enzyme levels and histopathological changes. Mice were monitored and weighed every other day. The maximum tolerable dose (MTD) was defined as the highest dose with < 15% body weight loss, not causing death, nor any prominent observable changes within one week of administration. Upon sacrifice, blood was collected by cardiac puncture. Briefly, toxicity was evaluated as follows: (a) lethal toxicity was defined as any death and/or body weight loss exceeding 15% in treated mice [lethal dose (LD)]; (b) the difference in mean body weight was calculated with respect to the beginning of treatment (day 1) as: (mean body weight on day x − mean body weight on day 1)/(mean body weight on day 1) × 100; (c) visual post-mortem inspection of peritoneal cavity was performed for any observations of macroscopic changes in organ morphology and the implantation site with surrounding tissues was examined for signs of infection, inflammation and fibrous encapsulation of the delivery system; (d) hepatotoxicity was assessed by the alanine aminotransferase (ALT) assay and histopathological analysis. Briefly, 500 μl of ALT reagent (Thermo Electron Corporation, Melbourne, Australia) was added to 50 μl serum, incubated at 37°C for a minute, following which the absorbance of the samples was read and recorded at 340 nm at times 0, 30, 60, 90 s. Peritoneal tissues, organs (liver, intestines, kidneys) and/or implants were harvested and fixed in 4% paraformaldahyde (PFA), paraffin embedded, sectioned at 5 μm and hematoxylin and eosin (H&E) stained for histopathological assessment.
In vivo efficacy studies
The anti-tumour activity of PTXePC was evaluated in a human tumour xenograft model representing serous adenocarcinoma, which accounts for 40–50% of all ovarian carcinomas and has an extremely poor prognosis [16]. Female CD-1 nude mice were injected IP with 1 × 107 cells of the human ovarian adenocarcinoma SKOV-3 cell line, obtained from American Type Culture Collection (Rockville, MD, USA), suspended in 200 μl of RPMI-1640 medium and 1% fetal bovine serum (Invitrogen, Burlington, ON, Canada). Initial tumour burden was assessed in a group of mice (n = 4) sacrificed on day 7 or day 14 following SKOV-3 inoculation and IP therapy was initiated on day 7 or 14. Two sets of separate studies were carried out for both treatment initiation times. Mice were divided into four groups (n = 6 per group) for each study: (1) control—no treatment; (2) drug-free chitosan-ePC implant (12–13 mg); (3) PTXePC implant—sustained treatment, providing 20 mg/kg/week for a total of 3 weeks (60 mg/kg total PTX); (4) PTXCrEL bolus administration on a q7d × 3 schedule at 20 mg/kg for a total of 60 mg/kg. CrEL vehicle groups were not included due to high toxicity. Mice were monitored every other day and weighed weekly. Animals were euthanized if endpoints were reached as governed by the Canadian Council of Animal Care. Intraperitoneal tumour burden was assessed semi-qualitatively as previously reported (9). Excised tumours were fixed in 4% PFA, paraffin embedded, sectioned at 5 μm for histopathological analysis. Efficacy was assessed as follows: % tumour weight inhibition (TWI) = 100 − (mean tumour weight (TW) treated/mean TW control) × 100.
Assessment of apoptosis
PTX induced apoptosis was assessed in paraffin tumour sections (5 μm) by terminal deoxytransferase-mediated dUTP nick-end labeling (TUNEL), followed by immunocytochemical staining with peroxidase-coupled antidigoxigenin antibody and diaminobenzidine, Apoptag kit (Intergen, Purchase, NY, USA). After light staining with hematoxylin, nuclei that stained brown were scored as positive for apoptosis and those that stained blue were scored as negative. At least five 200× microscopic fields were scored and the apoptotic index was calculated as the percentage of cells that were scored positive.
HPLC
For the efficacy studies, plasma and tumour concentrations of PTX were determined by reverse phase HPLC and UV detection. Briefly, 150 μl of plasma or tumour homogenate (0.3 g of tumour homogenized in 1.2 ml of water) was mixed with 15 μl of the internal standard docetaxel (10 μg/ml in acetonitrile) and evaporated under nitrogen airflow. To extract docetaxel and paclitaxel, 5.0 ml of tert-methyl butyl-ether was added and vortexed, followed by mechanical shaking for 10 min. The mixture was then centrifuged at 5,000 rpm for 10.0 min. The organic layer was transferred into another clean test tube and quickly evaporated under nitrogen. The residue was reconstituted in150.0 μl of HPLC mobile phase (50% ACN: 50% phosphate buffer pH = 10.0) and 100.0 μl was injected into the system, Agilent 1100 (Mississauga, ON, Canada). The HPLC stationary phase consisted of a Waters Xterra® MS C18 4.6 × 250 mm column, 5 μm particle size (Milford, MA, USA) protected by a 2.1 × 20 mm guard column (5 μm particle size). Samples were injected and eluted with the mobile phase consisting of acetonitrile −10 mM sodium phosphate buffer (pH = 10.0) (50:50, v/v) at a flow rate of 0.6 ml/min. Detection of PTX was at 227 nm, Waters Dual λ Absorbance Detector 2487 (Milford, MA, USA). The limit of detection for PTX was 5.0 ng/ml for plasma and 20.0 ng/g for tumor samples.
Statistical analysis
Results are presented as means ± SE. Data were analyzed using one-way ANOVA and unpaired Student’s t test for comparison between groups. Differences between groups were considered statistically significant at P < 0.05.
Results
Safety and biocompatibility of chitosan-ePC drug delivery system
Based on the total amount of PTX contained within the drug delivery system, the implant provided a sustained, zero-order release of 5.0 ± 0.5% PTX per day, which is consistent with our previous in vitro and in vivo findings [5, 17]. PTX doses of up to 280 mg/kg/week (40 mg/kg/day) were well tolerated when administered as PTXePC. The MTD and LD of the PTXePC implants could not be determined as the ethical limitation of implantable formulation size posed a constraint to the administration of larger doses. Mice implanted with the drug-free or PTXePC formulations appeared healthy and did not exhibit significant weight loss or mortality throughout the study period (Table 1, Fig. 1a). Upon sacrifice, visual and histopathological assessment revealed normal organ morphology and no signs of infection, inflammation, local irritation or fibrous encapsulation of the implant. No hepatotoxicities were observed in the drug-free and PTXePC treated animals, as they displayed normal serum ALT enzyme levels and normal liver histopathology (Table 1, Fig. 2a, b).

Effect of treatments on body weight. Mice (n = 3–8 per group) were treated with various PTX doses administered as PTXePC or PTXCrEL and with drug-free chitosan-ePC implants or CrEL vehicle as controls for 21 days. Body weight changes were monitored over the treatment period. Dosages indicated correspond to weekly PTX. a Chitosan-ePC delivery system—PTX and drug-free. No significant weight loss was observed in these groups, indicating that the system was well tolerated. b IP bolus PTXCrEL and CrEL vehicle. Significant weight loss (25%) was observed in the group receiving 60 mg/kg, indicating lethal toxicity. Note that 60 mg/kg corresponds to animals dosed with 15 mg/kg PTX every other day and that, mice treated with doses of 20 mg/kg every other day or 30 mg/kg/week had lethal toxicity and are not displayed. Data expressed as mean ± SE

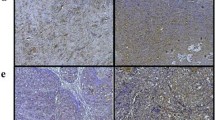
Histopathological assessment of hepatotoxicity. Mice (n = 6–8) were administered various doses of PTX as PTXePC or PTXCrEL, or drug-free chitosan-ePC or CrEL vehicle as controls for 21 days. At the end of the treatment period, livers were harvested, fixed (in 4% PFA), paraffin-embedded and sectioned (5 μm). a drug-free chitosan-ePC (20 mg implant); b PTXePC (70 mg/kg/week); c PTXCrEL (30 mg/kg/week); d CrEL (1 ml/kg/week), H&E stained sections of livers, 200× magnification
Safety and biocompatibility of commercial PTX formulation
Initially, 20 mg/kg of IP bolus PTXCrEL was administered to animals every other day since this dosage has been previously reported as tolerable [7, 10, 12, 14]. However, 67% (2/3) of deaths resulted following the first dose. Apathy and weight loss of 25% was also observed in the third animal after the fourth dose, which precluded further dosing. Mice treated with 15 mg/kg of IP bolus PTXCrEL every other day also resulted in 33% lethality, 25% body weight loss and hepatotoxicity (Fig. 1b). In order to mimic clinically relevant dosing schedules, IP bolus PTXCrEL was therefore administered at doses of 15, 20, 25 and 30 mg/kg on a q7d × 3 schedule. Lethal toxicity was observed at 25–30 mg/kg/week PTXCrEL as 37.5–67% mortality occurred following the first administration (Table 1). Moreover, all of these animals had signs of irritation within the peritoneal cavity with abnormal organ morphology (hypertrophy), hepatic inflammation and degeneration (Fig. 2c). Mice receiving 15 and 20 mg/kg/week PTXCrEL had extensive adipose tissue disposition within the peritoneal cavity and mild hepatic inflammation. Based on mortality, weight loss and histopathological changes, the maximum tolerated dose of IP bolus PTXCrEL was therefore, established to be 20 mg/kg/week.
CrEL toxicity
IP administration of CrEL (25–50 μl) resulted in signs of neurotoxicity, consisting of neuromotory symptoms (tremours, ataxia) in 100% of the animals and a significantly high number of deaths (75%, 6/8 animals) following the first administration. Abnormal peritoneal organ morphology (hypotrophy of intestines and liver) was observed upon sacrifice with widespread adipose tissue disposition within the peritoneal cavity. Moreover, extensive hepatotoxicity was detected in these animals since serum ALT levels were significantly elevated (323.0 ± 48.9 U/l) and histopathological analysis revealed severe hepatic inflammation, degeneration and fibrosis (Table 1; Fig. 2d).
Anti-tumour efficacy studies
Efficacy studies were performed in the SKOV-3 IP xenograft model of ovarian cancer. Tumours were not macroscopically evident on day 7 following SKOV-3 inoculation, however, macroscopic evidence was present at day 14; at this time point, mean initial tumour burden was established to be 0.04 ± 0.004 g in one set of studies and 0.57 ± 0.08 g in the other (i.e. two separate sets of studies). Nearing the end of the study period, control animals displayed signs of physical wasting, however, changes in body weight were counteracted by the formation of ascites fluid, which was indicated by heavy abdominal distention and confirmed upon sacrifice (∼1 ml of bloody fluid in 90% of tumour-bearing mice was observed). Large solid tumours (>1 mg) adhered loosely to the adipose tissue in the pelvic region, intestines and/or omentum, liver and spleen with many other smaller tumour masses throughout the peritoneal cavity and/or diaphragm. Complete tumour inhibition was achieved with both formulations, PTXePC and PTXCrEL, when treatment was initiated on day 7 post-SKOV-3 inoculation. However, when treatment was commenced on day 14 following SKOV-3 inoculation, complete tumour responsiveness was only achieved with PTXePC as opposed to partial responsiveness (47%) with PTXCrEL when initial tumour burden was 0.04 ± 0.004 g (Table 2; Fig. 3). When treatment was initiated with heavier initial tumour burden (0.57 ± 0.08 g) on day 14 following SKOV-3 inoculation, 74% tumour reduction was observed with PTXePC, whereas no significant inhibition was seen with PTXCrEL (Table 2; Fig. 3). About 50% of the PTXCrEL-treated animals displayed autonomic neurotoxicity consisting of impaired intestinal motility as observed upon sacrifice. PTX plasma and tumour concentrations are summarized in Table 2.

PTXCrEL and PTXePC anti-tumour efficacy in SKOV-3 ovarian cancer xenograft model. Human ovarian tumours were induced by the SKOV-3 cell-line in female CD-1 nude mice. Treatment with PTXePC (20 mg/kg/week for a total of 60 mg/kg) or IP bolus PTXCrEL on q7d × 3 schedule at 20 mg/kg for a total of 60 mg/kg was initiated 14 days post SKOV-3 inoculation, with drug-free chitosan-ePC implants or no treatment as controls. Control represents combination of both untreated and drug-free implant groups. When tumour burden was 0.04 ± 0.004 g at time of treatment initiation, ANOVA analysis revealed significant difference between groups, P = 0.001 (*). However, when tumour burden was 0.57 ± 0.08 g at the time of treatment initiation, the only significance was found between the control and PTXePC group, student t test, P = 0.03 (**). Data presented as mean ± SE
Apoptosis
Figure 4 illustrates apoptotic cells in cancer tissue obtained from SKOV-3 inoculated mice that received PTXCrEL or PTXePC therapy or no treatment. Approximately 85% of cells were apoptotic in the PTXePC treated tumours, whereas only about 4% of apoptotic cells were observed in the PTXCrEL groups (Table 2).

Effect of PTXCrEL and PTXePC treatment on apoptosis. Tumours were obtained from the various treatment groups upon termination of the study and the TUNEL assay was performed to assess apoptosis. Nuclei stained brown scored as positive for apoptosis and those stained blue scored as negative. Representative tumour sections: a untreated control; b PTXCrEL; c PTXePC (treatment commenced on day 14 following SKOV-3 inoculation with initial tumour burden of 0.57 ± 0.08 g). Five micrometre paraffin sections, 200× magnification
Discussion
Overall results from our studies demonstrate that the recently developed implantable PTXePC formulation provides sustained and localized release of PTX, is less toxic and more biocompatible than commercially available PTXCrEL. This novel formulation was capable of providing PTX doses of up to 280 mg/kg/week in mice, with limited toxicity and no signs of adverse effects, thereby significantly increasing the MTD. Furthermore, this biodegradable and biocompatible system displayed superior anti-tumour efficacy in comparison to PTXCrEL in the SKOV-3 induced human ovarian adenocarcinoma xenograft model.
PTX tolerability varies in experimental animals [7, 10, 12, 14]. In our studies, the MTD for PTXCrEL was established to be 20 mg/kg/week based on weight changes and lethality, although at this dose the majority of animals still displayed some signs of apathy, temporary respiratory distress and mild hepatic inflammation. Mice subjected to higher doses displayed abnormal peritoneal organ morphology, significant lethality and hepatotoxicity. The severity of hepatic inflammation and degeneration escalated with increasing PTXCrEL dose. Therefore, this limited the maximum dose which could be used in our efficacy studies. In contrast, the chitosan-ePC formulation was capable of delivering PTX doses of up to 280 mg/kg/week, thereby significantly exceeding the MTD for PTX by more than 14-fold. No observable toxicities, nor indications of pathological changes were detected, suggesting that the toxicities observed in the PTXCrEL treated animals were due to CrEL and not PTX. Likewise, Abraxane (ABI-007), a recently FDA-approved injectable CrEL-free PTX formulation, has demonstrated significantly reduced neutropenia and hypersensitivity reactions and superior efficacy in breast cancer in comparison to standard PTXCrEL, again suggesting that the observed toxicities are accredited to CrEL [3, 8].
Although the drawbacks of CrEL as a PTX solvent are recognized, we found limited information on CrEL toxicity alone. Therefore, we examined the effects of CrEL itself in conjunction with the PTXCrEL formulation. Our observations indicate that CrEL is poorly tolerated and severely toxic in mice. CrEL administration resulted in severe hepatotoxicity as indicated by elevated serum ALT levels, which is a sign of hepatocyte membrane damage. Furthermore, histopathological analysis of liver sections revealed severe inflammation, fatty acid infiltration, necrosis and fibrosis, indicative of hepatic injury and degeneration. To date the effects of CrEL on hepatotoxicity have not been reported. As PTX undergoes extensive hepatic metabolism, hepatotoxicity induced by CrEL may alter PTX metabolism, resulting in reduced PTX clearance, elevated systemic PTX and increased PTX-associated toxicities. Since CrEL is utilized clinically, it is important to address these CrEL-induced pathophysiological manifestations. Although a number of other CrEL-free injectable formulations have been developed, such as albumin nanoparticles, polyglutamates, emulsions, liposomes, taxene analogues and pro-drugs, none of them have been reported to provide both sustained and localized intraperitoneal delivery of PTX for the treatment of ovarian cancer.
At equivalent doses, our efficacy studies demonstrated enhanced tumour responsiveness with PTXePC in comparison to PTXCrEL in the SKOV-3 xenograft model of ovarian cancer. Tumour burden at the time of treatment initiation appeared to be an important factor in the determination of responsiveness. When treatment was commenced on day 7 following SKOV-3 inoculation and no macroscopic evidence of tumours was present at this time point, complete tumour inhibition was achieved with both PTXePC and PTXCrEL. However, when treatment was initiated at a later date and macroscopic evidence of tumours was present (day 14 following SKOV-3 inoculation), significantly improved tumour responsiveness was achieved with PTXePC in comparison to PTXCrEL. Because tumours are heterogeneous in nature, and are composed of areas of edema and necrosis, macroscopic measurements of tumour mass alone are not always the most accurate marker of efficacy, therefore microscopic evaluation of tumours is required to improve assessment of efficacy. In this context, we observed a significantly extensive degree of apoptosis in tumours obtained from the PTXePC treatment group in comparison to PTXCrEL, indicating that PTXePC therapy was more effective. This enhanced tumour responsiveness with PTXePC may be a result of the combination of both regional and continuous drug administration. As ovarian tumours are predominantly confined to the peritoneal cavity, local administration would allow for high PTX concentrations directly at the tumour site, while alleviating systemic toxicities [2, 13]. Indeed, significantly higher PTX concentrations were observed in tumour tissues than plasma in the PTXePC treated groups. Although to date, there are various reports on IP therapy, this strategy was never clinically implicated as a gold-standard treatment method, until recently, in light of the latest clinical trial, the National Cancer Institute announced that IP chemotherapy is now the preferred method of treatment for advanced ovarian cancer [1].
In addition, along with the benefits of localized administration, sustained drug delivery may also enhance efficacy as duration of drug exposure is a critical factor for tumour penetration. In this regard, it has been shown that prolonging PTX exposure time enhances its cytotoxic effects [6, 11]. Indeed, we observed dramatically higher PTX concentrations in the tumours obtained from the PTXePC-treated groups, which is also reflected by the significantly higher degree of apoptosis occurring in these tumours versus PTXCrEL. PTX exerts its cytotoxic effects mainly though apoptosis, therefore continuous exposure to PTX steadily increases the proportion of apoptotic cells [9]. In fact, it has been previously reported that in 3-dimensional breast cancer histocultures only limited penetration of PTX occurs following a 4 hour exposure time, however, prolonged PTX exposure (48 h) resulted in uniform PTX distribution throughout the 3-dimensional histoculture and increased apoptosis [6]. Therefore, bolus PTXCrEL dosing may not allow for sufficient tumour penetration as reflected in our efficacy studies. In addition to local PTX delivery, PTXePC is also capable of targeting vascularized tumour tissue, since PTX levels were also detected in plasma. However, plasma concentrations were below detection limits in PTXCrEL treated animals, likely due to drug-free intervals and rapid drug clearance. Thus it is possible that only minimal populations of tumour cells are killed with each PTXCrEL treatment as tumour penetration is limited. Moreover, sustained exposure to PTX may enhance the therapeutic index by providing constant therapeutic drug levels which may in turn decrease adverse events associated with peak plasma concentrations. Overall our studies demonstrated enhanced efficacy with equivalent doses of PTXePC, possibly as a result of direct and sustained drug delivery. In addition, it is important to note that as the MTD was dramatically increased, an even greater degree of efficacy may be achieved when PTX doses are further increased.
Conclusions
The PTX possesses anti-tumour activity for a variety of solid tumours, however, its current formulation in CrEL appears to limit its therapeutic potential due to toxicities. Additionally, maintaining cytotoxic concentrations of PTX may not occur with conventional dosing, therefore a novel approach is required in order to improve PTX efficacy. Our novel chitosn-ePC formulation is a more tolerable, biocompatible and safer method for PTX administration, capable of providing higher dosages without adverse effects. Furthermore, this delivery system demonstrated greater therapeutic efficacy in the human SKOV-3 ovarian cancer xenograft model. Although additional studies are necessary in order to fully characterize the nature of tumour uptake, biodistribution and cytotoxic mechanism of the PTXePC drug delivery system, these initial findings demonstrate superior tolerability and therapeutic efficacy. Hence further investigation of the chitosan-ePC delivery system is warranted in order to assess its possible clinical use in the treatment of ovarian cancer.
References
Armstrong D, Bundy B, Wenzel L, Huang H, Baergen R, Lele S, Copeland L, Walker J, Burger R, Group GO (2006) Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 354:34-43
Dedrick RL, Myers CE, Bungay PM, DeVita VTJ (1978) Pharmacokinetic rationale for peritoneal drug administration in the treatment of ovarian cancer. Cancer Treat Rep 62:1–11
Gradishar WJ, Tjulandin S, Davidson N, Shaw H, Desai N, Bhar P, Hawkins M, O’Shaughnessy J (2005) Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol 23:7794–7803
Grant J, Blicker M, Piquette-Miller M, Allen C (2005) Hybrid films from blends of chitosan and egg phosphatidylcholine for localized delivery of paclitaxel. J Pharm Sci 94:1512–1527
Ho EA, Vassileva V, Allen C, Piquette-Miller M (2005) In vitro and in vivo characterization of a novel biocompatible polymer-lipid implant system for the sustained delivery of paclitaxel. J Control Release 104:181–191
Jang SH, Wientjes MG, Au JL (2001) Determinants of paclitaxel uptake, accumulation and retention in solid tumors. Invest New Drugs 19:113–123
Le Garrec D, Gori S, Luo L, Lessard D, Smith DC, Yessine MA, Ranger M, Leroux JC (2004) Poly(N-vinylpyrrolidone)-block-poly(d,l-lactide) as a new polymeric solubilizer for hydrophobic anticancer drugs: in vitro and in vivo evaluation. J Control Release 99:83–101
Micha JP, Goldstein BH, Birk CL, Rettenmaier MA, Brown JVr (2006) Abraxane in the treatment of ovarian cancer: the absence of hypersensitivity reactions. Gynecol Oncol 100:437–438
Mori T, Kinoshita Y, Watanabe A, Yamaguchi T, Hosokawa K, Honjo H (2006) Retention of paclitaxel in cancer cells for 1 week in vivo and in vitro. Cancer Chemother Pharmacol (Epub ahead of print)
Nicoletti MI, Lucchini V, D’Incalci M, Giavazzi R (1994) Comparison of paclitaxel and docetaxel activity on human ovarian carcinoma xenografts. Eur J Cancer 30A:691–696
O’Shaughnessy JA, Fisherman JS, Cowan KH (1994) Combination paclitaxel (Taxol) and doxorubicin therapy for metastatic breast cancer. Semin Oncol 21:19–23
Polizzi D, Pratesi G, Tortoreto M, Supino R, Riva A, Bombardelli E, Zunino F (1999) A novel taxane with improved tolerability and therapeutic activity in a panel of human tumor xenografts. Cancer Res 59:1036–1040
Rowinsky EK, Donehower RC (1995) Paclitaxel (taxol). N Engl J Med 332:1004–1014
Sharma D, Chelvi TP, Kaur J, Chakravorty K, De TK, Maitra A, Ralhan R (1996) Novel Taxol formulation: polyvinylpyrrolidone nanoparticle-encapsulated Taxol for drug delivery in cancer therapy. Oncol Res 8:281–286
Sparreboom A, van Tellingen O, Nooijen WJ, Beijnen JH (1996) Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer Res 56:2112–2115
Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K (2000) Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 88:2584–2589
Vassileva V, Allen C, Piquette-Miller M (2004) Release profile characterization of a novel implantable paclitaxel loaded lipid polymer system in cd-1 mice. AAPS Journal 6
Acknowledgments
The authors would like to thank Mr. Ji Zhang for his technical assistance. This work was supported by grants from the Ontario Cancer Reseach Network and the National Cancer Institute of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vassileva, V., Grant, J., De Souza, R. et al. Novel biocompatible intraperitoneal drug delivery system increases tolerability and therapeutic efficacy of paclitaxel in a human ovarian cancer xenograft model. Cancer Chemother Pharmacol 60, 907–914 (2007). https://doi.org/10.1007/s00280-007-0449-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-007-0449-0