
Abstract
In acute myeloid leukemia (AML), mutations of the Fms-like tyrosine kinase 3 receptor (FLT3) and its overexpression are related with hyperleukocytosis, higher risk of relapse, and decrease of both disease-free survival and overall survival. It has been suggested that this phenomenon confers proliferative and survival advantages to the malignant blast cells. As a consequence, it is an attractive therapeutic target. As the best treatment strategy for mutated FLT3 AML remains to be defined, the addition of FLT3 inhibitor drugs to chemotherapy or to the bone marrow transplant approach has become a growing strategy. With encouraging results, this combination seems to be an attractive option. Relevant data regarding the current treatment trends on mutated FLT3 AML is reviewed here.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Regardless of the increased molecular comprehension of acute myeloid leukemia (AML), there have been few advances in its treatment [1]. Mutations of Fms-like tyrosine kinase 3 receptor (FLT3) and its overexpression have been identified as ominous markers in this disease and are related with hyperleukocytosis, higher risk of relapse, and decrease of both disease-free survival (DFS) and overall survival (OS) [2].
Wild-type FLT3 (FLT3-WT) is a cell membrane receptor expressed in primitive hematopoietic elements and is required for the activation of diverse cell survival pathways. Naturally, it is activated by the FLT3 ligand, which is produced by neighboring cells, progenitor cells, and by the microenvironment [3]. In 1996, Nakao et al. identified a tandem mutation in the internal juxtamembranal region of the receptor (FLT3-ITD), that drives a constitutive activation and the amplification of the FL-induced signal [3, 4]. The FLT3 gene is located on chromosome 13q12 and encodes the FLT3 tyrosine kinase receptor. Activating mutations including internal-tandem duplications (ITDs) of the juxtamembranal region (head to tail duplication of 3–400 base pairs in exons 14 or 15), tyrosine kinase domain 1, and mutations involving the D835/I836 residues and others of the tyrosine kinase domain (TKD) [47••] are identified up to 30% of adults [3] and 16.5% of children with AML [5, 6].
Driven by the success of BCR-ABL tyrosine kinase inhibition in Chronic Myeloid Leukemia (CML) [7], diverse FLT3 inhibitor drugs (FLT3ID) with spectrum against FLT3-WT, FLT3-ITD, and FLT3-TKD, have been tested [8]. Some of these drugs were initially assayed in solid malignancies of kidney, liver, pancreas, and lung, and subsequently tested in AML with aberrant FLT3 expression. As monotherapy, they all have shown a variable, limited, and brief activity. Thereafter, some of them have been integrated in combination schemes with either conventional chemotherapy, hypomethylating drugs, protein-synthesis inhibitors, or in the context of bone marrow transplant (BMT) [9, 10••, 11, 12, 13].
Conventional Chemotherapy in AML with Mutant FLT3
The results of conventional chemotherapy regimens used in patients with mutated FLT3 AML are greatly unsatisfactory although the complete responses (CR) are comparable to the CR achieved in cases with FLT3-WT (79.6%). A difference exists with an increased relapse risk (25.6% FLT3-WT vs 75% FLT3 ITD) and decreased DFS and OS [14, 15]. Nevertheless, in the >60-year-old group, this variable has not worsened the results obtained when conventional chemotherapy alone is used [16].
In an effort for improving the conventional induction regimen results, the Eastern Cooperative Oncology Group E1900 study explored Daunorrubicin 60 vs. 90 mg/m2 in the first induction in AML patients younger than 50 years old, and albeit the study was prematurely closed because of an excessive mortality risk, an intention-to-treat reanalysis found out that patients with FLT3-ITD were benefited by the 90 mg/m2 dosage with better DFS and OS when compared to the lower dosage group (DFS 45 vs. 33% and OS 54 vs. 34%). This finding suggests an advantageous role for a 90 mg/m2 dose in younger FLT3-ITD AML patients, particularly in those with a higher allele burden [17].
The clinical trials that explore the efficacy of conventional chemotherapy in mutated FLT3 AML are scarce. In this setting for a period of 15 years, the MD Anderson group studied the evolving therapeutic strategies that include the incorporation FLT3ID to conventional chemotherapy regimens. In 173 patients treated with conventional chemotherapy alone compared with 51 patients receiving a combination regimen that included FLT3ID, the CR plus CR with incomplete platelet recovery (CRp) achieved was 67 vs. 72.5% (p = 0.45). Notably, the OS showed a clear improvement throughout the time, with a rise from 9.6 months in 2000 to 17.8 months in 2014. This was probably due to the better therapeutic strategies used, including, but not limited to the FLT3ID in combined regimes and the increased usage of BMT [18•].
Tyrosine Kinase Inhibitors against FLT3
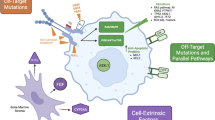
In AML, it is established that FLT3 mutations FLT3-ITD/FLT3-TKD or the overexpression of FLT3-WT confers proliferative and survival advantages to the malignant blast cells [19]. This has turned the receptor in a therapeutic target [20]. With a vision driven by the development and success of the tyrosine kinase inhibitors (TKI) used in CML, TKIs with spectrum against FLT3 have been studied [7]. Structurally, the majority of these inhibitor drugs are heterocyclic compounds that competitively inhibit the ATP-binding site in the ATP-binding pocket of the TKD. Functionally, these are multi-kinase inhibitors. As with other drugs, FLT3ID can bind to the active, inactive, and transitional state of FLT3, causing cell-cycle arrest, inhibiting proliferation and inducing apoptosis in a variety of FLT3 dependent cell lines (Fig. 1 ) [22•]. In AML early clinical trials, FLT3ID showed a not sustained and not robust response against FLT3 [16].

Schematic representation of the wild type FLT3 receptor activation by ATP and downstream stimulation of proliferative and blocking apoptosis pathways. Also, FLT3-ITD and FLT3-TKD mutant blockage by FLT3-ID and its negative effect on proliferation and anti-apoptosis pathways are shown. FL indicates FLT3 ligand; ITD internal tandem duplication; TKD 835 tyrosine kinase domain 835 mutation
These drugs are characterized by a relatively wide spectrum of inhibition to different kinases, phenomenon that in clinical trials is associated with an incidence of several adverse effects. Although theoretically, this property would make them more able to inhibit other survival and/or proliferative pathways; in acute leukemia, this biological advantage has not been demonstrated. Actually, two generations of FLT3ID have been categorized; meanwhile, additional inhibitors are in active investigation. Each generation has different specificity, inhibition spectrum, potency, and adverse effects [8]. The first-generation FLT3ID were originally developed with other tyrosine kinases in mind, and most require a half-maximum inhibitory concentration (IC50) above 400 nM [23•]. Second generation inhibitors have been designed specifically against FLT3 with a reduced IC50 and a minor impact in other kinases.
Selected First Generation Inhibitors
Sunitinib
First FLT3ID assayed in 2005, belongs to the first generation and is a derivative of indolinone. It has unspecific activity against FLT3-ITD and FLT3-TKD, also against KIT, PDGFR, and KDR [8, 24].
Monotherapy
At doses of 50 and 75 mg for 4-week cycles, followed by a 2-week rest period in 2005, a clinical trial evaluated Sunitib as monotherapy in 16 primary or secondary relapsed/refractory AML advanced age patients, 4 of them with mutated FLT3 (ITD and TKD), with a median of 72 years old, and no candidates for induction chemotherapy. Partial responses were achieved, and best responses occurred in patients with mutated FLT3. The responses were short-sustained with a maximum of 16 weeks. The 75-mg trial was suspended for an excessive toxicity [24].
Combined Therapy
In 2015, a Phase I/II Trial, Sunitinib efficacy, was studied in 22 de novo and untreated mutated FLT3 (FLT-ITD and FLT3-TKD) AML patients older than 60 years old. They received one or two induction cycles with intermediate doses of Cytarabine plus standard doses of Daunorrubicin, and then three consolidation cycles with intermediate doses of Cytarabine were added. Sunitinib 25-mg dose was administered on days 1–7 of each chemotherapy cycle and posterior to the consolidation cycles as maintenance. CR/CR incomplete (CRi) were attained in 59% (13 of 22 evaluable patients in Phase II). The principal observed toxicities were myelosuppression and associated fever [25].
Midostaurin
Compound derived from staurosporin, an alkaloid from Streptomyces staurosporeys. It is a multitargeted kinase, against FLT3-ITD, FLT3-TKD, and FLT3-WT as well as PKC, PDGF alpha and beta, Cdk1, Src, Fgr, Syk, c-KIT, and VEGF. Its IC50 for FLT3ITD is less than 10 nM [26].
Monotherapy
One of the first clinical trials with Midostaurin was run in 20 advanced age patients with relapsed/refractory mutated FLT3 AML or high risk myelodysplastic syndrome (MDS), who received the drug as monotherapy at 75 mg three times in a day. Two fatalities occurred because of pulmonary events of uncertain etiology. With a median of 13 weeks, the drug response was discrete and of short duration with a reduction in peripheral blood blast count in 70% of the patients. The inhibition of autophosphorylation mediated by FLT3 was documented in most of the responding patients, in whom the in vivo activity of the drug could be noted. This opened a new path for diverse clinical trials with combination therapy [27].
Combined Therapy
So far, one of the most extensive clinical trials is the international CALBG10603/RATIFY study with a cohort of 3279 newly diagnosed AML adult patients, 717 were identified with FLT3-ITD or FLT3-TKD, in whom conventional chemotherapy induction was followed by a high dose Cytarabine plus Midostaurin or Placebo, and then maintenance therapy with orally administered Midostaurin or Placebo was given.
With a median follow-up of 57 months, CR was 59% for the Midostaurin arm vs. 54% in the Placebo arm (p = 0.18). OS was 74.7 vs. 26 months respectively (p = 0.007); DFS was 8 vs. 3 months (p = 0.004). Notably, there was no significant difference on grades 3 or 4 therapy related adverse effects. Thereby, these results suggest that in AML patients with mutated FLT3 regardless of the allelic burden, Midostaurin can be combined with conventional chemotherapy in a secure manner and allows to achieve better results compared when it is not administered [10••].
Lestaurtinib
It is an orally bioavailable indocarbazolic alkaloid, derived from K-252a, an indocarbazole proceeding from a culture broth composed by Nocardiopsis sp. and Nonomuraea longicatena [21]. In 2001, Levis et al. demonstrated its potent inhibitory properties in leukemia cell lines. In vitro experiments showed that Lestaurtinib inhibits autophosphorylation of the receptor in a dose-dependent manner, with an IC50 of 2–3 nM. Additionally, BFa3 cells with Asp835 mutation were inhibited with a similar IC50. Recent data suggests that this FTL3ID has activity against JAK2-WT and JAK2-V617F [28].
Monotherapy
Lestaurtinib 60 mg twice a day dose was evaluated in a clinical trial that included 14 relapsed/refractory high risk mutated FLT3 AML patients. Five patients had clinical evidence of biological activity and measurable clinical response, including significant reductions in peripheral blood and bone marrow blast cells. Minimal toxicity was found [29].
Combined Therapy
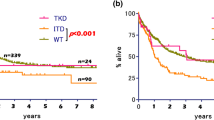
Derived from UK MRC AML15 and UK NCRI AML1 studies, a randomized clinical trial involving 500 mutated FLT3 AML adult patients, evaluated Lestaurtinib vs. Placebo. As a first line treatment for a 5-year period, Lestaurtinib or Placebo was added to conventional chemotherapy. Lestaurtinib dosage was 80 mg two times a day following each chemotherapy cycle for up to 28 days during 4 cycles. With a median age of 49 years, 381 patients had FLT3-ITD, 127 had FLT3-TKD, and 2 had both mutations. There was no difference in remission achieved between the two arms with CR/CRi of 92% in Lestaurtinib vs. 94% in control group. Five years DFS was 40 vs. 37% in the control group, and OS was 46 vs. 43% in the control group without significant difference. Yet, a variable analysis identified a patient subgroup with azoles concomitant administration (which has been proven to significantly raise lestaurtinib plasma levels) and Gentuzumab Ozogamizin who achieved an OS of 61% [28].
Selected Second Generation Inhibitors
Quizartinib
As a member of the second-generation FLT3ID, Quizartinib has a specific activity against FLT3, with an IC50 of 1 nM. It is 25 times more potent than other inhibitors. In mutated FLT3, it has shown an inhibition capacity against FLT3-ITD but not over FLT3-TKD. It also has an activity on KIT, PDGF alpha, PDGF beta, and CD1FR [8, 23•].
Monotherapy
The first clinical trial on refractory/relapsed AML patients was a Phase I carried out by the MD Anderson group (explored doses 12 to 450 mg daily). It included 76 adult patients with mutated FLT3 (n 17), indeterminate FLT3 status (n 22), and FLT3 negative (n 37). Patient’s median age was 60 years and had three previous therapy lines. Thirty percent of them achieved a response, including 13% CR and 17% partial responses. Responses were attained in 53% of 17 mutated FLT3 patients (n 9), 41% of the 22 indeterminate FLT3 status group (n 9), and 14% of the 37 negative FLT3 patients (n 5). The median response duration was 13.3 weeks, and the median OS was 14 weeks. Drug associated adverse events included nausea 16% and QT prolongation 12%. The best-tolerated dose was 200 mg/day with toxicity limiting dose for QT prolongation [31].
Combined Therapy
A Phase I clinical trial in elderly AML patients explored Quizartinib in combination with conventional chemotherapy. Fifty-five adult patients with de novo AML were enrolled, only 4 of them had FLT3-ITD. Six cohorts with scaled doses of 60, 90, or 135 mg were categorized. Drug was administered for 7 or 14 days with a toxicity limiting dose restriction. A regimen of 40 mg/day for 14 days resulted from a de-escalation cohort due to grades 3 and 4 adverse events. Quizartinib for a 14-day period was given sequentially 2 days after each of the two courses of Cytarabine, Daunorrubicin, and Etoposide combined therapy followed by one course of Daunorrubicin/Cytarabine (2 + 5). CR was achieved in 33 of the 42 evaluable patients (79%). Reported OS is 15 months. Authors concluded that Quizartinib at 40 mg for 14 days can safely be given sequentially after chemotherapy in older patients with newly diagnosed AML [30].
Another trial tested Quizartinib integrated to a salvage chemotherapy regimen in pediatric AML patients. A dose of 60 mg/m2/day was studied, being administered 5 days after intensive chemotherapy with 17 evaluable patients. 2CR, 1CRp, and 10 stable diseases were achieved. The near complete inhibition of autophosphorylation was evident in all patients. The drug was more efficient in FLT3-ITD than FLT3-WT [32].
Sorafenib
It is a multi-kinase inhibitor with spectrum against RAF-1, VEGFR, PDGFR, KIT and FLT3 [33]. Its clinical efficacy in AML has been explored in diverse scenarios: as monotherapy, in combination with chemotherapy during induction, consolidation and as maintenance in the post-BMT setting [16].
Monotherapy
Sorafenib was studied for the first time in 16 refractory/relapsed AML patients (7 FLT3-WT, 6 FLT3-ITD, 1 FLT3 TKD/ITD and 2 FLT3-TKD) treated in a Phase I trial. Patients were randomly assigned to receive Sorafenib in 21-day cycles of 5 days per week (n = 7 patients; arm A) or of 14 days (n = 9 patients; arm B). In both arms, the starting dose level was 200 mg twice daily. Subsequent dose levels were 600, 800 and 1200 mg daily in cohorts of three subjects at each dose level. No limiting toxicity dose was seen. Efficacy was only limited to a transitory blast reduction both in peripheral blood and bone marrow in all FLT3-ITD patients [34].
Combined Therapy
A Sorafenib Phase I/II Clinical Trial in 51 untreated, younger than 65 years AML patients, evaluated the feasibility of administering 400 mg of the drug twice a day on days 1 to 7 in combination with a continuous infusion of intermediate dose Cytarabine for 3 to 4 days plus Idarrubicin at conventional dosage for 3 days as induction chemotherapy. CR was found in 75%, nevertheless when the group was divided regarding the mutational status of FLT3, 14 of the 15 patients with FLT3-ITD achieved a CR of 94% (the 15th patient achieved a CRp), whereas the FLT3-WT patients obtained a 66% of CR. With a median follow-up of 54 weeks, the survival probability in 1 year was estimated in 74% [14].
FLT3ID (Midostaurin) Plus Hypomethylating Agents
The combination of Midostaurin with hypomethylating agents has also been explored. In a clinical trial that included a preclinical phase, AML cellular cultures were exposed to Decitabine and Midostaurin. The combination proved to be effective in vitro on cell lines that expressed the FLT3-ITD mutation. This strategy was translated into a clinical phase to a group of 16 newly diagnosed elderly or relapsed/refractory adult patients with AML, from which only 13% had FLT3-ITD. Nevertheless, the concomitant administration of drugs resulted in unacceptable pulmonary and cardiac toxicities; consequently, it was found that the best sequential administration was Decitabine followed by Midostaurin. In an intention-to-treat analysis, 75% of patients achieved a stable disease as a minimum and 25% achieved CR. These results require additional studies to evaluate their security in advanced age patients [35].
The MD Anderson group evaluated Midostaurin in combination with Azacitidine in 54 AML and high-risk MDS patients. They established two cohorts of patients with Midostaurin doses of 25 and 50 mg in combination with Azacitidine at a fixed dose of 75 mg/m2. Overall response at a median of 12 weeks was 26%. Responses occurred in 33%. Patients with FLT3 mutations but not previously transplanted or exposed to other FLT3 inhibitors achieved the greatest benefit. Toxicity was notorious because G3–4 non-hematological toxicity was reported in 38 (70%) patients, most frequently infections (56%), ejection fraction reduction (11%), and diarrhea or nausea/vomiting (9% each) [13].
Resistance to FLT3ID
Diverse mechanisms involved in the acquired resistance to FLT3ID have been described. Notoriously, punctual mutations in the binding loop and in the activation domain, amplification of the mutated receptor, co-expression of FLT3-WT and utilization of alternative pathways to cellular survival have been reported [36•, 37, 38, 39, 40]. The MD Anderson recently found that 22% of the patients previously exposed to FLT3ID, developed punctual mutation of the TKD [48]. In this setting, Crenolanib has been identified as a potent FLT3ID able to overcome resistance induced by punctual mutations in D835 of the mutated FLT3. Besides, it has a narrow spectrum minimizing the effect on erythropoiesis; these data could position Crenolanib as a potential effective and less myelotoxic drug [41•].
BMT in Mutated FLT3 AML
In FLT3 mutated AML patients, BMT role is not totally elucidated, although several publications support its use [42, 43, 44]; others have not shown an improved outcome [45].
Brunet et al. evaluated the repercussion of FLT3-ITD mutation and a “normal” karyotype in AML patients who underwent allogeneic BMT in first CR. They studied 206 patients; 120 (58%) presented FLT3-ITD; and among these, two had a primary graft failure. When compared to the negative mutated FLT3 group, no difference in the rate of acute or chronic graft vs. host disease was noted. At 2 years, a superior relapse accumulated incidence (30 vs. 16% p = 0.006) in the group of FLT3-ITD patients was observed. Other factors that favored a higher trend to relapse were patients older than 42 years, more than one chemotherapy course before achieving CR, shorter interval between CR and BMT, and female gender. At 2 years, DFS was lower in FLT3-ITD patients (58 vs. 71% p = 0.04) [46].
In this scenario, the integration of Sorafenib 400 mg twice a day as a maintenance therapy following allogeneic BMT was proved on 22 patients in first or second CR. The drug was started between 45 and 125 days after BMT. One year follow-up DFS was 85%, and OS was 95% with an OS of 100% of patients in first CR, with just one case of relapse [11]. When compared with trials limited to BMT alone [46], the results seem encouraging.
Following BMT, FLT3-ITD AML relapses carry a poor prognosis. Sorafenib has proven to be useful also in this scenario. De Freitas et al. carried out a retrospective trial of Sorafenib in 13 adult patients. Sorafenib was used as monotherapy or in association with hypomethylating agents or T cell donator infusion. Hematological response was documented in 12 of the 13 patients (92%), with 5 (38%) achieving a CR in BM. Manageable adverse effects were presented, being the most common severe cytopenias and hand-foot syndrome. The OS at day 100 from relapse was 65% and at 1 year 22%. These findings suggest that Sorafenib could represent a future therapeutic option for patients with relapse after BMT, for bridging them to an additional transplant [47••].
Conclusions
When AML is treated with conventional chemotherapy, mutated FLT3 confers a poor prognosis. Its identification has driven to the development of a family of drugs capable of inhibiting its expression. Used as monotherapy, these drugs have limited and brief anti-leukemic activity. Their best contribution has been proved in combination with sequential chemotherapy or as maintenance after BMT (Table 1 ); in this setting, the most promising inhibitors are Midostaurin and Sorafenib.
As a predictable phenomenon, emergence of resistance to FLT3ID has already been identified. Other FLT3ID as Crenolanib are capable to overcome this condition and are being actively explored.
Until now, the best therapeutic strategy to treat mutated FLT3 AML remains to be defined. Nevertheless, available data indicates that in young patients and in the post-BMT setting, integrating FLT3ID to treatment strategies is beneficial, but further research is required.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Catherine C. Coombs, Martin S. Tallman and Ross L. Levine. Molecular therapy for acute myeloid leukaemia. Nature reviews. Clinical oncology. 2015. Advance on line publication. doi:10.1038/nrclinonc.2015.210
Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML10 and 12 trials. Blood. 2001;98(number 6):1752–9. doi:10.1182/blood.V98.6.1752.
Gary Gilliland D, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(number 5):1532–42. doi:10.1182/blood-2002-02-0492.
Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the FLT3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8.
Iwai T, Yokota S, Nakao M, et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children’s cancer and leukemia study group, Japan. Leukemia. 1999;13:38–43.
Meshinchi S, Woods WG, Stirewalt DL, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001;97:89–94. doi:10.1182/blood.V97.1.89.
O’Brien SG, Guilhot F, Larson RA. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004.
Kiyoi H. FLT3 inhibitors: recent advances and problems for clinical application. Nagoya J Med Sci. 2015;77:7–17.
Xu G, Mao L, Liu H, Yang M, Jin J, Qian W. Sorafenib in combination with low-dose-homoharringtonine as a salvage therapy in primary refractory FLT3-ITD-positive AML: a case report and review of literature. Int J Clin Exp Med. 2015;8(11):19891–4.
•• Stone RM, Mandrekar S, Sanford BL, et al. The multi-kinase inhibitor Midostaurin (M) prolongs survival compared with placebo (P) in combination with Daunorubicin (D)/Cytarabine (C) induction (ind), high-dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with FLT3 mutations (muts): an international prospective randomized (rand) P-controlled double-blind trial (CALGB 10603/RATIFY [alliance]). Blood. 2015;126(23):6. August 29, 2016. Established the feasibility and efficacy of the combination of chemotherapy and Midostaurin in one of the most extensive cohorts of patients studied to date.
Chen Y-B, Li S, Lane AA, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for FLT3-ITD AML. Biol Blood Marrow Transplant. 2014 December;20(12):2042–8. doi:10.1016/j.bbmt.2014.09.007.
Sandmaier BM, Khaled SK, Oran B, Gammon G, Trone D, Frankfurt O. Results of a phase 1 study of quizartinib (AC220) as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic cell transplantation. Blood. 2014;124:428.
Strati P, Kantarjian H, Ravandi F. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am J Hematol. 2015 April;90(4):276–81. doi:10.1002/ajh.23924.
Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, et al. Phase I/II study of combination therapy with Sorafenib, Idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(Number 11) doi:10.1200/JCO.2009.25.4888.
Rombouts WJC, Blokland I, Löwenberg B, Ploemacher RE. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplications in the Flt3 gene. Leukemia. 2000;14:675–83.
Hassanein M, Almahayni MH, Gaballa S, Ahmed SO, Fakih RE. FLT3 inhibitors for treating acute myeloid leukemia, clinical lymphoma. Myeloma and Leukemia. 2016; doi:10.1016/j.clml.2016.06.002.
Burnett AK, et al. Higher Daunorubicin exposure benefits FLT3 mutated acute myeloid leukemia. Blood. 2016;128(Number 3):449–52. doi:10.1182/blood-2016-04-712091.
• Talha B, Kantarjian Hagop M, Nogueras-Gonzalez Graciela M, et al. Improvement in clinical outcome of FLT3 ITD mutated acute myeloid leukemia over the last one and half decade. Am J Hematol. 90(11) doi:10.1002/ajh.24140. Describes the evolution of treatment and results over 6 eras along 14 years in a single institution.
Carow CE, Levenstein M, Kaufmann SH, Chen J, Amin S, Rockwell P, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-UFIk2) in human Leukemias. Blood. 1996;87(3):1089–96.
Pawar R, Bali OPS, Malhotra BK, Lamba G. Recent advances and novel agents for FLT3 mutated acute myeloid leukemia. Stem Cell Investig. 2014;1:7. doi:10.3978/j.issn.2306-9759.2014.03.03.
Kase H, Iwahashi K, Matsuda Y. K-252a, a potent inhibitor of protein kinase C from microbial origin. J Antibiot (Tokyo). 1986;39(8):1059–65.
• Francisco Alejandro Lagunas-Rangel, Carlos Cortes-Penagos, Martha Eva Viveros-Sandoval. FLT3: beyond good and evil. Hematol Oncol 2016; 1–9. DOI 10.1002/hon.2330 A concise review of pharmacology of FLT3ID and physiology of FLT3 mutations.
• Seth A. Wander, Mark J. Levis and Amir T. Fathi. The evolving role of FLT3 inhibitors in acute myeloid leukemia: Quizartinib and beyond. 2014, Vol. 5(3) 65–77. DOI: 10.1177/2040620714532123 Describes the experience and future perspectives of FLT3ID.
Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O’Farrell A-M. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(Number 3) doi:10.1182/blood-2004-05-1846.
Fiedler W, Kayser S, Kebenko M, Janning M, Krauter J, et al. A phase I/II study of Sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169:694–700. doi:10.1111/bjh.13353.
Gallogly MM, Lazarus HM. Midostaurin: an emerging treatment for acute myeloid leukemia patients. Journal of Blood Medicine. 2016;7:73–83. doi:10.2147/JBM.S100283.
Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(Number 1):54–60. doi:10.1182/blood-2004-03-0891.
Shabbir M, Stuart R. Lestaurtinib, a multitargeted tyrosinse kinase inhibitor: from bench to bedside. Expert Opin Investig Drugs. 2010;19(3):427–36. doi:10.1517/13543781003598862.
Douglas Smith B, Levis M, Beran M, Giles F, Kantarjian H, Berg K. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(Number 10):3669–76. doi:10.1182/blood-2003-11-3775.
Burnett AK, Bowen D, Russell N, Knapper S, Milligan D, Hunter AE, et al. AC220 (Quizartinib) can be safely combined with conventional chemotherapy in older patients with newly diagnosed acute myeloid leukaemia: experience from the AML18 pilot trial. Blood. 2013;122:622.
Jorge E. Cortes, Hagop Kantarjian, James M. Foran, Darejan Ghirdaladze, Mamia Zodelava. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3–internal tandem duplication status. J Clin Oncol 31:3681–3687. DOI: 10.1200/JCO.2013.48.8783.
Tood M. Cooper, Jeannette Cassar, Elena Eckroth, Jemily Malvar, Richard Sposto, Paul Gaynon, Bill H. Chang, Lia Gore, Keith August. Et. al. A Phase I study of quizartinib combined with chemotherapy in relapsed childhood leukemia: therapeutic advances in childhood leukemia & lymphoma (TACL) study. February 27, American Association for Cancer Reserch. DOI: 10.1158/1078-0432.CCR-15-1998.
Wilhelm SM, Carter C, Tang LY, et al. BAY 43-9006 exhibits broad Spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109.
Zhang W, Konopleva M, Shi Y-x, et al. Mutant FLT3: a direct target of sorafenib in acute Myelogenous leukemia. J Natl Cancer Inst. 2008;100:184–98. doi:10.1093/jnci/djm328.
Williams CB, Kambhampati S, Fiskus W, Wick J, Dutreix C, Ganguly S. Preclinical and phase I results of decitabine in combination with midostaurin (PKC412) for newly diagnosed elderly or relapsed/refractory adult patients with acute myeloid leukemia. Pharmacotherapy. 2013;33(12):1341–52. doi:10.1002/phar.1316.
• Linblad O, Cordero E, Puissant A, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to Sorafenib in AML. Oncogene. 2016; doi:10.1038/onc.2016.41. Describes the aberrant activation of cell survival and proliferative pathways as a mechanism to overcome inhibition of FLT3.
Grimm D, Lieb J, Weyer V et al. Organic Cation Transporter 1 (OCT1) mRNA expression in hepatocellular carcinoma as a biomarker for Sorafenib treatment. BMC Cancer. 2016.1–8. DOI 10.1186/s12885-016-2150-3.
Smith CC, Lin K, Stecula A. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia. 2015 December;29(12):2390–2. doi:10.1038/leu.2015.165.
Chen F, Ishikawa Y, Akashi A, et al. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget. 2016;7(30):47018–32.
Huang A, Huaiqiang J, Liu K, et al. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016;1-9 doi:10.1016/j.canlet.2016.04.040.
• Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, Levis M. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123(1):94–100. doi:10.1182/blood-2013-10-529313. Describes the potential capacity of Crenolanib to overcome acquired resistance to FLT3ID.
Meshinchi S, Arceci RJ, Sanders JE, et al. Role of allogeneic stem cell transplantation in FLT3/ITD-positive AML. Blood. 2006;108:400.
Bornhauser M, Illmer T, Schaich M, et al. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood. 2007;109:2265–6.
Brunet S, Perea G, Esteve J, et al. Adverse impact of FLT3 internal tandem duplication in patients with poor-risk acute myeloid leukaemia allocated to autologous transplantation. Bone Marrow Transpl. 2004;33:S3. (Suppl 1; abstr 71)
Gale R, Hills R, Kottaridis P, et al. No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML10 and 12 trials. Blood. 2005;106:3658–65. doi:10.1182/blood-2005-03-1323.
Brunet S, Labopin M, Esteve J, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. J Clin Oncol. 2012;30:735–41. doi:10.1200/JCO.2011.36.9868.
•• De Freitas T, Marktel S, Piemontese S, et al. High rate of hematological responses to Sorafenib in FLT3-ITD acute myeloid leukemia relapsed after allogeneic hematopoietic stem cell transplantation. Eur J Haematol. 2015; doi:10.1111/ejh.12647. Explores the feasibility of Sorafenib as maintenance therapy following allogenic BMT in FLT3-ITD AML patients.
Alvarado Y, Kantarjian H, Luthra R, Ravandi F, Borthakur G. Treatment with FLT3 inhibitor in patients with FLT3-mutated AML is associated with development of secondary FLT3-TKD mutations. Cancer. 2014;120(14):2142–9. doi:10.1002/n28705.
Acknowledgments
The authors wish to thank Dr. Mónica Vazquez del Mercado (CUCS, Universidad de Guadalajara) for her invaluable support in English grammar review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Carlos Best-Aguilera has received compensation from Avillion Development 1, Ltd., Novartis, AbbVie Farmacéuticos S.A. de C.V., and Celgene for service as a consultant.
O Rodrigo Gómez-Vázquez declares that he has no conflict of interest.
A. Elizabeth Guzmán-Hernández declares that she has no conflict of interest.
R. Monserrat Rojas-Sotelo has received compensation from Janssen and Avillion Development 1, Ltd. for service as a consultant.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Leukemia
Rights and permissions
About this article

Cite this article
Best-Aguilera, C., Rodrigo Gómez-Vázquez, O., Elizabeth Guzmán-Hernández, A. et al. Treatment of Acute Myeloid Leukemia with the FLT3 Gene Mutation. Curr Oncol Rep 19, 21 (2017). https://doi.org/10.1007/s11912-017-0573-x
Published:
DOI: https://doi.org/10.1007/s11912-017-0573-x