
Abstract
The primary function of the glycoprotein hormone erythropoietin (Epo) is to promote red cell production by inhibiting apoptosis of erythrocytic progenitors in hemopoietic tissues. However, functional Epo receptors (Epo-R) have recently been demonstrated in various nonhemopoietic tissues indicating that Epo is a more pleiotropic viability and growth factor. Herein, in vitro and in vivo effects of Epo in the brain and the cardiovascular system are reviewed. In addition, the therapeutic impact of Epo in oncology is considered, including the question of whether Epo might promote tumor growth. Convincing evidence is available that Epo acts as a neurotrophic and neuroprotective factor in the brain. Epo prevents neuronal cells from hypoxia-induced and glutamate-induced cell death. Epo-R is expressed by neurons and glia cells in specific regions of the brain. Epo supports the survival of neurons in the ischemic brain. The neuroprotective potential of Epo has already been confirmed in a clinical trial on patients with acute stroke. With respect to the vasculature, Epo acts on both endothelial and smooth muscle cells. Epo promotes angiogenesis and stimulates the production of endothelin and other vasoactive mediators. In addition, Epo-R is expressed by cardiomyocytes. The role of Epo as a myocardial protectant is at the focus of present research. Epo therapy in tumor patients is practiced primarily to maintain the hemoglobin concentration above the transfusion trigger and to reduce fatigue. In addition, increased tumor oxygenation may improve the efficacy of chemotherapy and radiotherapy. However, tumor cells often express Epo-R. Therefore, careful studies are required to fully exclude that recombinant human Epo (rHuEpo) promotes tumor growth.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Erythropoietin (Epo) has been well characterized as the renal glycoprotein hormone which promotes the survival, proliferation, and differentiation of erythrocytic progenitors in hemopoietic tissues. Recombinant human Epo (rHuEpo) and related compounds have proved most useful for treatment of the anemia associated with chronic renal failure and, more restrictedly, certain types of nonrenal anemias (for references see [97, 134]). Epo maintains erythropoiesis by binding to specific transmembrane receptors (Epo-R) which are expressed primarily by erythrocytic progenitors such as the burst-forming units–erythroid (BFU-E) and the colony-forming units–erythroid (CFU-E). The functional human Epo-R is a member of the cytokine class I receptor superfamily and presents as a homodimer of two identical glycoprotein chains of 484 amino acids [169]. Each of the chains is composed of an extracellular domain with conserved cysteines and a WSXWS motif, a hydrophobic transmembrane sequence, and a cytoplasmic domain to which the protein tyrosine kinase JAK2 (Janus kinase 2) is affiliated. The Epo molecule binds to both Epo-R subunits [157], whereby the dissociation constants for the two binding sites differ greatly (1 μM vs 1 nM). With respect to novel recombinant Epo analogues, it is important to note that the affinity for the receptor decreases with Epo glycosylation [61]. The carbohydrate portion of Epo is thought to prevent Epo-R binding through electrostatic forces [53]. Epo binding induces a conformational change and a tighter connection of the two Epo-R subunits [44, 160, 183]. Consequently, the two JAK2 molecules of the cytoplasmic regions of the Epo-R subunits undergo autophosphorylation [160, 207] and catalyze the phosphorylation of tyrosine residues of Epo-R, thereby providing docking sites for signaling proteins containing SRC homology 2 (SH2) domains [13, 185]. The complex network of Epo-R signaling involves (1) the expression of the antiapoptotic protein bcl-xL [81], (2) the activation of mitogen-activated protein kinase (MAPK) and phosphatidyl-inositol 3-kinase (PI-3K/Akt), and (3) homodimerization of the signal transducer and activator of transcription 5 (STAT5). The concerted action of these mediators increases the rates of survival, proliferation, and differentiation of the erythrocytic progenitors [109, 151, 219]. In vitro studies have shown that the Epo-induced signaling pathways return to nearly basal levels after 30–60 min of stimulation [196]. The effect of Epo is terminated by the action of the hemopoietic cell phosphatase (HCP) which catalyzes JAK2 dephosphorylation [110, 216]. The Epo/Epo-R complex is then internalized and degraded [181, 196].
Whereas most of the erythrocytic progenitors die without Epo [54, 113], an increasing number of BFU-E and CFU-E escape from apoptosis and proliferate in the presence of Epo, thereby producing large progeny of proerythroblasts and normoblasts. The time from the CFU-E to the reticulocyte is about 7 days and involves several cell divisions. Therefore, there is a lag of a few days before reticulocytosis occurs following a rise in the plasma Epo concentration due to an increase in endogenous Epo gene expression in association with hypoxic stress or on the administration of rHuEpo.
Originally, stimulation of erythropoiesis was thought to be the sole physiological function of Epo [93]. However, recent advances in analytical techniques have led to the demonstration of Epo-R mRNA, Epo-R protein, Epo binding to Epo-R, and intracellular signaling in a variety of nonhemopoietic cells and organs, including the brain, cardiovascular tissues (endothelium, vascular smooth muscle, cardiomyocytes), the liver, gastrointestinal tissues, pancreatic islands, the kidney, the testis, and the female reproductive organs (for references see [101, 127, 138, 168]). Thus, Epo is a more pleiotropic survival and growth factor than initially thought (Fig. 1). It is assumed that Epo has neurotrophic and neuroprotective [43, 49, 98, 99, 126, 128], vascular [128, 177], and cardioprotective [155, 177] functions. Herein, relevant experimental studies and preliminary clinical observations are summarized with respect to the value of rHuEpo therapy in cerebral and cardiac disorders. On the other hand, given that Epo is a pleiotropic growth factor, the worrying question has to be raised as to whether tumor cells express Epo-R and whether Epo can induce or promote tumor growth. This aspect is highly relevant to the rationale of rHuEpo treatment of renal and nonrenal anemias, particularly the anemia associated with cancer.

Target tissues of erythropoietin (EPO). Circulating EPO acts primarily on erythropoietic progenitors. In addition, the hormone stimulates angiogenesis. Brain-derived EPO exerts its action in a paracrine way. Neuroprotective and cardioprotective effects of the pharmacological administration of rHu-EPO have been reported. Whether EPO stimulates tumor growth is a matter of debate
Effects of Epo on neuronal cells
Expression of Epo-R by neurons and neurotrophic effects
Masuda et al. [127] first detected low affinity binding sites for Epo in rat PC12 pheochromocytoma and mouse basal forebrain SN6 cell cultures. PC12 cells respond to exogenously added Epo with a rapid increase in the cytosolic Ca2+ concentration and the release of monoamines. More recent studies with PC12 cells have confirmed that Epo modulates dopamine release and NO production [107, 112] and affects the rate of the expression of several genes as assessed by cDNA array screening [161]. Another seminal finding was reported by Konishi et al. [111] who showed that Epo increases choline acetyltransferase activity in primary cultures of mouse neurons and the survival of septal cholinergic neurons in rats with fimbria-fornix transections. Several groups of investigators have subsequently demonstrated Epo-R mRNA and protein in distinct areas of rodent and mammalian brain [57, 119, 124]. Epo binding sites are mainly located in the hippocampus, capsula interna, cortex, and midbrain of mice [57]. Hypoxia exposure leads to an increase in Epo mRNA levels in human and mouse brain [18, 45, 119, 165, 176]. In human embryos, both Epo-R and Epo become detectable in the brain 5 weeks post-conception [52, 103, 105, 118]. Both neurons and astrocytes express Epo-R [18, 19, 105, 176, 180]. In addition, brain capillary endothelial cells express Epo-R, as shown in rat [213] and human [26] biopsies. Interestingly, rat capillary endothelial cells express two different forms of Epo-R mRNA [213]. Epo-R and Epo have also been detected by immunohistochemistry in peripheral rat nerves [35]. The histological assignment has been confirmed in several cell culture studies. Epo-R expression has been demonstrated in primary cultures or cell lines of neuronal origin [18, 19, 45, 127, 137, 143, 210], astrocytes [18, 124, 143, 180], microglia [143], and brain-derived capillary endothelial cells [18, 213].
Trophic effects of Epo have been demonstrated in cultures of cortical and cholinergic neurons [111, 137]. Epo also stimulates the proliferation and differentiation of neuronal stem and progenitor cells [173, 179]. Clearly, however, while the Epo/Epo-R system is required for prenatal and postnatal erythropoiesis, this does not appear to be the case with respect to the neuronal Epo/Epo-R system. Suzuki et al. [182] have established a transgenic mouse line which expresses Epo-R exclusively in hemopoietic tissues. The transgenic mice develop normally and show no neurologic abnormalities despite the lack of Epo-R in the brain and other nonhemopoietic tissues.
Neuroprotective effects of Epo in vitro
Glutamate is considered one of the major mediators of neuronal cell death due to hypoxia [46]. Hypoxic neurons release large amounts of glutamate, which can act both on ionotropic receptors (NMDA-R, AMPA-R/kainate-R) and metabotropic receptors [130]. Preconditioning of rat hippocampal and cerebral cortical cells in primary culture with Epo has been shown to reduce glutamate cytotoxicity [137]. In fact, preconditioning with Epo protects neurons from both NMDA-induced and NO-induced apoptosis [18, 56]. When acutely added, Epo fails to prevent rat hippocampal and cerebral cortical neurons in primary culture from NO-induced death although it inhibits the NMDA-R-mediated increase in the cytosolic Ca2+ concentration [166]. Recent in vitro studies indicate that the PI-3K/Akt pathway is of primary importance in the neuroprotective action of Epo by maintaining mitochondrial membrane potential in anoxic primary hippocampal neuronal cell cultures [48]. Destabilization of the mitochondrial membrane potential leads to the release of cytochrome C, which activates the caspases 8, 1, and 3 that promote DNA fragmentation [50]. Other studies regarding Epo/Epo-R signaling in neuronal cells are described elsewhere [47, 49].
Neuroprotective effects of Epo in vivo
Evidence for a neuroprotective action of Epo in the brain was first provided by the group of Sasaki in 1998 [165, 166]. Infusion of rHuEpo into the lateral ventricles of Mongolian gerbils with experimental cerebral ischemia prevents ischemia-induced learning disability and rescues hippocampal CA1 neurons from death, whereas infusion of soluble Epo-R augments neuronal degeneration and impairs learning ability [166]. rHuEpo infused into the cerebral ventricles of rats with permanent middle artery occlusion reduces ischemia-induced place navigation disability, cortical infarction, and thalamic degeneration [165]. Further studies have shown that locally administered rHuEpo upregulates the expression of bcl-xL in the hippocampal CA1 field [203], inhibits NO production [33], and maintains cognitive functions [40] in gerbils with experimental cerebral ischemia. The role of NO is not fully understood, however, since Genc et al.[70, 71] have reported that Epo exerts neuroprotective effects in vivo by increasing NO production. NO can be cytotoxic for neuronal cells, but it may also improve O2 supply by means of vasodilation. In addition, NO induces Epo-R expression in neuronal cell cultures [148].
Preconditioning stressors such as hypoxia render tissues more tolerant to subsequent stress events. Accordingly, brief periods of sublethal cerebral ischemia protect rats from subsequent stroke caused by permanent middle cerebral artery occlusion [174]. Hypoxic preconditioning also reduces hypoxia-induced ischemic brain injury in neonatal rats [191] and kainate-induced neuronal damage in adult rats [64]. The beneficial effect of hypoxia preconditioning is partly mediated by Epo which is increasingly produced in the hypoxic brain. Indeed, a recent study has shown that the protective effect of hypoxic preconditioning is significantly reduced in mice when Epo signaling is locally blocked by infusion of soluble Epo-R into the cerebral ventricle [158].
It was earlier assumed that systemically administered Epo would not enter the brain because of the blood–brain barrier [102, 104, 125]. However, when biotinylated rHuEpo was administered intraperitoneally (i.p.) to rats, peroxidase reaction product was observed in surrounding capillaries and later localized to scattered neurons in the brain [26]. Jumbe [99] has shown the cerebrospinal fluid to serum concentration ratios to be about 1×10−3 following the intravenous (i.v.) administration of rHuEpo (5000 U/kg) or darbepoetin alfa (25 μg/kg) in rats. The calculated mean area under the concentration–time curve (AUC0–8), by noncompartmental analysis, was 340 mU h/ml for rHuEpo and 3.6 ng h/ml for darbepoetin alfa in cerebrospinal fluid vs 370,000 mU h/ml and 4500 ng h/ml in serum, respectively. The i.p. administration of rHuEpo (25–100 U) has been reported to reduce postischemic malonyl dialdehyde levels, NO formation, brain edema, and hippocampal CA1 neuronal loss in gerbils with bilateral carotid occlusion [33]. Other investigators have reported that the systemic administration of high doses of rHuEpo to experimental animals reduces the volume of infarction 24 h after middle cerebral artery occlusion [175], reduces mortality rate [29], prevents neuronal damage [6], increases cerebral blood flow [20, 78], and reduces neurologic deficits [79, 80]. Investigations in rats, in whom crush injury was mimicked by the aneurysm clamp technique or in whom traumatic brain injury was induced, have shown that rHuEpo prevents motor neuron apoptosis and neurologic disability [42] and improves recovery of motor function [77]. Possibly due to the prevention of death of neuronal cells rHuEpo reduces the inflammatory reaction in hypoxic brain [197]. A recent, elegant gene therapy study in rats has shown that i.v. injected naked plasmid DNA encoding Epo induces neuroprotective effects similar to rHuEpo [202]. Based on animal studies, investigators have also put forward the concept that rHuEpo may be of value in retinal diseases involving apoptosis of photoreceptors or of retinal ganglionic cells [82, 100].
Furthermore, the Epo/Epo-R system has been proposed to play a role in peripheral nerves. Epo-R immunoreactivity has been detected in the somata and axons as well as in Schwann cells of rat sciatic nerves [35]. rHuEpo protects mechanically injured dorsal root ganglionic neurons from undergoing apoptosis [170]. In an experimental model of diabetes, rHuEpo has proved to partially reverse the alterations in nociception and to restore Na+/K+-ATPase activity in nerve fibers [22].
Clinical experience with rHuEpo in stroke patients
Knowing that brain ischemia is a leading cause of death and disability in humans and that there is need for additional stroke therapy strategies, Ehrenreich et al. [62] performed a clinical trial with rHuEpo in patients with acute stroke. Initially, a safety study was carried out in which 13 patients received rHuEpo i.v. (3.3×104 U) once daily for the first 3 days after stroke. The mean concentration of Epo in the cerebral spinal fluid of the patients increased to 17 U/l (compared to the normal value of about 1 U/l [21, 125, 144]). Serum Epo levels in the patients approximated 5000 U/l 3 h after rHuEpo infusion [62] (compared to a normal serum level of about 15 U/l in nonanemic humans [96]). Thereafter, a double-blind randomized proof-of-concept study was carried out on 40 patients who received either rHuEpo or saline. Study inclusion criteria were age <80 years, ischemic stroke within the middle cerebral artery territory, symptom onset <8 h before rHuEpo or saline administration, and deficits on stroke scales. The results of the trial indicated a strong trend for reduction in infarct size in the rHuEpo-treated patients as indicated by magnetic resonance imaging. The reduction in infarct size was associated with a marked neurological recovery and clinical outcome 1 month after stroke. Thus, rHuEpo therapy may add to the beneficial effect of conventional clot-dissolving strategies in stroke patients [62].
Résumé and future directions
Epo produced in the brain exerts a local function that is distinct from that in erythropoiesis. Epo and Epo-R are expressed both by neurons and astrocytes. Similar to renal Epo, cerebral Epo production increases on hypoxic stress. The response to hypoxia is accomplished by the stabilization and activation of the hypoxia-inducible transcription factors 1 and 2 (HIF-1 and HIF-2), which induce the expression of several genes encoding proteins that protect tissues from O2 and energy deprivation. There is convincing evidence that Epo is an antiapoptotic and mitogenic factor for neuronal cells. In experimental animals, locally or systemically administered rHuEpo is neuroprotective against a variety of insults, including cerebral ischemia, subarachnoid hemorrhage, head injury, and experimental autoimmune encephalomyelitis. With respect to the therapeutic use of rHuEpo as a neuroprotective agent in humans, the blood–brain barrier is a critical issue. The concept has been put forward that there are specific transport mechanisms to ship Epo from the systemic circulation into the central nervous system [26]. In addition, Epo may even get easier access to the brain after hypoxic lesion of the blood–brain barrier. Along these lines, hypoxia stimulates the production of vascular endothelial growth factor, which is one of the main mediators of the leakage of the blood–brain barrier in brain ischemia and trauma [66, 140]. In view of the limited penetration of intact Epo across the blood–brain barrier, Erbayraktar et al. [65] have tested the efficacy of asialo-rHuEpo, which has a plasma half-life of 1.4 min and exerts no erythropoiesis-stimulating activity. This drug could be administered at high doses without producing erythrocytosis. Reportedly, asialo-rHuEpo crosses the blood–brain barrier after i.v. administration, binds to neurons within the hippocampus and cortex in a pattern corresponding to the distribution of Epo-R, and is neuroprotective in cerebral ischemia, spinal compression, and sciatic nerve crush [65].
Effects of Epo in the cardiovascular system
Effects of Epo on endothelial cells
Epo-R is expressed on human vascular endothelial cells from coronary, pulmonary, and cerebral arteries, the umbilical vein, and dermal vessels [9, 12]. Epo stimulates in vitro the proliferation and migration of human, murine, and bovine endothelial cells [8, 141, 162]. In addition, Epo induces a proangiogenic phenotype of endothelial cells and neovascularization [39, 92]. It also promotes blood vessel formation in the uterus of ovariectomized mice [214]. Very recent studies have shown that rHuEpo administration in humans produces an increase in the number of circulating endothelial progenitor cells, which may be beneficial in augmenting the neovascularization of ischemic tissues [11, 86]. In vitro, rHuEpo causes a rapid tyrosine phosphorylation of cytosolic proteins and the translocation of STAT5 in human umbilical vein endothelial cell (HUVEC) cultures [84]. A recent differential display analysis of HUVEC extracts has revealed four groups of genes that are upregulated by rHuEpo, including those encoding proteins in the control of vascular function (e.g., thrombospondin-1), gene transcription (e.g., c-myc purine-binding transcription factor PuF), mitochondrial function (e.g., cytochrome C oxidase subunit 1), and regulators of signal transduction [68]. However, it must be noted—and this holds true for most of the endothelial cell culture studies cited below—that the concentration of rHuEpo added to the cultures was much higher than that generally reached in the vascular bed.
Epo can stimulate the production of several endothelial-derived modulators of the vascular tone, including some with vasoconstrictive and some with vasorelaxant properties. The highly potent vasoconstrictor endothelin is released [37, 106] via a Ca2+-dependent mechanism [24, 38, 198]. This effect has been implicated clinically in the arterial hypertension observed in rHuEpo-treated patients with chronic renal failure [37]. Clearly, however, rHuEpo therapy-induced increases in arterial blood pressure result primarily from the elevated blood viscosity and the abolishment of tissue hypoxia-associated vasodilation [51]. Indeed, while tenfold overexpression of Epo in transgenic mice activates the tissue endothelin system [159], the plasma endothelin concentration is not consistently elevated in rHuEpo-treated patients with chronic renal failure [28]. Studies in partially nephrectomized uremic rats have shown that immunoreactive endothelin-1 is increased in arterial walls on rHuEpo therapy, but not in circulation [115]. In the same experimental model, selective endothelin-1 receptorA (ETA) blockade but not nonselective ETA/B blockade prevents the aggravation of hypertension [27]. Interestingly, the acute administration of rHuEpo produces increases in plasma endothelin-1 levels and in blood pressure in spontaneously hypertensive rats (SHR), but not in normotensive Wistar-Kyoto rats (WKR; [188]). Thus, rHuEpo may aggravate a preexisting state of hypertension.
Other mediators reported to be released in response to rHuEpo treatment of cultured endothelial cells include the vasoconstricting prostanoids prostaglandin F2α and thromboxane B2 [24], plasminogen activator inhibitor-1 [142], and the vasodilating vascular endothelial growth factor (VEGF) [147]. The vasodilatory action of VEGF is known to be mediated by NO [90, 154]. The exact nature of the interference of Epo with the endothelial NO system remains to be clarified, since the induction of NO synthase protein and activity [12, 190, 206, 212] as well as no change [146] or depression [149, 201] have been reported. In vivo, the Epo-induced rise of the red blood cell count increases blood viscosity and endothelial shear stress, which is the major regulating stimulus for the endothelial NO system. Thus, in Epo transgenic mice (hematocrit 0.85), the intravascular NO bioavailability is increased due to marked induction of the endothelial NO synthase [85, 199].
Effects of Epo on vascular smooth muscle
Vascular smooth muscle cells also have been shown to express Epo-R [4, 7] and been investigated extensively in relation to the rHuEpo therapy-associated arterial hypertension. In reduced complexity experiments using isolated vessels and cell cultures, Epo induced vasoconstriction [87] and vascular smooth muscle contraction [136], whereas in the isolated hemoglobin-perfused kidney setting no increase in renal vascular resistance was observed [153]. Epo signaling in vascular smooth muscle cells is Ca2+ dependent [5, 114, 145] and includes the activation of the phospholipase C cascade and the activation of oncogenes (myc, jun, fos) [76] promoting DNA replication and cellular growth. Thus, by endorsing vascular smooth muscle cell growth, Epo could be a factor in vascular hypertrophy and arterial hypertension. Other intracellular signals activated by Epo include the MAPK [5, 7] and PI-3K/Akt pathways [4], which are intimately involved in the inhibition of apoptosis [60, 208].
Myocardial protection by Epo
To foster new therapeutic approaches for the treatment of myocardial disease, it is critical to dissect the underlying subcellular pathways used by potential cytoprotectants. In this regard, rHuEpo has become especially attractive. The prerequisite of Epo action, the expression of Epo-R, has been proven for murine [211] as well as human embryonic cardiomyocytes [103], and for human adult cardiomyocytes [189] and cardiac tissue (own unpublished observation). Epo has been shown to act as a mitogen on neonatal rat cardiomyocytes, a process which involves tyrosine kinase and protein kinase C [200]. Of note, the murine knockout of either the Epo gene or the Epo-R gene results in a phenotype of severe cardiac malformations with embryonic lethality at embryonic day 13.5 [211]. However, more recent studies have shown that a tissue-specific knockout of Epo-R selectively in nonhemopoietic tissues does not result in a defect or abnormality in the myocardium [182]. Thus, the impaired heart development seen in earlier studies was likely caused by the decrease in red cell numbers, and thereby, reduced O2 transport capacity of the blood.
Recent experimental studies have provided strong evidence that Epo protects the myocardium (Table 1). In hypoxia-exposed mice, treatment with rHuEpo enhances cardiac contractility, while treatment with anti-Epo-antibody has the opposite effect [178]. In addition, it has been reported that Epo administration reduces myocardial infarction volume, protects against ischemia reperfusion injury, and promotes beneficial ventricular remodeling in mice, rats, and rabbits [32, 34, 135, 155, 156]. Importantly, beneficial effects were not only seen with preemptive Epo administration 24 h prior to the coronary artery occlusion but also when Epo was given after reperfusion was started. Thus, in a real-life setting with delayed access of patients to reperfusion treatment, i.e., emergency coronary bypass grafting for acute coronary occlusion, rHuEpo administration several hours later still might have a beneficial effect on infarct size, extent of reperfusion injury, and functional recovery of the myocardium. Importantly, cardioprotective effects of Epo were seen without an increase in hematocrit, thus eliminating O2 delivery as an etiologic factor in myocyte survival and function.
The myocardial protection by Epo is based on the antiapoptotic effect of Epo on cardiac myocytes and possibly cardiac fibroblasts [156]. Epo-induced myocardial survival involves a PI-3/Akt-dependent process leading to a reduction of the number of apoptotic myocytes [48, 156, 189]. Since apoptotic cell death of myocytes is a key feature of myocardial damage from infarction and ischemia/reperfusion [60, 133], the reduction of the number of apoptotic myocytes might result in improved myocardial survival and function.
Impact of rHuEpo therapy in oncology
Rationale for the use of rHuEpo
Patients with malignant tumors often present with normochromic and normocytic anemia. Although red cell survival may also be shortened, the anemia is primarily of the hypoproliferative type. Iron availability is reduced despite normal iron stores. Proinflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), and interferons inhibit the proliferation of erythrocytic progenitors [129] and suppress Epo gene expression [94]. The concentration of serum Epo is relatively low for the degree of anemia in many cancer patients as first shown by Miller et al. [131]. Anemia (hemoglobin <120 g/l) is a negative prognostic factor for successful tumor therapy and disease-free survival [73, 187, 193]. A thoughtful review of the role of tumor hypoxia on tumor progression has been provided very recently [194]. rHuEpo or its analogue, darbepoetin alfa [75], can substitute for the lacking endogenous Epo and counteract the cytotoxic effects of chemotherapy and radiotherapy on erythrocytic progenitors. In vitro studies with Epo-producing human hepatic cells [209] and isolated perfused canine kidneys [67] have shown that cytotoxic drugs may inhibit Epo synthesis, apart from their suppressing erythropoiesis directly. On the other hand, however, clinical studies indicate that Epo production is maintained on both platinum and nonplatinum chemotherapy [36].
The primary goals of rHuEpo therapy are to maintain the patient’s hemoglobin concentration above the transfusion trigger, to reduce asthenia or fatigue, and to increase exercise tolerance [55, 74, 150]. The alleviation of tumor and tumor therapy-associated anemia may also restore brain functions [98, 117]. Although it is generally taken for granted that rHuEpo treatment leads to anemia improvement in cancer patients, a recent critical review has emphasized the use of inappropriate quality of life instruments in almost all clinical studies reported so far and suggested more reliable research [25]. In general, 10–30% of the tumor patients still have to be transfused despite rHuEpo therapy [14]. An increase in hemoglobin concentration ≥10 g/l or in reticulocyte count ≥40×109/l after 4 weeks of rHuEpo treatment is considered a reliable indicator of response [41]. According to evidence-based clinical practice guidelines published by the American Society of Clinical Oncology and the American Society of Hematology [163], the use of rHuEpo is recommended as a treatment option for patients with chemotherapy-associated anemia with a hemoglobin concentration below 100 g/l. The dosage of rHuEpo (or its analogue darbepoetin alfa) should be titrated to maintain a hemoglobin concentration of 120 g/l to avoid cardiovascular disorders.
Two recent clinical trials have shown that hemoglobin concentrations in cancer patients should not be raised into the normal range. First, a multicenter trial that sought to assess the effect of rHuEpo treatment to maintain normal hemoglobin concentrations on survival in breast cancer patients under chemotherapy was terminated early because of an increase in mortality in the first 4 months of the study [116]. However, there were difficulties in the interpretation of that study which showed imbalances of risk factors between the rHuEpo and the placebo groups, and the lack of occurrence of anemia in the placebo group [116]. Second, a randomized, double-blind, placebo-controlled trial on 351 patients with head and neck cancers receiving radiotherapy has shown an advantage in progression-free survival in the placebo-treated compared to the rHuEpo-treated group [89]. However, apart from some imbalances in prognostic factors (such as smoking) and in patient numbers, a main problem of this study seems to have been the overcorrection of anemia [192]. The mean hemoglobin concentration was 154 g/l (±17 g/l) in patients treated with rHuEpo for 9 weeks [89]. Evidence suggests that the hemoglobin concentration leading to the highest tumor oxygenation ranges from 130 g/l to 140 g/l, because intratumoral blood flow is disturbed at higher hemoglobin levels [192]. The association between O2 capacity of the blood and tumor oxygenation, on the one side, and tumor growth and efficiency of radiotherapy and chemotherapy, on the other side, is plausible [59, 69, 72, 91, 186, 195]. As discussed elsewhere [95], however, neither the experimental nor the clinical support for this association is fully convincing. Even less well understood is the mechanism underlying the recent observation that rHuEpo treatment improves tumor oxygenation independently of its effect on erythropoiesis [23]. In this study rHuEpo was administered to rats before or after mammary adenocarcinoma transplantation. At similar blood hemoglobin concentrations, O2 measurement histograms revealed significantly less hypoxic tumor areas in animals during rHuEpo therapy [23].
Direct influence of Epo on tumor cell growth
Since rHuEpo was introduced as a drug for treatment of renal anemia almost 20 years ago, several groups of investigators have carefully studied whether Epo can induce or promote tumor growth. Clinically, no evidence has been reported so far indicating that the erythrocytic growth factor Epo directly stimulates tumor cell proliferation. In addition, an elegant study in transgenic mice transfected with a construct that linked the human Epo gene to an erythroid-specific regulatory element has shown that the continuous stimulation of erythropoiesis leads to erythrocytosis but not to erythroleukemia [122]. However, there is at least one case report of Epo-dependent leukemic transformation of myelodysplastic syndrome (MDS) to acute monoblastic leukemia (AML) [31]. A careful examination has shown Epo-R expression on leukemia cells in 60% of patients with all French–American–British types of AML and in 29% of acute lymphoblastic leukemia (ALL) cases [184]. In vitro a proliferative response to Epo was observed in 16% of patients. Patients with both Epo-R expression and in vitro response to Epo had shorter remission duration than those without Epo-R [184]. Thus, close observation for leukemic transformation is necessary in patients with MDS on rHuEpo therapy.
In light of the demonstration of Epo-R expression in various nonerythroid tissues, it is important to regard studies aimed at investigating direct effects of Epo on tumor cells. Kayser and Gabius [108] first suggested that human tumors may express Epo-R. In their study 81% of human lung carcinoma tissues possessed Epo-binding sites as detected by use of biotinylated rHuEpo. Epo-R transcripts and Epo-R protein were subsequently demonstrated in human renal carcinoma [204], tumors of the cervix and other organs of the female reproductive tract [3, 215, 216], and in various specimens of common pediatric tumors such as neuroblastomas, brain tumors, hepatoblastomas, and Wilms’ tumors [15]. By immunohistochemistry, Epo-R has been shown to be expressed in breast carcinoma [2, 10, 88] and in vestibular schwannoma [58].
Of major concern are studies indicating that local inhibition of Epo-R signaling results in tumor regression. Yasuda et al. [215] first showed that the application of anti-Epo-antibody or of soluble Epo-R into transplants of uterine or ovarian tumors in nude mice produces a decrease in tumor size. Similarly, the administration of anti-Epo-antibody, soluble Epo-R, or an inhibitor of JAK2 resulted in a delay in tumor growth with 45% reduction in maximal tumor depth in a tumor-Z chamber model with rat mammary adenocarcinoma cells [10]. Yasuda et al. [215] have put forward the interesting hypothesis that Epo could be essential for the development of tumor progression through its antiapoptotic effect on the endothelium. In addition, in the absence of an intact Epo/Epo-R system, the increase in the number of cells undergoing apoptotic death may promote local immune reactions by attracting neutrophilic granulocytes and monocytes [215]. Mittelman et al. [132], in studying murine myeloma models, have shown that rHuEpo treatment induced complete tumor regression in 30–60% of mice with a syngeneic progressively growing tumor. This regression was related to a tumor-specific immune response to the myeloma cells which was mediated by T cells [132]. The effects of Epo on antitumor immune responses are an important field for future investigations.
Previous reports on the expression of Epo-R and the effects of rHuEpo on cultured tumor cells are also conflicting. Epo-R has been detected in cultures of various malignant human cell lines [1, 3, 10, 58, 152, 171, 204, 205, 217]. Some of these studies suggested a link between Epo-R expression and tumor cell proliferation [1, 2, 10, 204]. For example, rHuEpo (range: 0.5–100 U/ml) stimulated the growth of human renal carcinoma cells in culture [204]. The addition of rHuEpo (≥10 U/ml) produced a significant increase in the release of angiogenic growth factors from tumor cell cultures, namely VEGF and placenta growth factor, from pediatric tumor cell lines [15]. The authors have suggested that Epo antagonists (with transfusion support) could be potentially used in conjunction with antiangiogenic agents and chemotherapy [15]. In other studies no relationship between Epo-R expression and tumor growth was apparent [17, 58, 83, 164, 171, 205, 210]. In fact, no growth stimulation of human primary tumor specimens [16] or of permanent hemopoietic or nonhemopoietic malignant cell lines [17, 139, 164] was observed, even when tumor cells were incubated with Epo at concentrations that were by several orders of magnitude higher than those achieved physiologically or by the administration of rHuEpo. Westphal et al. [205] recently investigated Epo-R and granulocyte colony-stimulating factor (G-CSF) receptor expression in various human benign and malignant cell lines. Treatment with rHuEpo (up to 1000 U/ml) had no effect on the rate of proliferation and tyrosine phosphorylation when Epo-R-positive tumor cell lines were tested. In studying various tumor cell lines, Liu et al. [120] found that neither rHuEpo nor granulocyte-monocyte colony-stimulating factor (GM-CSF) influence basal viability. However, pretreatment with these growth factors resulted in a reduction of the cytotoxic effects of cisplatin in cell lines with high growth factor receptor expression [120].
Conclusions
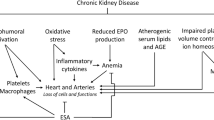
The primary function of Epo is to inhibit apoptosis of erythrocytic progenitors and, thereby, to stimulate the growth of young red blood cells. rHuEpo has proven to be a most useful replacement therapy for the prevention of the anemia associated with chronic kidney disease. The drug raises hematocrit and blood hemoglobin concentration in a dose-dependent and predictable way and abolishes the need for red cell transfusion with its risks of incompatibility reactions, infections, and iron overload. Present pharmacological interest focuses on the use and development of recombinant erythropoiesis-stimulating drugs with prolonged survival in the circulation by producing analogues with additional carbohydrate chains [61, 63] or with attached polyethylene glycol polymers [121]. Next to the renal anemias, possible indications for the administration of rHuEpo may be the anemias associated with autoimmune diseases, acquired immunodeficiency syndrome (AIDS), malignancies, and surgical interventions. The detection of functional Epo-R in nonhemopoietic tissues has recently aroused experimental and clinical interest in the use of rHuEpo as a survival factor for nonhemopoietic cells and tissues.
Convincing evidence has accumulated that Epo acts as a neurotrophic and neuroprotective factor in the central nervous system. In vitro, Epo protects neuronal cells from hypoxia-induced and glutamate-induced cell death. In animal models, Epo promotes the survival of neurons and synapses and reduces the size of infarct areas in the ischemic brain. A first clinical trial has shown neuroprotective potential of rHuEpo in patients with acute stroke [62]. It is hoped that the drug may prove useful for treatment of other neuronal disorders such as brain trauma, inflammatory diseases, and degenerative diseases [30, 98, 123, 167].
Epo exerts mitotic effects on vascular tissues and induces the production and release of vasoactive mediators from the endothelium. Of major clinical relevance are recent observations showing that rHuEpo promotes the mobilization of endothelial progenitor cells which may be beneficial in the vascularization of ischemic tissues, including the heart [11, 86]. Knowledge concerning the ability of Epo to function as a specific myocardial protectant has recently started to emerge and the present experimental and clinical evidence mandate further, thorough investigation of the therapeutic potential of rHuEpo for myocardial protection. At present, it seems clear that cardiomyocytes express Epo-R and respond to Epo with activation of signaling pathways resembling those known from erythropoietic cells.
The role of Epo in tumor therapy needs to be further explored. Anemia-associated tissue hypoxia promotes angiogenesis, growth, and metastasis of tumors [172,194]. In addition, the efficacy of radiotherapy and chemotherapy depends on the availability of O2. In most cancer patients rHuEpo therapy increases the blood hemoglobin concentration and tumor oxygenation, thereby increasing the sensitivity of the tumor cells to radiotherapy and chemotherapy [73, 187]. The rHuEpo doses should be titrated to maintain a hemoglobin concentration of 120 g/l in tumor patients [163] because a further increase bears the risk of cardiovascular disorders. In vitro studies have shown that tumor cells may express Epo-R. These findings merit further exploration. However, based on almost 20 years of the use of rHuEpo in the clinical routine and on the observation of anemic persons with high endogenous Epo levels, there is at present no reason to give patients a fear that Epo may induce or promote tumor growth.
References
Acs G, Acs P, Beckwith SM, Pitts RL, Clements E, Wong K, Verma A (2001) Erythropoietin and erythropoietin receptor expression in human cancer. Cancer Res 61:3561–3565
Acs G, Zhang PJ, Rebbeck TR, Acs P, Verma A (2002) Immunohistochemical expression of erythropoietin and erythropoietin receptor in breast carcinoma. Cancer 95:969–981
Acs G, Zhang PJ, McGrath CM, Acs P, McBroom J, Mohyeldin A, Liu S, Lu H, Verma A (2003) Hypoxia-inducible erythropoietin signaling in squamous dysplasia and squamous cell carcinoma of the uterine cervix and its potential role in cervical carcinogenesis and tumor progression. Am J Pathol 162:1789–1806
Akimoto T, Kusano E, Inaba T, Iimura O, Takahashi H, Ikeda H, Ito C, Ando Y, Ozawa K, Asano Y (2000) Erythropoietin regulates vascular smooth muscle cell apoptosis by a phosphatidylinositol 3 kinase-dependent pathway. Kidney Int 58:269–282
Akimoto T, Kusano E, Ito C, Yanagiba S, Inoue M, Amemiya M, Ando Y, Asano Y (2001) Involvement of erythropoietin-induced cytosolic free calcium mobilization in activation of mitogen-activated protein kinase and DNA synthesis in vascular smooth muscle cells. J Hypertens 19:193–202
Alafaci C, Salpietro F, Grasso G, Sfacteria A, Passalacqua M, Morabito A, Tripodo E, Calapai G, Buemi M, Tomasello F (2000) Effect of recombinant human erythropoietin on cerebral ischemia following experimental subarachnoid hemorrhage. Eur J Pharmacol 406:219–225
Ammarguellat F, Llovera M, Kelly PA, Goffin V (2001) Low doses of EPO activate MAP kinases but not JAK2-STAT5 in rat vascular smooth muscle cells. Biochem Biophys Res Commun 284:1031–1038
Anagnostou A, Lee ES, Kessimian N, Levinson R, Steiner M (1990) Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proc Natl Acad Sci U S A 87:5978–5982
Anagnostou A, Liu Z, Steiner M, Chin K, Lee ES, Kessimian N, Noguchi CT (1994) Erythropoietin receptor mRNA expression in human endothelial cells. Proc Natl Acad Sci U S A 91:3974–3978
Arcasoy MO, Amin K, Karayal AF, Chou SC, Raleigh JA, Varia MA, Haroon ZA (2002) Functional significance of erythropoietin receptor expression in breast cancer. Lab Invest 82:911–918
Bahlmann FH, De Groot K, Spandau JM, Landry AL, Hertel B, Duckert T, Boehm SM, Menne J, Haller H, Fliser D (2004) Erythropoietin regulates endothelial progenitor cells. Blood 103:921–926
Banerjee D, Rodriguez M, Nag M, Adamson JW (2000) Exposure of endothelial cells to recombinant human erythropoietin induces nitric oxide synthase activity. Kidney Int 57:1895–1904
Barber DL, Beattie BK, Mason JM, Nguyen MHH, Yoakim M, Neel BG, D’Andrea AD, Frank DA (2001) A common epitope is shared by activated signal transducer and activator of transcription-5 (STAT5) and the phosphorylated erythropoietin receptor: implications for the docking model of STAT activation. Blood 97:2230–2237
Barosi G, Marchetti M, Liberato NL (1998) Cost-effectiveness of recombinant human erythropoietin in the prevention of chemotherapy-induced anaemia. Br J Cancer 78:781–787
Batra S, Perelman N, Luck LR, Shimada H, Malik P (2003) Pediatric tumor cells express erythropoietin and a functional erythropoietin receptor that promotes angiogenesis and tumor cell survival. Lab Invest 83:1477–1487
Bauer E, Danhauser-Riedl S, De Riese W, Raab H-R, Sandner S, Meyer H-J, Neukam D, Hanauske U, Freund M, Poliwoda H, Rastetter J, Hanauske A-R (1992) Effects of recombinant human erythropoietin on clonogenic growth of primary human tumor specimens in vivo. Onkologie 15:254–258
Berdel WE, Oberberg D, Reufi B, Thiel E (1991) Studies on the role of recombinant human erythropoietin in the growth regulation of human nonhematopoietic tumor cells in vitro. Ann Hematol 63:5–8
Bernaudin M, Marti HH, Roussel S, Divoux D, Nouvelot A, MacKenzie ET, Petit E (1999) A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J Cereb Blood Flow Metab 19:643–651
Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, Duchatelle I, MacKenzie ET, Petit E (2000) Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia 30:271–278
Bertram Springborg J, Ma X, Rochat P, Knudsen GM, Amtorp O, Paulson OB, Juhler M, Vidiendal Olsen N (2002) A single subcutaneous bolus of erythropoietin normalizes cerebral blood flow autoregulation after subarachnoid haemorrhage in rats. Br J Pharmacol 135:823–829
Bertram Springborg J, Sonne B, Frederiksen HJ, Foldager N, Poulsgaard L, Klausen T, Steen Jorgensen S, Vidiendal Olsen N (2003) Erythropoietin in the cerebrospinal fluid of patients with aneurysmal subarachnoid haemorrhage originates from the brain. Brain Res 12:143–148
Bianchi R, Buyukakilli B, Brines M, Savino C, Cavaletti G, Oggioni N, Lauria G, Borgna M, Lombardi R, Cimen B, Comelekoglu U, Kanik A, Tataroglu C, Cerami A, Ghezzi P (2004) Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proc Natl Acad Sci U S A 101:823–828
Blackwell KL, Kirkpatrick JP, Snyder SA, Broadwater G, Farrell F, Jolliffe L, Brizel DM, Dewhirst MW (2003) Human recombinant erythropoietin significantly improves tumor oxygenation independent of its effects on hemoglobin. Cancer Res 63:6162–6165
Bode-Boger SM, Boger RH, Kuhn M, Radermacher J, Frolich JC (1996) Recombinant human erythropoietin enhances vasoconstrictor tone via endothelin-1 and constrictor prostanoids. Kidney Int 50:1255–1261
Bottomley A, Thomas R, van Steen K, Flechtner H, Djulbegovic B (2002) Human recombinant erythropoietin and quality of life: a wonder drug or something to wonder about? Lancet Oncol 3:145–153
Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, Itri LM, Cerami A (2000) Erythropoietin crosses the blood–brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A 97:10526–10531
Brochu E, Lacasse S, Lariviere R, Kingma I, Grose JH, Lebel M (1999) Differential effects of endothelin-1 antagonists on erythropoietin-induced hypertension in renal failure. J Am Soc Nephrol 10:1440–1446
Brunet P, Lorec AM, Leonetti F, Roubicek C, Jaber K, Roux F, Berland Y (1994) Plasma endothelin in haemodialysis patients treated with recombinant human erythropoietin. Nephrol Dial Transplant 9:650–654
Buemi M, Grasso G, Corica F, Calapai G, Salpietro FM, Casuscelli T, Sfacteria A, Aloisi C, Alafaci C, Sturiale A, Frisina N, Tomasello F (2000) In vivo evidence that erythropoietin has a neuroprotective effect during subarachnoid hemorrhage. Eur J Pharmacol 392:31–34
Buemi M, Cavallaro E, Floccari F, Sturiale A, Aloisi C, Trimarchi M, Corica F, Frisina N (2003) The pleiotropic effects of erythropoietin in the central nervous system. J Neuropathol Exp Neurol 62:228–236
Bunworasate U, Amouk H, Mindeman H, O’Loughlin KL, Sait SNJ, Barcos M, Stewart CC, Baer MR (2001) Erythropoietin-dependent transformation of myelodysplastic syndrome to acute monoblastic leukemia. Blood 98:3492–3494
Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL (2003) Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation 108:79–85
Calapai G, Marciano MC, Corica F, Allegra A, Parisi A, Frisina N, Caputi AP, Buemi M (2000) Erythropoietin protects against brain ischemic injury by inhibition of nitric oxide formation. Eur J Pharmacol 401:349–356
Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A, Brines M (2003) Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci U S A 100:4802–4806
Campana WM, Myers RR (2001) Erythropoietin and erythropoietin receptors in the peripheral nervous system: changes after nerve injury. FASEB J 15:1804–1806
Canaparo R, Casale F, Muntoni E, Zara GP, Della-Pepa C, Berno E, Pons N, Fornari G, Eandi M (2000) Plasma erythropoietin concentrations in patients receiving intensive platinum or nonplatinum chemotherapy. Br J Clin Pharmacol 50:146–153
Carlini RG, Dusso AS, Obialo CI, Alvarez UM, Rothstein M (1993) Recombinant human erythropoietin (rHuEPO) increases endothelin-1 release by endothelial cells. Kidney Int 43:1010–1014
Carlini RG, Gupta A, Liapis H, Rothstein M (1995) Endothelin-1 release by erythropoietin involves calcium signaling in endothelial cells. J Cardiovasc Pharmacol 26:889–892
Carlini RG, Reyes AA, Rothstein M (1995) Recombinant human erythropoietin stimulates angiogenesis in vitro. Kidney Int 47:740–745
Catania MA, Marciano MC, Parisi A, Struriale A, Buemi M, Grasso G, Squadrito F, Caputi AP, Calapai G (2002) Erythropoietin prevents cognition impairment induced by transient brain ischemia in gerbils. Eur J Pharmacol 437:147–150
Cazzola M, Mercuriali F, Brugnara C (1997) Use of recombinant human erythropoietin outside the setting of uremia. Blood 89:4248–4267
Celik M, Gokmen N, Erbayraktar S, Akhisaroglu M, Konakc S, Ulukus C, Genc S, Genc K, Sagiroglu E, Cerami A, Brines M (2002) Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc Natl Acad Sci U S A 99:2258–2263
Cerami A, Brines M, Ghezzi P, Cerami C, Itri LM (2002) Neuroprotective properties of epoetin alfa. Nephrol Dial Transplant 17:8–12
Cheetham JC, Smith DM, Aoki KH, Stevenson JL, Hoeffel TJ, Syed RS, Egrie J, Harvey TS (1998) NMR structure of human erythropoietin and a comparison with its receptor bound conformation. Nat Struct Biol 5:861–866
Chin K, Yu X, Beleslin-Cokic B, Liu C, Shen K, Mohrenweiser HW, Noguchi CT (2000) Production and processing of erythropoietin receptor transcripts brain. Mol Brain Res 81:29–42
Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623–634
Chong ZZ, Kang JQ, Maiese K (2002) Hematopoietic factor erythropoietin fosters neuroprotection through novel signal transduction cascades. J Cereb Blood Flow Metab 22:503–514
Chong ZZ, Kang JQ, Maiese K (2003) Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation 106:2973–2979
Chong ZZ, Kang JQ, Maiese K (2003) Erythropoietin: cytoprotection in vascular and neuronal cells. Curr Drug Targets Cardiovasc Haematol Disord 3:141–154
Chong ZZ, Lin SH, Kang JQ, Maiese K (2003) Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J Neurosci Res 71:659–669
Cirillo M, Capasso G, DeSanto NG (1993) Relationship between hematocrit and blood pressure: implications for primary hypertension. Nephron 65:505–510
Dame C, Bartmann P, Wolber E-M, Fahnenstich H, Hofmann D, Fandrey J (2000) Erythropoietin gene expression in different areas of the developing human central nervous system. Dev Brain Res 125:69–74
Darling RJ, Kuchibhotla U, Glaesner W, Micanovic R, Witcher DR, Beals JM (2002) Glycosylation of erythropoietin affects receptor binding kinetics: role of electrostatic interactions. Biochemistry 41:14524–14531
De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, Srinivasula SM, Alnemri ES, Testa U, Peschle C (1999) Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 401:489–493
Demetri GD, Kris M, Wade J, Degos L, Cella D (1998) Quality-of-life benefit in chemotherapy patients treated with epoetin alfa is independent of disease response or tumor type: results from a prospective community oncology study. Procrit Study Group. J Clin Oncol 16:3412–3425
Digicaylioglu M, Lipton SA (2001) Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 412:641–647
Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C, Gassmann M (1995) Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proc Natl Acad Sci U S A 92:3717–3720
Dillard DG, Venkatraman G, Cohen C, Delgaudio J, Gal AA, Mattox DE (2001) Immunolocalization of erythropoietin and erythropoietin receptor in vestibular schwannoma. Acta Otolaryngol 121:149–152
Dunst J (2000) Hemoglobin level and anemia in radiation oncology: prognostic impact and therapeutic implications. Semin Oncol 27:4–8
Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, Lips DJ, Doevendans PA (2004) Role of apoptosis in reperfusion injury. Cardiovasc Res 61:414–426
Egrie JC, Browne JK (2001) Development and characterization of novel erythropoiesis stimulating protein (NESP). Br J Cancer 84:3–10
Ehrenreich H, Hasselblatt M, Dembrowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck H-H, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Rüther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Sirén A-L (2002) Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med 8:495–505
Elliott S, Lorenzini T, Asher S, Aoki K, Brankow D, Buck L, Busse L, Chang D, Fuller J, Grant J, Hernday N, Hokum M, Hu S, Knudten A, Levin N, Komorowski R, Martin F, Navarro R, Osslund T, Rogers G, Rogers N, Trail G, Egrie J (2003) Enhancement of therapeutic protein in vivo activities through glycoengineering. Nat Biotechnol 21:414–421
Emerson MR, Samson FE, Pazdernik TL (2000) Effects of hypoxia preconditioning on expression of metallothionein-1,2 and heme oxygenase-1 before and after kainic acid-induced seizures. Cell Mol Biol 46:619–626
Erbayraktar S, Grasso G, Sfacteria A, Xie Q, Coleman T, Kreilgaard M, Torup L, Sager T, Erbayraktar Z, Gokmen N, Yilmaz O, Ghezzi P, Villa P, Fratelli M, Casagrande S, Leist M, Helboe L, Gerwein J, Christensen S, Geist MA, Pedersen LO, Cerami-Hand C, Wuerth JP, Cerami A, Brines M (2003) Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Natl Acad Sci U S A 100:6741–6746
Fischer S, Wobben M, Marti HH, Renz D, Schaper W (2002) Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res 63:70–80
Fisher JW, Roh BL (1964) Influence of alkylating agents on kidney erythropoietin production. Cancer Res 24:983–988
Fodinger M, Fritsche-Polanz R, Buchmayer H, Skoupy S, Sengoelge G, Horl WH, Sunder-Plassmann G (2000) Erythropoietin-inducible immediate-early genes in human vascular endothelial cells. J Investig Med 48:137–149
Fyles AW, Milosevic M, Pintilie M, Syed A, Hill RP (2000) Anemia, hypoxia and transfusion in patients with cervix cancer: a review. Radiother Oncol 57:13–19
Genc S, Kuralay F, Genc K, Akhisaroglu M, Fadiloglu S, Yorukoglu K, Fadiloglu M, Gure A (2001) Erythropoietin exerts neuroprotection in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated C57/BL mice via increasing nitric oxide production. Neurosci Lett 298:139–141
Genc S, Akhisaroglu M, Kuralay F, Genc K (2002) Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci Lett 321:73–76
Glaser CM, Millesi W, Kornek GV, Lang S, Schull B, Watzinger F, Selzer E, Lavey RS (2001) Impact of hemoglobin level and use of recombinant erythropoietin on efficacy of preoperative chemoradiation therapy for squamous cell carcinoma of the oral cavity and oropharynx. Int J Radiat Oncol Biol Phys 50:705–715
Glaspy J (1997) The impact of epoetin alfa on quality of life during cancer chemotherapy: a fresh look at an old problem. Semin Hematol 34:20–26
Glaspy J, Bukowski R, Steinberg D, Taylor C, Tchekmedyian S, Vadhan RS (1997) Impact of therapy with epoetin alfa on clinical outcomes in patients with nonmyeloid malignancies during cancer chemotherapy in community oncology practice. J Clin Oncol 15:1218–1234
Glaspy JA, Jadeja JS, Justice G, Kessler J, Richards D, Schwartzberg L, Tchekmedyian NS, Armstrong S, O’Byrne J, Rossi G, Colowick AB (2002) Darbepoetin alfa given every 1 or 2 weeks alleviates anaemia associated with cancer chemotherapy. Br J Cancer 87:268–276
Gogusev J, Zhu DL, Herembert T, Ammarguellat F, Marche P, Drueke T (1994) Effect of erythropoietin on DNA synthesis, proto-oncogene expression and phospholipase C activity in rat vascular smooth muscle cells. Biochem Biophys Res Commun 199:977–983
Gorio A, Gokmen N, Erbayraktar S, Yilmaz O, Madaschi L, Cichetti C, Di Giulio AM, Vardar E, Cerami A, Brines M (2002) Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc Natl Acad Sci U S A 99:9450–9455
Grasso G (2001) Neuroprotective effect of recombinant human erythropoietin in experimental subarachnoid hemorrhage. J Neurosurg Sci 45:7–14
Grasso G, Buemi M, Alafaci C, Sfacteria A, Passalacqua M, Sturiale A, Calapai G, De Vico G, Piedimonte G, Salpietro FM, Tomasello F (2002) Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc Natl Acad Sci U S A 99:5627–5631
Grasso G, Passalacqua M, Sfacteria A, Conti A, Morabito A, Mazzullo G, De Vico G, Buemi M, Macri B, Tomasello F (2002) Does administration of recombinant human erythropoietin attenuate the increase of S-100 protein observed in cerebrospinal fluid after experimental subarachnoid hemorrhage? J Neurosurg 96:565–570
Gregory T, Yu C, Ma A, Orkin SH, Blobel GA, Weiss MJ (1999) GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94:87–96
Grimm C, Wenzel A, Groszer M, Mayser H, Seeliger M, Samardzija M, Bauer C, Gassmann M, Remé CE (2002) HIF-1-induced erythropoietin in the hypoxic retina protects against light-induced retinal degeneration. Nature Med 8:718–724
Grossi A, Vannucchi AM, Bacci P, Caporale R, Cappelli G, Visconti G, Pagliai G, Ferrini PR (1998) Erythropoietin upregulates the expression of its own receptor in TF-1 cell line. Leuk Res 22:145–151
Haller H, Christel C, Dannenberg L, Thiele P, Lindschau C, Luft FC (1996) Signal transduction of erythropoietin in endothelial cells. Kidney Int 50:481–488
Hasegawa J, Wagner KF, Karp D, Li D, Shibata J, Heringlake M, Bahlmann L, Depping R, Fandrey J, Schmucker P, Uhlig S (2004) Altered pulmonary vascular reactivity in mice with excessive erythrocytosis. Am J Respir Crit Care Med 169:829–835
Heeschen C, Aicher A, Lehmann R, Fichtlscherer S, Vasa M, Urbich C, Mildner-Rihm C, Martin H, Zeiher AM, Dimmeler S (2003) Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood 102:1340–1346
Heidenreich S, Rahn KH, Zidek W (1991) Direct vasopressor effect of recombinant human erythropoietin on renal resistance vessels. Kidney Int 39:259–265
Hengartner MO, Horvitz HR (1994) C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76:665–676
Henke M, Laszing R, Rübe C, Schäfer U, Haase K-D, Schilcher B, Mose S, Beer KT, Burger U, Dougherty C, Frommhold H (2003) Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet 362:1255–1260
Horowitz JR, Rivard A, van der Zee R, Hariawala M, Sheriff DD, Esakof DD, Chaudhry GM, Symes JF, Isner JM (1997) Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol 17:2793–2800
Höckel M, Schlenger K, Mitze M, Schäffer U, Vaupel P (1996) Hypoxia and radiation response in human tumors. Semin Radiat Oncol 6:3–9
Jaquet K, Krause K, Tawakol-Khodai M, Geidel S, Kuck KH (2002) Erythropoietin and VEGF exhibit equal angiogenic potential. Microvasc Res 64:326–333
Jelkmann W (1986) Renal erythropoietin: properties and production. Rev Physiol Biochem Pharmacol 104:139–215
Jelkmann W (1998) Proinflammatory cytokines lowering erythropoietin production. J Interferon Cytokine Res 18:555–559
Jelkmann W (2000) Use of recombinant human erythropoietin as an antianemic and performance enhancing drug. Curr Pharm Biotechnol 1:11–31
Jelkmann W (2003) Biochemistry and assays of Epo. In: Jelkmann W (ed) Erythropoietin: molecular biology and clinical use. Graham, Johnson City, pp 35–63
Jelkmann W (2003) Erythropoietin: molecular biology and clinical use. Graham, Johnson City
Jelkmann W (2004) Effects of erythropoietin on brain function. Curr Pharm Biotechnol (in press)
Jumbe NL (2002) Erythropoietic agents as neurotherapeutic agents: what barriers exist? Oncology 16:91–107
Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM (2002) Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci U S A 99:10659–10664
Juul SE (2000) Nonerythropoietic roles of erythropoietin in the fetus and neonate. Clin Perinatol 27:527–541
Juul SE, Harcum J, Li Y, Christensen RD (1997) Erythropoietin is present in the cerebrospinal fluid of neonates. J Pediatr 130:428–430
Juul SE, Yachnis AT, Christensen RD (1998) Tissue distribution of erythropoietin and erythropoietin receptor in the developing human fetus. Early Hum Dev 52:235–249
Juul SE, Stallings SA, Christensen RD (1999) Erythropoietin in the cerebrospinal fluid of neonates who sustained CNS injury. Pediatr Res 46:543–547
Juul SE, Yachnis AT, Rojiani AM, Christensen RD (1999) Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatr Dev Pathol 2:148–158
Katoh K, Mizuno K, Hashimoto S, Okazaki K, Asahi K, Kuriki M, Yamada D, Fukuchi S (1994) Direct evidence for erythropoietin-induced release of endothelin from peripheral vascular tissue. Life Sci 54:L253–L259
Kawakami M, Iwasaki S, Sato K, Takahashi M (2000) Erythropoietin inhibits calcium-induced neurotransmitter release from clonal neuronal cells. Biochem Biophys Res Commun 279:293–297
Kayser K, Gabius HJ (1992) Analysis of expression of erythropoietin-binding sites in human lung carcinoma by the biotinylated ligand. Zentralbl Pathol 138:266–270
Klingmuller U (1997) The role of tyrosine phosphorylation in proliferation and maturation of erythroid progenitor cells—signals emanating from the erythropoietin receptor. Eur J Biochem 249:637–647
Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF (1995) Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 80:729–738
Konishi Y, Chui DH, Hirose H, Kunishita T, Tabira T (1993) Trophic effect of erythropoietin and other hematopoietic factors on central cholinergic neurons in vitro and in vivo. Brain Res 609:29–35
Koshimura K, Murakami A, Sohmiya M, Tanaka I, Katagiri Y (1999) Effects of erythropoietin on neuronal activity. J Neurochem 72:2565–2572
Koury MJ, Bondurant MC (1992) The molecular mechanism of erythropoietin action. Eur J Biochem 210:649–663
Kusano E, Akimoto T, Umino T, Yanagiba S, Inoue M, Ito C, Ando Y, Asano Y (2001) Modulation of endothelin-1-induced cytosolic free calcium mobilization and mitogen-activated protein kinase activation by erythropoietin in vascular smooth muscle cells. Kidney Blood Press Res 24:192–200
Lebel M, Lacasse M, Lariviere R, Kingma I, Grose JH (1998) Plasma and blood vessel endothelin-1 concentrations in hypertensive uremic rats treated with erythropoietin. Clin Exp Hypertens 20:939–951
Leyland-Jones B (2003) Breast cancer trial with erythropoietin terminated unexpectedly. Lancet Oncol 4:459–460
Leyland-Jones B, O’Shoughnessy JA (2003) Erythropoietin as a critical component of breast cancer therapy: survival, synergistic, and cognitive applications. Semin Oncol 30:174–184
Li Y, Juul SE, Morris-Wiman JA, Calhoun DA, Christensen RD (1996) Erythropoietin receptors are expressed in the central nervous system of mid-trimester human fetuses. Pediatr Res 40:376–380
Liu C, Shen K, Liu Z, Noguchi CT (1997) Regulated human erythropoietin receptor expression in mouse brain. J Biol Chem 272:32395–32400
Liu WM, Powles T, Shamash J, Propper D, Oliver T, Joel S (2004) Effect of haemopoietic growth factors on cancer cell lines and their role in chemosensitivity. Oncogene 23:981–990
Macdougall L, Bailon P, Tare N, Pahlke W, Pill J, Brandt M (2003) CERA (Continuous Erythropoiesis Receptor Activator) for the treatment of renal anemia: an innovative agent with unique receptor binding characteristics and prolonged serum half-life. J Am Soc Nephrol 14:769A
Madan A, Lin C, Wang Z, Curtin PT (2003) Autocrine stimulation by erythropoietin in transgenic mice results in erythroid proliferation without neoplastic transformation. Blood Cells Mol Dis 30:82–89
Marti HH, Bernaudin M (2003) Function of erythropoietin in the brain. In: Jelkmann W (ed) Erythropoietin: molecular biology and clinical use. Graham, Johnson City, pp 195–215
Marti HH, Wenger RH, Rivas LA, Straumann U, Digicaylioglu M, Henn V, Yonekawa Y, Bauer C, Gassmann M (1996) Erythropoietin gene expression in human, monkey and murine brain. Eur J Neurosci 8:666–676
Marti HH, Gassmann M, Wenger RH, Kvietikova I, Morganti-Kossmann MC, Kossmann T, Trentz O, Bauer C (1997) Detection of erythropoietin in human liquor: intrinsic erythropoietin production in the brain. Kidney Int 51:416–418
Marti HH, Bernaudin M, Petit E, Bauer C (2000) Neuroprotection and angiogenesis: dual role of erythropoietin in brain ischemia. News Physiol Sci 15:225–229
Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas F, Tabira T, Sasaki R (1993) Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J Biol Chem 268:11208–11216
Masuda S, Nagao M, Sasaki R (1999) Erythropoietic, neurotrophic, and angiogenic functions of erythropoietin and regulation of erythropoietin production. Int J Hematol 70:1–6
Means RT, Krantz SB (1992) Progress in understanding the pathogenesis of the anemia of chronic disease. Blood 80:1639–1647
Michaelis EK (1998) Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol 54:369–415
Miller CB, Jones RJ, Piantadosi S, Abeloff MD, Spivak JL (1990) Decreased erythropoietin response in patients with the anemia of cancer. N Engl J Med 322:1689–1692
Mittelman M, Neumann D, Peled A, Kanter P, Haran-Ghera N (2001) Erythropoietin induces tumor regression and antitumor immune responses in murine myeloma models. Proc Natl Acad Sci U S A 98:5181–5186
Mocanu MM, Baxter GF, Yellon DM (2000) Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br J Pharmacol 130:197–200
Molineux G, Foote M-A, Elliot SG (2003) Erythropoietins and erythropoiesis. Birkhäuser, Basel
Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, Talan MI (2003) Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sci U S A 100:11612–11617
Morakkabati N, Gollnick F, Meyer R, Fandrey J, Jelkmann W (1996) Erythropoietin induces Ca2+ mobilization and contraction in rat mesangial and aortic smooth muscle cultures. Exp Hematol 24:392–397
Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R (1997) Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 76:105–116
Moritz KM, Lim GB, Wintour EM (1997) Developmental regulation of erythropoietin and erythropoiesis. Am J Physiol 273:R1829–R1844
Mundt D, Berger MR, Bode G (1992) Effect of recombinant human erythropoietin on the growth of human tumor cell lines in vitro. Micro-titertec-tetrazolium assay. Arzneimittelforschung 42:92–95
Nag S, Takahashi JL, Kilty DW (1997) Role of vascular endothelial growth factor in blood–brain barrier breakdown and angiogenesis in brain trauma. J Neuropathol Exp Neurol 56:912–921
Nagai T, Akizawa T, Nakashima Y, Kohjiro S, Nabeshima K, Kanamori N, Takayama K, Kinugasa E, Koshikawa S (1995) Effects of rHuEpo on cellular proliferation and endothelin-1 production in cultured endothelial cells. Nephrol Dial Transplant 10:1814–1819
Nagai T, Akizawa T, Kohjiro S, Koiwa F, Nabeshima K, Niikura K, Kino K, Kanamori N, Kinugasa E, Ideura T (1996) rHuEPO enhances the production of plasminogen activator inhibitor-1 in cultured endothelial cells. Kidney Int 50:102–107
Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S, Kim SU (2001) Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. J Neuropathol Exp Neurol 60:386–392
Nakamura T, Ebihara I, Shimada N, Koide H (1998) Elevated levels of erythropoietin in cerebrospinal fluid of depressed patients. Am J Med Sci 315:199–201
Neusser M, Tepel M, Zidek W (1993) Erythropoietin increases cytosolic free calcium concentration in vascular smooth muscle cells. Cardiovasc Res 27:1233–1236
Ni Z, Wang XQ, Vaziri ND (1998) Nitric oxide metabolism in erythropoietin-induced hypertension: effect of calcium channel blockade. Hypertension 32:724–729
Nitta K, Uchida K, Kimata N, Honda K, Kobayashi H, Kawashima A, Yumura W, Nihei H (1999) Recombinant human erythropoietin stimulates vascular endothelial growth factor release by glomerular endothelial cells. Eur J Pharmacol 373:121–124
Noguchi CT, Chen ZY (2003) Nitric oxide mediates hypoxia-induced erythropoietin receptor expression on neurons. Ann Hematol 82:S112
Noguchi K, Yamashiro S, Matsuzaki T, Sakanashi M, Nakasone J, Miyagi K, Sakanashi M (2001) Effect of 1-week treatment with erythropoietin on the vascular endothelial function in anaesthetized rabbits. Br J Pharmacol 133:395–405
Nowrousian MR (1998) Recombinant human erythropoietin in the treatment of cancer-related or chemotherapy-induced anaemia in patients with solid tumours. Med Oncol 15 [Suppl 1]:S19–S28
Oda A, Sawada K-I (2000) Signal transduction in primary cultured human erythroid cells. J Hematother Stem Cell Res 9:417–423
Ohigashi T, Yoshioka K, Fisher JW (1996) Autocrine regulation of erythropoietin gene expression in human hepatocellular carcinoma cells. Life Sci 58:421–427
Pagel H, Jelkmann W, Weiss C (1989) Erythropoietin and blood pressure. Horm Metab Res 21:224
Parenti A, Morbidelli L, Cui XL, Douglas JG, Hood JD, Granger HJ, Ledda F, Ziche M (1998) Nitric oxide is an upstream signal of vascular endothelial growth factor-induced extracellular signal-regulated kinase 1/2 activation in postcapillary endothelium. J Biol Chem 273:4220–4226
Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, Thompson RB, Petrofski JA, Annex BH, Stamler JS, Koch WJ (2003) A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest 112:999–1007
Parsa CJ, Kim J, Riel RU, Pascal LS, Thompson RB, Petrofski JA, Matsumoto A, Stamler JS, Koch WJ (2004) Cardioprotective effects of erythropoietin in the reperfused ischemic heart: a potential role for cardiac fibroblasts. J Biol Chem 279:20655–20662
Philo JS, Aoki KH, Arakawa T, Narhi LO, Wen J (1996) Dimerization of the extracellular domain of the erythropoietin (EPO) receptor by EPO: one high-affinity and one low-affinity interaction. Biochemistry 35:1681–1691
Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, Victorov I, Kapinya K, Dirnagl U, Meisel A (2003) Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 34:1981–1986
Quaschning T, Ruschitzka F, Stallmach T, Shaw S, Morawietz H, Goettsch W, Hermann M, Slowinski T, Theuring F, Hocher B, Luscher TF, Gassmann M (2003) Erythropoietin-induced excessive erythrocytosis activates the tissue endothelin system in mice. FASEB J 17:259–261
Remy I, Wilson IA, Michnick SW (1999) Erythropoietin receptor activation by a ligand-induced conformation change. Science 283:990–993
Renzi MJ, Farrell FX, Bittner A, Galindo JE, Morton M, Trinh H, Jolliffe LK (2002) Erythropoietin induces changes in gene expression in PC-12 cells. Mol Brain Res 104:86–95
Ribatti D, Presta M, Vacca A, Ria R, Giuliani R, Dell’Era P, Nico B, Roncali L, Dammacco F (1999) Human erythropoietin induces a pro-angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Blood 93:2627–2636
Rizzo JD, Lichtin AE, Woolf SH, Seidenfeld J, Bennett CL, Cella D, Djulbegovic B, Goode MJ, Jakubowski AA, Lee SJ, Miller CB, Rarick MU, Regan DH, Gordon MS, Gordon MS (2002) Use of epoetin in patients with cancer: evidence-based clinical practice guidelines of the American Society of Clinical Oncology and the American Society of Hematology. Blood 100:2303–2320
Rosti V, Pedrazzoli P, Ponchio L, Zibera C, Novella A, Lucotti C, Della-Cuna GR, Cazzola M (1993) Effect of recombinant human erythropoietin on hematopoietic and non-hematopoietic malignant cell growth in vitro. Haematologica 78:208–212
Sadamoto Y, Igase K, Sakanaka M, Sato K, Otsuka H, Sakaki S, Masuda S, Sasaki R (1998) Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochem Biophys Res Commun 253:26–32
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R (1998) In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A 95:4635–4640
Sasaki R (2003) Pleiotropic functions of erythropoietin. Intern Med 42:142–149
Sasaki R, Masuda S, Nagao M (2000) Erythropoietin: multiple physiological functions and regulation of biosynthesis. Biosci Biotechnol Biochem 64:1775–1793
Sawyer ST (1989) The two proteins of the erythropoietin receptor are structurally similar. J Biol Chem 264:13343–13347
Sekiguchi Y, Kikuchi S, Myers RR, Campana WM (2004) ISSLS prize winner: erythropoietin inhibits spinal neuronal apoptosis and pain following nerve root crush. Spine 28:2577–2584
Selzer E, Wacheck V, Kodym R, Schlagbauer-Wadl H, Schlegel W, Pehamberger H, Jansen B (2000) Erythropoietin receptor expression in human melanoma cells. Melanoma Res 10:421–426
Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D (2003) Tumor hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev 29:297–307
Shingo T, Sorokan ST, Shimazaki T, Weiss S (2001) Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J Neurosci 21:9733–9743
Simon RP, Niiro M, Gwinn R (1993) Prior ischemic stress protects against experimental stroke. Neurosci Lett 163:135–137
Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan S, Gleiter C, Pasquali C, Capobianco A, Mennini T, Heumann R, Cerami A, Ehrenreich H, Ghezzi P (2001) Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci U S A 98:4044–4049
Siren AL, Knerlich F, Poser W, Gleiter CH, Bruck W, Ehrenreich H (2001) Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol (Berl) 101:271–276
Smith KJ, Bleyer AJ, Little WC, Sane DC (2003) The cardiovascular effects of erythropoietin. Cardiovasc Res 59:538–548
Sterin-Borda L, Barcelo AC, Bozzini CE (2003) Erythropoietin improves cardiac contractility in post-hypoxic mice. Br J Haematol 121:180–186
Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay C (2000) Enhanced proliferation, survival, and dopaminergic differentation of CNS precursors in lowered oxygen. J Neurosci 20:7377–7383
Sugawa M, Sakurai Y, Ishikawa-Ieda Y, Suzuki H, Asou H (2002) Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci Res 44:391–403
Supino-Rosin L, Yoshimura A, Altaratz H, Neumann D (1999) A cytosolic domain of the erythropoietin receptor contributes to endoplasmic reticulum-associated degradation. Eur J Biochem 263:410–419
Suzuki N, Ohneda O, Takahashi S, Higuchi M, Mukai HY, Nakahata T, Imagawa S, Yamamoto M (2002) Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood 100:2279–2288
Syed RS, Reid SW, Li C, Cheetham JC, Aoki KH, Liu B, Zhan H, Osslund TD, Chirino AJ, Zhang J, Finer-Moore J, Elliott S, Sitney K, Katz BA, Matthews DJ, Wendoloski JJ, Egrie J, Stroud RM (1998) Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature 395:511–516
Takeshita A, Shinjo K, Naito K, Ohnishi K, Higuchi M, Ohno R (2002) Erythropoietin receptor in myelodysplastic syndrome and leukemia. Leuk Lymphoma 43:261–264
Tauchi T, Feng GS, Shen R, Hoatlin M, Bagby GCJr, Kabat D, Lu L, Broxmeyer HE (1995) Involvement of SH2-containing phosphotyrosine phosphatase Syp in erythropoietin receptor signal transduction pathways. J Biol Chem 270:5631–5635
Teicher BA (1995) Physiologic mechnaisms of therapeutic resistance. Drug Resistance Clin Oncol Hematol 9:475–506
Thomas G (2001) The effect of hemoglobin level on radiotherapy outcomes: the Canadian experience. Semin Oncol 28:60–65
Tojo A, Doumoto M, Oka K, Numabe A, Kimura K, Yagi S (1996) Endothelin-mediated effect of erythropoietin on blood pressure and renal hemodynamics in hypertensive rats. Am J Physiol 270:R744–R748
Tramontano AF, Muniyappa R, Black AD, Blendea MC, Cohen I, Deng L, Sowers JR, Cutaia MV, El Sherif N (2003) Erythropoietin protects cardiac myocytes from hypoxia-induced apoptosis through an Akt-dependent pathway. Biochem Biophys Res Commun 308:990–994
Tsukahara H, Hiraoka M, Hori C, Hata I, Okada T, Gejyo F, Sudo M (1997) Chronic erythropoietin treatment enhances endogenous nitric oxide production in rats. Scand J Clin Lab Invest 57:487–493
Vannucci RC, Towfighi J, Vannucci SJ (1998) Hypoxic preconditioning and hypoxic-ischemic brain damage in the immature rat: pathologic and metabolic correlates. J Neurochem 71:1215–1220
Vaupel P, Mayer A (2004) Erythropoietin to treat anaemia in patients with head and neck cancer. Lancet 363:992
Vaupel P, Mayer A , Briest S, Hockel M (2003) Oxygenation gain factor: a novel parameter characterizing the association between hemoglobin level and the oxygenation status of breast cancers. Cancer Res 63:7634-7637
Vaupel P, Mayer A , Hockel M (2004) Tumor hypoxia and malignant progression. Methods Enzymol 381:335-354
Vaupel P, Thews O, Kelleher DK, Hoeckel M (1998) Oxygenation of human tumors: the Mainz experience. Strahlenther Onkol 174 [Suppl 4]:6–12
Verdier F, Walrafen P, Hubert N, Chretien S, Gisselbrecht S, Lacombe C, Mayeux P (2000) Proteasomes regulate the duration of erythropoietin receptor activation by controlling down-regulation of cell surface receptors. J Biol Chem 275:18375–18381
Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M, Ghezzi P (2003) Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med 198:971–975
Vogel V, Kramer HJ, Backer A, Meyer-Lehnert H, Jelkmann W, Fandrey J (1997) Effects of erythropoietin on endothelin-1 synthesis and the cellular calcium messenger system in vascular endothelial cells. Am J Hypertens 10:289–296
Wagner K, Katschinski DM, Hasegawa J, Schumacher D, Meller B, Gembruch U, Schramm U, Jelkmann W, Gassmann M, Fandrey J (2001) Chronic inborn erythrocytosis leads to cardiac dysfunction and premature death in mice overexpressing erythropoietin. Blood 97:536–542
Wald MR, Borda ES, Sterin-Borda L (1996) Mitogenic effect of erythropoietin on neonatal rat cardiomyocytes: signal transduction pathways. J Cell Physiol 167:461–468
Wang XQ, Vaziri ND (1999) Erythropoietin depresses nitric oxide synthase expression by human endothelial cells. Hypertension 33:894–899
Wang CH, Liang CL, Huang LT, Liu JK, Hung PH, Sun A, Hung KS (2004) Single intravenous injection of naked plasma DNA encoding erythropoietin provides neuroprotection in hypoxia–ischemia rats. Biochem Biophys Res Commun 314:1064–1071
Wen TC, Sadamoto Y, Tanaka I, Zhu PX, Nakata K, Ma YJ, Hata R, Sakanaka M (2002) Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J Neurosci Res 67:795–803
Westenfelder C, Baranowski RL (2000) Erythropoietin stimulates proliferation of human renal carcinoma cells. Kidney Int 58:647–657
Westphal G, Niederberger E, Blum C, Wollman Y, Knoch TA, Rebel W, Debus J, Friedrich E (2002) Erythropoietin and G-CSF receptors in human tumor cells: expression and aspects regarding functionality. Tumori 88:150–159
Wilcox CS, Deng X, Doll AH, Snellen H, Welch WJ (1993) Nitric oxide mediates renal vasodilation during erythropoietin-induced polycythemia. Kidney Int 44:430–435
Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN (1993) JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 74:227–236
Wojchowski DM, Gregory RC, Miller CP, Pandit AK, Pircher TJ (1999) Signal transduction in the erythropoietin receptor system. Exp Cell Res 253:143–156
Wolff M, Jelkmann W (1993) Effects of chemotherapeutic and immunosuppressive drugs on the production of erythropoietin in human hepatoma cultures. Ann Hematol 66:27–31
Wollman Y, Westphal G, Blum M, Simantov R, Blumberg S, Peer G, Chernihovsky T, Friedrich E, Iaina A (1996) The effect of human recombinant erythropoietin on the growth of a human neuroblastoma cell line. Life Sci 59:315–322
Wu H, Lee SH, Gao J, Liu X, Iruela-Arispe ML (1999) Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development 126:3597–3605
Wu XC, Johns EJ, Richards NT (1999) Relationship between erythropoietin and nitric oxide in the contraction of rat renal arcuate arteries and human umbilical vein endothelial cells. Clin Sci (Lond) 97:413–419
Yamaji R, Okada T, Moriya M, Naito M, Tsuruo T, Miyatake K, Nakano Y (1996) Brain capillary endothelial cells express two forms of erythropoietin receptor mRNA. Eur J Biochem 239:494–500
Yasuda Y, Masuda S, Chikuma M, Inoue K, Nagao M, Sasaki R (1998) Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J Biol Chem 273:25381–25387
Yasuda Y, Musha T, Tanaka H, Fujita Y, Fujita H, Utsumi H, Matsuo T, Masuda S, Nagao M, Sasaki R, Nakamura Y (2001) Inhibition of erythropoietin signalling destroys xenografts of ovarian and uterine cancers in nude mice. Br J Cancer 84:836–843
Yasuda Y, Fujita Y, Masuda S, Musha T, Ueda K, Tanaka H, Fujita H, Matsuo T, Nagao M, Sasaki R, Nakamura Y (2002) Erythropoietin is involved in growth and angiogenesis in malignant tumours of female reproductive organs. Carcinogenesis 23:1797–1805
Yasuda Y, Fujita Y, Matsuo T, Koinuma S, Hara S, Tazaki A, Onozaki M, Hashimoto M, Musha T, Ogawa K, Fujita H, Nakamura Y, Shiozaki H, Utsumi H (2003) Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis 24:1021–1029
Yi T, Zhang J, Miura O, Ihle JN (1995) Hematopoietic cell phosphatase associates with erythropoietin (Epo) receptor after Epo-induced receptor tyrosine phosphorylation: identification of potential binding sites. Blood 85:87–95
Yoshimura A, Misawa H (1998) Physiology and function of the erythropoietin receptor. Curr Opin Hematol 5:171–176
Acknowledgments
Thanks are due to Ms. Lisa Zieske for expert secretarial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jelkmann, W., Wagner, K. Beneficial and ominous aspects of the pleiotropic action of erythropoietin. Ann Hematol 83, 673–686 (2004). https://doi.org/10.1007/s00277-004-0911-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-004-0911-6