
Abstract
WT1 is a transcription factor involved in differentiation and proliferation of acute myeloid leukemia (AML) blasts and is expressed in 90% of cases, as determined by nested reverse transcription polymerase chain reaction (RT-PCR). It is proposed to be a key molecule in leukemia promotion. To assess the relevance of WT1 expression, we analyzed blood and bone marrow samples from 58 AML patients (37 at diagnosis, 8 in hematological remission, and 13 at relapse) for the level of WT1 expression, using quantitative real-time RT-PCR. In addition, 21 randomly chosen samples were also analyzed for the quantitative expression of the main WT1 splice variants. As expected, samples from patients at the time of diagnosis or relapse showed significantly higher WT1 expression compared to samples from patients in remission or control samples. No striking difference in expression levels was found between various French-American-British (FAB) subtypes. The level of WT1 expression observed in patients at the time of initial diagnosis was similarly high in patients at relapse. Expression of the four main isoforms (E5+/KTS+, E5−/KTS+, E5+/KTS−, and E5−/KTS−) was found in all samples with significantly higher expression levels of the E5+ variants. Together, these findings support the potential of WT1 as a target for novel treatment approaches in AML.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is characterized by a variety of complex molecular genetic abnormalities of partly still unresolved significance for gene expression and regulation. One common feature, however, is a strong overexpression of the Wilms’ tumor protein 1 (WT1), which can be found in up to 90% of AML cases [8, 15, 17].
WT1 is an embryonic zinc finger transcription factor, which was originally identified for its involvement as a tumor suppressor gene in the pathogenesis of Wilms’ tumors [5]. In addition to acute leukemias, WT1 is expressed in a variety of other malignant tumors, including malignant mesothelioma, lung, breast, and renal cell cancer [13, 27]. WT1 has therefore been suggested to play a role as an oncogene, although its functional significance within the leukemogenesis process still remains to be elucidated.
The WT1 gene, which is located on the short arm of chromosome 11, consists of ten exons, encoding a complex pattern of at least 24 protein isoforms, which exhibit different DNA-binding specificities [25]. Mainly four distinct mRNA transcripts are expressed, reflecting the presence or absence of exon 5 (E5) and the KTS− region. In human 20-week-old kidney the relative ratio of the four splice variants KTS−/E5−, KTS−/E5+, KTS+/E5−, and KTS+/E5+ is 1:2.5:3.8:8.3 [6]. WT1 plays a pivotal role in the development of the urogenital tract during embryogenesis [11, 23]. In adults it is expressed at very low levels in kidney, ovary, testis, spleen, and normal hematopoietic progenitor cells [8].
Since WT1 is considered a therapeutic target, it is important to know its expression level and its splice variant composition in various AML subtypes and during the course of the disease. In our study we determined the splice variant composition and compared the expression levels of WT1 in AML blasts of different French-American-British (FAB) subtypes at the time of diagnosis and at the time of relapse.
Materials and methods
Blood and bone marrow samples
Fifty-eight blood (n=20) or bone marrow samples (n=38) from 37 AML patients at diagnosis, 8 patients in hematological remission, and 13 patients at relapse were analyzed. The median age of the patients was 62 (27–86) years; there were 45 patients with de novo AML and 13 patients with secondary AML. There were 48 fresh samples; 10 samples were cryopreserved on liquid nitrogen. Patients were classified according to the FAB classification. The proportion of blasts in the samples was determined by microscopy or automated cell counter.
Bone marrow samples of patients who underwent hip replacement surgery (n=5) or were diagnosed with nonmalignant disorders (n=8) served as controls.
Mononuclear cells (MNC) were fractionated by Ficoll-Isopaque density gradient centrifugation (Pharmacia, Erlangen, Germany). MNCs from leukemia patients at the time of diagnosis or relapse were predominantly (>60%) blast cells.
To assess WT1 expression levels in peripheral blood stem cell (PBSC) preparations, we further analyzed unpurged PBSC from healthy stem cell donors (n=10), as well as purged PBSC from lymphoma patients (n=3). All PBSC were harvested by leukapheresis after stimulation of healthy donors with granulocyte colony-stimulating factor (G-CSF) (2×5 μg/kg s.c. per day; day −5 until leukapheresis) or after mobilization chemotherapy and subsequent stimulation with G-CSF (2×5 μg/kg s.c. per day; from day 6 after start of chemotherapy until leukapheresis) of the lymphoma patients. Purged PBSC were prepared using immunomagnetic beads coupled with CD34+ antibodies (CliniMACS, Miltenyi Biotec, Bergisch Gladbach, Germany). PBSC samples were only used for analysis if they contained at least 1×106 CD34+ hematopoietic progenitor cells and their proportion in purged PBSC was at least 90%. Informed consent was obtained from all patients and healthy donors.
RNA extraction and reverse transcription (RT)
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) as described by the supplier. RNA integrity was checked electrophoretically and quantified spectrophotometrically.
Reverse transcription was carried out on 2 μg total RNA diluted in a 22.5-μl mixture containing 2 μl oligo p(dT)15 primer (0.8 μg/μl), 1 μl dNTP (25 mM), 1 μl RNasIN (40 U/μl), 1 μl Omniscript reverse transcriptase (4 U/μl), 2 μl RT buffer (×10), and 15.5 μl H2O. After incubation at 37°C for 1 h, the reverse transcriptase was inactivated for 5 min at 95°C and cDNA was stored at −20°C or immediately used for real-time polymerase chain reaction (PCR). All reagents, except oligo p(dT)15 primers and RNasIN (Roche Diagnostics, Mannheim, Germany), were purchased from Qiagen (Hilden, Germany).
Quantification of WT1 expression
Quantification of WT1 mRNA was performed on a LightCycler system (Roche Diagnostics, Mannheim, Germany). To compensate for variations of pre-PCR steps in all samples the housekeeping gene porphobilinogen deaminase (PBGD) mRNA was also quantified. Then, 2 µl of each cDNA was diluted to a volume of 18 μl PCR mix (LightCycler Faststart DNA Master Hybridization Probes Kit, Roche Diagnostics, Mannheim, Germany), containing 0.5 pmol of each primer and 0.2 pmol each of two marker cDNA-specific oligonucleotide probes with either a donor fluorophore at its 3′-end (fluorescein) or an acceptor fluorophore (LC Red 640) at the 5′-end. The LC Red 640-labeled probes were also dephosphorylated at the 3′-end to avoid probe extension. Specific primers and probes were carefully designed using the LC probe design software (Roche Diagnostics, Mannheim, Germany). Primers and optimal probe sequences purchased from Metabion (Munich, Germany) are listed in Table 1. PCR conditions and target-specific final MgCl2 concentrations are listed in Table 2. For each target an initial denaturation cycle at 95°C for 10 min and a final extension cycle at 72°C for 2 min and at 40°C for 1 min was performed. Specificity of the PCR products was confirmed electrophoretically.
TOPO 2.1 (Invitrogen, Groningen, Netherlands) recombinant vectors, carrying either WT1 or PBGD amplicon-specific sequences were serially diluted in water containing 0.4 μg/μl polyadenylic acid (Pharmacia Biotech, Freiburg, Germany). All samples were tested in duplicate using the average value of both duplicates for quantification. The amount of recombinant TOPO 2.1 marker plasmid equivalents was calculated by the LightCycler Software using these standard curves. The relative amount of WT1 transcript is expressed as ratio of TOPO 2.1-WT1 (pg/μl): TOPO 2.1-PBGD (pg/μl).
Quantification of WT1 splice variants
Real-time quantitative RT-PCR for WT1 splice variants was performed on an Applied Biosystems GeneAmp 5700 Sequence Detection System using TaqMan technology: 5 μl of cDNA was diluted to a final PCR volume of 25 μl, containing 12.5 μl Universal Mastermix (Applied Biosystems, Foster City, Calif., USA), 2.5 μl of each primer (final concentration 200 nM), and 2.5 μl of the TaqMan probe (final concentration 100 nM). Primers and TaqMan probes were designed using Primer Express software (Applied Biosystems, Foster City, Calif., USA) and purchased from TIB Molbiol (Berlin, Germany). The sequences are listed in Table 1. PCR conditions are given in Table 2. Prior to cycling there was an initial step at 95°C for 1 min and 50°C for 2 min. Three reactions were performed independently on each sample using a different Mastermix and a different PCR cycling. A dilution series of full length WT1 mRNA amplified in pcDNA3.1(+) (Invitrogene, Karlsruhe, Germany) with an insert of the WT1 gene was used as reference for the expression of either total WT1 or E5+ and KTS+. The number of total WT1, E5+, or KTS+ copies in the dilution series and samples were calculated using GeneAmp 5700 SDS software. As internal standard GAPDH cDNA was measured analogous to WT1 using the TaqMan GAPDH control reagents (Applied Biosystems, Foster City, Calif., USA) as described by the supplier. Comparison of total WT1 expression determined by RT-PCR in the LightCycler system with PBGD as housekeeping gene and by RT-PCR in the TaqMan system with GAPDH as housekeeping gene showed a good correlation of the results (r=0.77, p=0.001, Pearson correlation; data not shown).
The ratios of E5+ or KTS+ variants were calculated by dividing the copy counts of E5 or KTS by the copy counts of total WT1, the ratio of E5− or KTS− variants by subtraction of the ratio of E5+ or KTS+ from 1. As the splicing of E5 and KTS should be independent from each other, it can be assumed that the proportions of E5+, E5−, KTS+, or KTS− variants are similar in all four different splice variants. With this assumption the ratios of the four main splice variants can be calculated by multiplication of the ratios of the E5+, E5−, KTS+, or KTS− variants with each other. The expression of the four main splice variants was therefore calculated by multiplication of the ratios of E5+ with KTS+ (+/+ variant), E5− with KTS+ (−/+ variant), E5+ with KTS− (+/− variant), or E5− with KTS− (−/− variant). The results are given as ratio of total WT1 expression.
To reduce the risk of contamination, thermocycling and post-PCR steps were performed in separate laboratories. PCR mixtures were set up in a template tamer (Oncor Appligene, Heidelberg, Germany). Negative controls were performed for all RT-PCR steps according to European Organization of Research and Treatment of Cancer (EORTC) recommendations, including a reverse transcriptase negative control for every sample [9].
Statistical analysis
Statistical significance was tested by the Mann–Whitney U test when comparing two different groups or by the Kruskal–Wallis H test when comparing more than two different groups. Tests were performed using WinSTAT Version 2001.1 software.
Results
As shown in Fig. 1, 46 of 50 (92%) AML patients tested at diagnosis or relapse had blast cells expressing WT1 mRNA at a considerable level. The median amount was 0.017 pg WT1 per pg PBDG (range: 0.00005–7.20; 25 percentile: 0.004, 75 percentile: 0.09). No significant difference was found between samples from patients at diagnosis and patients at relapse (p=0.79, U test). Compared to control bone marrow/PBSC samples, however, patients at diagnosis or relapse of AML had a significantly higher expression of WT1 (Fig. 1; p<0.001, U test). The difference between control bone marrow and PBSC samples was not significant (p=0.12, U test).

WT1 mRNA levels in AML samples from patients at diagnosis or relapse as well as in control bone marrow samples and PBSC samples. Only positive PCR results are shown. The difference between AML samples at diagnosis/relapse and control bone marrow/PBSC samples is significant (p<0.001, U test). The differences between patients at diagnosis and relapse and between control bone marrow samples and PBSC samples are not significant (solid bar median, BM bone marrow)
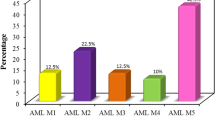
Median and variation within FAB subtypes for WT1 expression in patients at diagnosis or relapse did not differ (p=0.69, H test). The median amount of WT1 in AML M0 (n=4) was 0.004 pg per pg PBDG, in AML M1 (n=6) 0.01 pg per pg PBDG, in AML M2 (n=18) 0.001 pg per pg PBDG, in AML M4 (n=12) 0.01 pg per pg PBDG, and in AML M5 (n=9) 0.02 pg per pg PBDG; the one patient with AML M6 had a WT1 expression of 0.008 pg per pg PBGD (Fig. 2).

Quantitative WT1 expression in AML patients at diagnosis or relapse depending on the FAB subtype. Only positive PCR results are shown. There is no significant difference of WT1 expression between these FAB subtypes (p=0.69, H test; solid bar median)
Samples from patients in remission [median: 0.009 (range: 0.00002–0.051; 25 percentile: 0.00006, 75 percentile: 0.01) pg WT1 per pg PBDG] had a significantly higher WT1 expression compared to control bone marrow/PBSC samples (Fig. 3; p=0.02, U test). Compared to samples from patients at diagnosis or relapse of AML, the median WT1 expression in samples from patients at remission was significantly lower (Fig. 3; p=0.008, U test) with some, however, reaching similarly high WT1 expression levels as patients at diagnosis or relapse.

WT1 mRNA levels in AML samples from patients at diagnosis/relapse, at hematological remission as well as in control bone marrow/PBSC samples. Only positive PCR results are shown. Samples from patients at hematological remission have a significantly lower WT1 mRNA level than samples from patients at diagnosis/relapse (p=0.008, U test) and a significantly higher WT1 mRNA level than control bone marrow/PBSC samples (p=0.02, U test; solid bar median, BM bone marrow)
The 21 patients analyzed for WT1 splice variants expressed the E5 as well as the KTS region. The median copy count of total WT1 was 809 (range: 77–3125; 25 percentile: 232, 75 percentile: 1641) copies per 105 copies GAPDH; the median copy count of E5+ was 499 (range: 10–2987; 25 percentile: 171, 75 percentile: 1117) and of KTS+ 358 (range: 15–1,729; 25 percentile: 358, 75 percentile: 690) copies per 105 copies GAPDH. The median ratio of E5+/total WT1 expression was 65% (range: 8–100; 25 percentile: 60, 75 percentile: 85), of KTS+/total WT1 expression 48% (range: 6–100; 25 percentile: 31, 75 percentile: 57), of E5−/total WT1 expression 35% (range: 0–92; 25 percentile: 15, 75 percentile: 40), and of KTS−/totalWT1 expression 52% (range: 0–94; 25 percentile: 43, 75 percentile: 69). The calculation of the four splice variants showed a significantly higher proportion of the E5+ variants (p<0.0001, H test) with 34% (range: 1–82; 25 percentile: 23, 75 percentile: 39) of E5+/KTS+, 14% (range: 0–25; 25 percentile: 5, 75 percentile: 22) E5−/KTS+, 35% (range: 0–69; 25 percentile: 25, 75 percentile: 48) E5+/KTS−, and 17% (range: 0–79; 25 percentile: 7, 75 percentile: 25) E5−/KTS− (Fig. 4).

Quantification of the four main WT1 splice variants (E5+/KTS+, E5−/KTS+, E5+/KTS−, or E5−/KTS−) in 21 AML patients. Results are given as ratio of total WT1 expression (solid bar median). There is a significantly higher expression of the E5+ splice variants (p<0.0001, H test)
Discussion
Expression of WT1 and its role in AML has been intensively investigated in recent years [16]. Most investigators report that WT1 is expressed in up to 90% of the patients with active disease as determined by qualitative RT-PCR [8, 15, 17], whereas WT1 expression in patients in hematological remission is rather rare [3, 26]. In correlation with these reports, we found substantial WT1 expression in more than 90% of patients with active disease. Importantly we observed similarly high WT1 expression levels at diagnosis or at relapse.
When we compared different FAB subtypes according to their quantitative WT1 expression, we found no statistically significant difference. This is in contrast to earlier studies with semiquantitative techniques which suggested an inverse correlation of WT1 expression and differentiation in leukemic cells [1, 4, 17, 18], but correlates with a recent study of Kreuzer et al., who also demonstrated that WT1 expression levels are similar across all FAB subtypes using real-time RT-PCR [12].
Several previous reports suggested that WT1 levels rise some months prior to hematological relapse and that WT1 levels correlate well with the levels of other MRD markers including CBFβ-MYH11 or AML1-ETO [2, 19]. In accordance with these studies, in our series some patients in remission had significantly higher WT1 levels than control bone marrow/PBSC samples, reaching similarly high WT1 expression levels as patients at diagnosis or relapse. Although the number of patients was too small for statistical analysis, these findings support designing proposals for WT1-targeted therapies. WT1 expression levels are not only similar at diagnosis and relapse, moreover quantification of WT1 might also help to identify those patients who might profit from a WT1-targeted therapy, but did not yet have a hematological relapse.
It has not yet been fully clarified whether there is a leukemia-specific expression of WT1 splice variants. To investigate the possibility of an imbalance in expression of splice variants, we assessed in AML the relative expression level of E5 and KTS, the contributors of the four main splice variants. We found an expression of all four splice variants (E5+/KTS+, E5−/KTS+, E5+/KTS−, and E5−/KTS−) with a significantly higher level of the E5+ variants. This correlates with results from Renshaw et al. who found—by using a semiquantitative optical method—a relative excess of exon 5-containing transcripts in leukemia cell lines and primary tumor material as well as in normal cells of the hematopoietic system [24]. A dominance of the unspliced variant (E5+/KTS+) was also found in WT1-expressing CD34+ CD38+ bone marrow single cells [7]. Moreover, an abundance of E5+ variants was also observed in de novo lung and head and neck squamous cell carcinomas [20, 21] and human embryonal kidney [6]. Our data therefore might indicate that the WT1 gene remains unchanged throughout the leukemogenic process and that relevant mutations rarely occur as already described previously [10].
Taken together, most AML blasts express WT1 and its major splice variants at considerable magnitude, and we do not have evidence for a loss of this molecule in leukemia relapse. Thus, there is no apparent selection for the regrowth of WT1-negative leukemia clones. This observation has important implications for WT1 as a therapeutic target, including recently activated peptide vaccination trials [14, 22, 28].
References
Bergmann L, Miething C, Maurer U, Brieger J, Karakas T, Weidmann E, Hoelzer D (1997) High levels of Wilms’ tumor gene (WT1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood 90:1217–1225
Cilloni D, Gottardi E, De Micheli D, Serra A, Volpe G, Messa F, Rege-Cambrin G, Guerrasio A, Divona M, Lo Coco F, Saglio G (2002) Quantitative assessment of WT1 expression by real time quantitative PCR may be a useful tool for monitoring minimal residual disease in acute leukemia patients. Leukemia 16:2115–2121
Gaiger A, Schmid D, Heinze G, Linnerth B, Greinix H, Kalhs P, Tisljar K, Priglinger S, Laczika K, Mitterbauer M, Novak M, Mitterbauer G, Mannhalter C, Haas OA, Lechner K, Jager U (1998) Detection of the WT1 transcript by RT-PCR in complete remission has no prognostic relevance in de novo acute myeloid leukemia. Leukemia 12:1886–1894
Garg M, Moore H, Tobal K, Liu Yin JA (2003) Prognostic significance of quantitative analysis of WT1 gene transcripts by competitive reverse transcription polymerase chain reaction in acute leukaemia. Br J Haematol 123:49–59
Haber DA, Housman DE (1992) Role of the WT1 gene in Wilms’ tumour. Cancer Surv 12:105–117
Haber DA, Sohn RL, Buckler AJ, Pelletier J, Call KM, Housman DE (1991) Alternative splicing and genomic structure of the Wilms’ tumor gene WT1. Proc Natl Acad Sci U S A 88:9618–9622
Hosen N, Sonoda Y, Oji Y, Kimura T, Minamiguchi H, Tamaki H, Kawakami M, Asada M, Kanato K, Motomura M, Murakami M, Fujioka T, Masuda T, Kim EH, Tsuboi A, Oka Y, Soma T, Ogawa H, Sugiyama H (2002) Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br J Haematol 116:409–420
Inoue K, Ogawa H, Sonoda Y, Kimura T, Sakabe H, Oka Y, Miyake S, Tamaki H, Oji Y, Yamagami T, Tatekawa T, Soma T, Kishimoto T, Sugiyama H (1997) Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood 89:1405–1412
Keilholz U, Willhauck M, Scheibenbogen C, de Vries TJ, Burchill S (1997) Polymerase chain reaction detection of circulating tumour cells. EORTC Melanoma Cooperative Group, Immunotherapy Subgroup. Melanoma Res 7 [Suppl 2]:133–141
King-Underwood L, Pritchard-Jones K (1998) Wilms’ tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood 91:2961–2968
Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R (1993) WT-1 is required for early kidney development. Cell 74:679–691
Kreuzer K, Saborowski A, Lupberger J, Appelt C, Na IK, le Coutre P, Schmidt CA (2001) Fluorescent 5′-exonuclease assay for the absolute quantification of Wilms’ tumour gene (WT1) mRNA: implications for monitoring human leukaemias. Br J Haematol 114:313–318
Loeb DM, Sukumar S (2002) The role of WT1 in oncogenesis: tumor suppressor or oncogene? Int J Hematol 76:117–126
Mailander V, Scheibenbogen C, Thiel E, Letsch A, Blau IW, Keilholz U (2004) Complete remission in a patient with recurrent acute myeloid leukemia induced by vaccination with WT1 peptide in the absence of hematological or renal toxicity. Leukemia 18:165–166
Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S, Reinhardt R, Thiel E (1995) Presence of Wilms’ tumor gene (WT1) transcripts and the wt1 nuclear protein in the majority of human acute leukemias. Leukemia 9:1060–1067
Menssen HD, Siehl JM, Thiel E (2002) Wilms’ tumor gene (WT1) expression is a panleukemic marker. Int J Hematol 76:103–109
Miwa H, Beran M, Saunders GF (1992) Expression of the Wilms’ tumor gene (WT1) in human leukemias. Leukemia 6:405–409
Miyagi T, Ahuja H, Kubota T, Kubonishi I, Koeffler HP, Miyoshi I (1993) Expression of the candidate Wilm’s tumor gene, WT1, in human leukemia cells. Leukemia 7:970–977
Ogawa H, Tamaki H, Ikegame K, Soma T, Kawakami M, Tsuboi A, Kim EH, Hosen N, Murakami M, Fufioka T, Masuda T, Taniguchi Y, Nishida S, Oji Y, Oka Y, Sugiyama H (2003) The usefulness of monitoring WT1 gene transcripts for the prediction and management of relapse following allogeneic stem cell transplantation in acute type leukemia. Blood 101:1698–1704
Oji Y, Miyoshi S, Maeda H, Hayashi S, Tamaki H, Nakatsuka S, Yao M, Takahashi E, Nakano Y, Hirabayashi H, Shintani Y, Oka Y, Tsuboi A, Hosen N, Asada M, Fujioka T, Murakami M, Kanato K, Motomura M, Kim EH, Kawakami M, Ikegame K, Ogawa H, Aozasa K, Kawase I, Sugiyama H (2002) Overexpression of the Wilms’ tumour gene WT1 in de novo lung cancers. Int J Cancer 100:297–303
Oji Y, Inohara H, Nakazawa M, Nakno Y, Akahani S, Nakatsuka SI, Koga S, Ikeba A, Abeno S, Honjo Y, Yamamoto Y, Iwai S, Yoshida K, Oka Y, Ogawa H, Yoshida JI, Aozasa K, Kubo T, Sugiyama H (2003) Overexpression of the Wilms’ tumour gene WT1 in head and neck squamous cell carcinoma. Cancer Sci 94:523–529
Oka Y, Tsuboi A, Murakami M, Hirai M, Tominaga N, Nakajima H, Elisseeva OA, Masuda T, Nakano A, Kawakami M, Oji Y, Ikegame K, Hosen N, Udaka K, Yasukawa M, Ogawa H, Kawase I, Sugiyama H (2003) Wilms tumor gene peptide-based immunotherapy for patients with overt leukemia from myelodysplastic syndrome (MDS) or MDS with myelofibrosis. Int J Hematol 78:56–61
Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D et al (1990) The candidate Wilms’ tumor gene is involved in genitourinary development. Nature 346:194–197
Renshaw J, King-Underwood L, Pritchard-Jones K (1997) Differential splicing of exon 5 of the Wilms tumour (WT1) gene. Genes Chromosomes Cancer 19:256–266
Scharnhorst V, Van der Eb A, Jochemsen A (2001) WT1 proteins: functions in growth and differentiation. Gene 273:141–161
Schmid D, Heinze G, Linnerth B, Tisljar K, Kusec R, Geissler K, Sillaber C, Laczika K, Mitterbauer M, Zochbauer S, Mannhalter C, Haas OA, Lechner K, Jager U, Gaiger A (1997) Prognostic significance of WT1 gene expression at diagnosis in adult de novo acute myeloid leukemia. Leukemia 11:639–643
Sugiyama H (2001) Wilms’ tumor gene WT1: its oncogenic function and clinical application. Int J Hematol 73:177–187
Tsuboi A, Oka Y, Osaki T, Kumagai T, Tachibana I, Hayashi S, Murakami M, Nakajima H, Elisseeva OA, Fei W, Masuda T, Yasukawa M, Oji Y, Kawakami M, Hosen N, Ikegame K, Yoshihara S, Udaka K, Nakatsuka S, Aozasa K, Kawase I, Sugiyama H (2004) WT1 peptide-based immunotherapy for patients with lung cancer: report of two cases. Microbiol Immunol 48:175–184
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors Jan M. Siehl and Mark Reinwald contributed equally to this paper.
Rights and permissions
About this article
Cite this article
Siehl, J.M., Reinwald, M., Heufelder, K. et al. Expression of Wilms’ tumor gene 1 at different stages of acute myeloid leukemia and analysis of its major splice variants. Ann Hematol 83, 745–750 (2004). https://doi.org/10.1007/s00277-004-0941-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-004-0941-0