
Abstract
Our understanding of how grain boundaries (GBs) can dramatically influence key mineral properties such as creep and diffusion depends on knowledge of their detailed atomic and electronic structures. For this purpose, we simulate different types of tilt GBs, (0l1)/[100], (1l0)/[001] and (012)/[100] modeled with stepped and non-stepped surfaces in Mg2SiO4 forsterite using a first-principles approach based on density functional theory. Our results suggest that several configurations arising from Mg-terminated planes with tilt angles ranging from 16° to 67° are energetically competitive over the entire pressure regime (0–17 GPa) studied. At the ambient pressure, the predicted important features of the boundaries include distorted bonds (Si–O and Mg–O distances changed by 1 and 4 %, respectively), coordination defects (four and fivefold Mg–O coordination), and void spaces (0.2–0.9 × 10−10 m3/m2). Also, the interface induces splitting of electronic states from the conduction band and kinks at the top of the valence band. These structural and electronic features continue to exist at higher pressures. The formation enthalpy and excess volume for each boundary configuration studied were shown to systematically increase and decrease, respectively, with pressure. The predicted energy range (0.8–1.7 J/m2 at zero pressure) widens by a factor of two at 17 GPa (1.1–2.8 J/m2). The presence of low-density and structurally distorted regions imply that these GBs can serve as primary impurity segregation sites, fast diffusion pathways, and electron-trapped regions, which all are relevant for mantle rheology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Material properties such as diffusion, conduction, and strength are known to be significantly influenced by the presence of surfaces and/or interfacial defects, which likely exist in naturally occurring as well as experimentally derived materials. Grain boundaries (GBs) in minerals and ceramics, aside from their technological relevance, are important from geophysical viewpoint in that their presence affects the polycrystalline properties and hence controls the behavior of rocks to a great extent (Kingery 1974; Poirier 1985). Mg2SiO4 forsterite along with its high pressure–temperature phases constitutes one of the most abundant materials of the Earth’s upper mantle. Experimental studies have reported inter-granular thin-film-like amorphous structures in natural mantle rocks and synthesized rocks (Wirth 1996; Drury and Fitz Gerald 1996), which are suggested to enhance the diffusion and creep rates (Duffy 1986; van Orman et al. 2003; Faul et al. 2011). The cationic (Al, Ca, Cr, Ti) enrichment of GBs occurs by segregation in natural and synthesized olivine samples (Hiraga et al. 2003, 2004). A transmission electron microscopy (TEM) study has reported the survival of the dislocation arrays for forsterite GBs up to tilt angle of 21.5° (Heinemann et al. 2005). The experimental approaches such as TEM and electron back-scattered diffraction may not be able to completely resolve three-dimensional structure, thereby limiting our knowledge of the structure and properties of GBs in minerals (e.g., Hartmann et al. 2010; Rohrer 2011).
Assessing the ability of GBs to incorporate point defects including impurities, facilitate diffusion and creep processes, and influence other physical properties requires the detailed knowledge of their atomic and electronic structures. The complexity of the inter-granular structures with their numerous compositional and orientation possibilities in minerals makes the modeling of GBs that mimic the natural samples very challenging. Successful interpretation of the properties depends on the appropriate description of the boundary structure based on specific models (Gleiter 1971; Duffy 1986). For example, low-angle boundaries can be described by dislocation model (Burgers 1939; Read and Shockley 1950) quite well, but individual dislocations in the case of high-angle boundaries are mostly indistinguishable due to interactions of the overlapping cores. Molecular dynamics simulations of GB-bearing Mg2SiO4 systems were previously performed using empirical potentials only at zero pressure (de Leeuw et al. 2000; Adjaoud et al. 2012). On the other hand, first principles simulations so far performed for simple oxides including MgO with/without point defects (McKenna and Shluger 2008; Verma and Karki 2010) are yet to be applied to complex silicate minerals. Here, we investigate the energetics and microscopic structures (at both the atomic and electronic level) of several tilt GBs in Mg2SiO4 forsterite at the upper mantle pressures by performing a series of first principles computer simulations.
Computational method
Our first principles simulations of Mg2SiO4 grain boundary systems are based on density functional theory using the local density approximation (Ceperley and Alder 1980) and the projector-augmented wave method (Kresse and Joubert 1999). The simulation program used is known as Vienna Ab-initio Simulation Package (VASP) (Kresse and Furthmüller 1996). A plane wave basis set with an energy cut-off of 400 eV was used to expand the wave functions at the Г point, and appropriate Pulay corrections were applied. The number of atoms in the simulation supercells varied from 392 to 1,344 (Table 1, Table S1) depending on the boundary type (stepped vs. non-stepped) and tilt angles (16°–67°) considered. Increasing the cell size (see Table S1) did not significantly affect the atomic arrangements near the boundaries and the boundary energetics. It is important to note that the first principles simulations of GBs with system sizes similar to the ones used here are still computationally intensive and rare.
In order to generate a supercell containing grain boundary defect (i.e., an input configuration), the interface was created by putting mirror image of one block by its side (across the interface plane), resulting in a bi-crystal with a boundary at the center. Application of the periodic boundary conditions introduces another oppositely oriented GB at the edges (parallel to the GB at the center) of the supercell and extends the boundary planes, thereby eliminating surface effects. The inter-grain distances should be large enough (≥15 Å here) to minimize any interactions between them. Unlike the case of MgO (Harris et al. 1999; Verma and Karki 2010), several configurations are possible for Mg2SiO4 in which isolated Si tetrahedra are connected by Mg ions (Adjaoud et al. 2012).
We create a series of Mg-terminated (keeping Si tetrahedral units intact) tilt boundaries of two types, (0l1)/[100] and (1l0)/[001], in the stepped and non-stepped arrangements (Figs. 1 and S1). The stepped boundaries consist of the terraces and step walls at right angles to each other. The (010) plane was taken to be the terrace as the (010) surface is considered to be the major cleavage plane in forsterite. The different planes, (001) and (100), were taken to be the step walls for the two types considered here, respectively. Such (010) stepped tilt boundaries were previously simulated at zero pressure using the MD method (de Leeuw et al. 2000). The key difference between the non-stepped and stepped boundaries lies in the atomic occupancy in the GB region. In the stepped surfaces, larger void regions can be present depending on the tilt angle and lateral positioning of the grains. However, the non-stepped boundaries minimize the degree of lattice defects (missing atoms in lattice sites) at the interface, so more atoms are present off the plane. We also model (012)/[100] boundary, which was previously studied at zero pressure by Adjaoud et al. (2012); note that the misorientation angle used there is related to the tilt angle used by a factor of 2. Each of our modeled configurations was structurally optimized as a function of pressure up to 17 GPa (Fig. 2; Table 1).

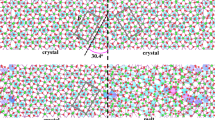
Illustration of the non-stepped (left) and stepped (right) configurations before relaxation for the (011)/[100] (top panel) and (110)/[001] (bottom panel) tilt grain boundaries. The tilt axes are [100] (equivalently a-axis) and [001] (equivalently c-axis), respectively. Multiple GB structural units along with the tilt angle and GB unit length (L GB) are shown. The images display golden Mg spheres, red O spheres, and blue Si–O tetrahedra. The axes a, b and c correspond to the GB supercell (the actual dimensions used in the simulations are given in Table 1)

Relaxed structures of the GB region for the (012)/[100] (top), (021)/[100] (middle) and (130)/[001] (bottom) non-stepped configurations at the zero pressure (left panel) and 17 GPa (right panel). Multiple GB structural units along with tilt angle and GB unit length (L GB) are shown. The images display golden Mg spheres, red O spheres, and blue Si–O tetrahedra. The axes a, b and c correspond to the GB supercell defined in Table 1
Results and analyses
Energetics
We calculate the formation energy (ΔE GB) of each grain boundary simulated for Mg2SiO4 forsterite using: ΔE GB = 0.5(E GB − E P)/A GB, where E GB and E P are the total energies of the defective (containing GB) and perfect (without GB) supercells, and A GB represents the area of the interface. The factor of 0.5 accounts for the presence of two oppositely pointing boundaries in the simulation supercell. First, we consider coherent and incoherent boundary types (Fig. S1). A coherent GB satisfies the condition of mirror symmetry at the boundary irrespective of the tilt angle. On the other hand, an incoherent GB is modeled by rigidly shifting the individual grains parallel to the boundary plane. For a particular tilt angle, the relative stability between the coherent boundary and various incoherent configurations was first determined from their energetics. For this purpose, tilt angles of 35.5° ((130)/[001]) and 60.4° ((031)/[100]) were chosen. In either case, a shift of one half of the GB structural unit length was found to correspond to the minimum energy configuration (Fig. S2). We do not consider shift in the other directions since the corresponding configurations are likely to be energetically unfavorable (Adjaoud et al. 2012).
The calculated grain boundary formation energies of various configurations modeled in this study are plotted against the tilt angle at zero pressure in Fig. 3. For all coherent configurations, the GB energies were found to be significantly high. For the incoherent configurations, our results suggest that the non-stepped boundaries are systematically more favorable than the stepped ones. The range of the calculated GB energies (considering only incoherent boundaries) is 0.8–3.0 J/m2, which compares favorably with the previously predicted ranges of 1.3–3.6 J/m2 (de Leeuw et al. 2000) and 1.0–2.6 J/m2 (Adjaoud et al. 2012) at zero pressure. For the (012)/[100] boundary, our calculated energy is 1.6 J/m2, compared with the MD value of 1.0 J/m2 (Adjaoud et al. 2012). The experimentally derived GB energy is 1.4 ± 0.4 J/m2 for natural olivine (Duyster and Stockhert 2001) and 0.9 ± 0.35 for the olivine-basalt system (Cooper and Kohlstedt 1982). It is remarkable that both the experimental values lie well within the calculated ranges.

Calculated variation of GB energy with tilt angle for different configurations at zero pressure: non-stepped incoherent (open symbols), stepped incoherent (solid symbols), and stepped coherent (symbols with cross inside) boundaries. Results from previous MD simulations (shaded small symbols) are shown for comparison: MD1: de Leeuw et al. (2000) and MD2: Adjaoud et al. (2012). Connecting lines (solid, dashed or dotted) are guide to the eye
For each of the grain boundary configurations simulated, the formation enthalpy was shown to increase monotonically with increasing pressure (Fig. 4). The enthalpy for more favorable configurations tends to increase more slowly with pressure than that for less favorable ones. As shown in Fig. 4, the calculated range of enthalpy (1.1–2.8 J/m2) at 17 GPa is two times wider than the zero pressure range (0.8–1.7 J/m2). Our results show that the (021)/[100] (49.5°) tilt boundary is energetically most favorable (0.8–1.1 J/m2) over the entire pressure range studied. The next most favorable one is (041)/[100] (66.9°) tilt boundary.

Calculated GB formation enthalpies for the selected (energetically competitive) non-stepped (open symbols and dashed lines) and stepped (denoted by “s”, solid symbols with solid lines) boundaries as a function of pressure
Structure
During structural relaxation, substantial structural rearrangement occurs in the immediate vicinity of the boundary region including rotation of the boundary Si tetrahedral units. One can see that the (relaxed) (012)/[100] structure contains more low-density (void) regions in between the two grains than the (021)/[100] and (130)/[001] configurations (Fig. 2, left). On compression, the boundary regions undergo further atomic rearrangement mainly by consuming available empty space (Fig. 2, right).
We analyze the bond length, bond length distortion index, and per-atom coordination state to characterize structural details in the boundary region. This analysis also serves as a crosscheck to the modeled GB system for the bulk region away from the boundary. The distortion index is calculated as \(\delta_{{{\uplambda}}} = \frac{1}{n}\sum_{i = 1}^{n} {\frac{{\lambda_{\text{i}} - \lambda_{\text{av}} }}{{\lambda_{\text{av}} }}}\), where λ i and λ av are, respectively, the individual and average bond lengths corresponding to any particular polyhedron with n Si–O or Mg–O bonds. At the ambient pressure, the average bond distances of λ SiO ~1.64 Å and λ MgO ~2.10 and ~2.07 Å (corresponding to two inequivalent octahedral Mg sites) are close to the corresponding crystalline values (Fig. 5). Both the Si–O and Mg–O distances and the corresponding distortion indices are almost dispersion-less except at the interface (Fig. 5), thereby affirming non-interacting nature of component grains. The distortion indices are close to 1 and 4 % for Si–O and Mg–O bonds, respectively, at the interface region. Compression shortens the average bond lengths, and the effects are less pronounced for the Si–O environment than they are for the Mg–O environment. Over the entire pressure range of 0–17 GPa studied, only 1 % reduction occurs in λ SiO, compared with 4 % decrease in λ MgO. Also, the difference in λ MgO between the two octahedral sites is suppressed with compression (Fig. 5). The trend is opposite in the case of bond distortions. For the bulk region, while there is a small decrease in δ λ for the Mg–O environment, tetrahedral environments (Si–O) display ~20 % increase in δ λ at 17 GPa with respect to their values at 0 GPa (Fig. 5). In contrary, interfacial Mg–O environments display up to 30 % increase in δ λ value at 17 GPa (compared with 0 GPa) when a small reduction in δ λ value occurs for the interfacial Si–O’s.

Variation of Si–O and Mg–O structural parameters along a distance perpendicular to the GB plane for the (021)/[100] (49.5°) tilt boundary at 0 and 17 GPa. Left panel shows average Si–O (top) and Mg–O (bottom) bond lengths (λ). Right panel shows the corresponding average bond length distortion indices (δ λ). Gray straight lines represent λ (or δ λ) calculated for the crystalline Mg2SiO4
Though by our construction the Si tetrahedral units remain intact, the boundary region undergoes substantial atomic relaxations. All Si atoms remain in tetrahedral state, i.e., they are fourfold coordinated with O atoms. However, the Mg coordination environment with respect to O at the interface region contains defects and also differs between the (0l1)/[100] and (1l0)/[001] types (Fig. S3). The fourfold coordinated Mg atoms are present in the (1l0)/[001] type in addition to the five and sixfold coordinated Mg atoms. The Mg and O atoms at the interface mostly accommodate the effects of pressure through coordination changes. With increasing pressure, the number of under-coordinated (<6) Mg atoms decreases, and the void spaces shift and get squeezed, thereby reducing the effective inter-atomic distances at the interface. Despite weak pressure-induced spreading of the Si–O distances, the Si atoms retain pure tetrahedral coordination over the entire pressure range studied.
We find that the structural disorder (rearrangement) in the GB region results in a reduced atomic density or an excess free volume in the system. Theoretically, the excess volume is expressed as ΔV GB = (V GB − V P)/2A GB, where V GB is the volume of simulation cell with GB, V P is the volume of the perfect supercell without GB, and A GB is the interfacial area. At the ambient pressure, we find that the lower the GB energy, the lower the GB excess volume (Fig. 6). Such a positive correlation between the boundary energy and excess volume was previously suggested (Adjaoud et al. 2012). The present first-principles values of (0.2–0.9) × 10−10 m3/m2 for ΔV GB compare well with the previous MD values of (0.3–1.2) × 10−10 m3/m2. The excess volumes tend to be smaller compared with MgO tilt boundaries [(0.81–0.93) × 10−10 m3/m2 reported by Verma and Karki 2010]. Although not directly comparable, excess volume as large as 1.7 × 10−10 m3/m2 has been reported experimentally for nanocrystalline metallic thin films (Kuru et al. 2009).

Calculated pressure dependence of the excess free volume for the selected (energetically competitive) non-stepped (open symbols and dashed lines) and stepped (denoted by “s”, solid symbols with solid lines) boundaries
The calculated pressure profiles of the excess volume display a rather consistent decreasing trend (Fig. 6). In all cases, the pressure suppresses the excess volume significantly. The excess volume for energetically more favorable configurations decreases at smaller rate compared with that for less favorable configurations. The overall range of excess volume at 17 GPa is much narrower than that at the zero pressure. The energy–volume positive correlation seen at the zero pressure does not hold at high pressure. Note that the (011) configuration has the smallest excess volume, whereas the (021) configuration has the lowest enthalpy at 17 GPa.
Electronic structures
The altered properties of materials containing GBs arise as a consequence of the changes in the electronic properties of the interfacial region. We now elucidate on the interfacial properties at the electronic level by examining the calculated electronic structures with and without GB and how they depend on pressure. The consequences of the presence of the GB are widening of the valence band and a noticeable reduction in the band gap relative to the perfect case (Fig. 7). Note that a substantial underestimation of the band gap is a well-known deficiency of the density functional theory that we do not attempt to address here.

Calculated total density of states (DOS) for the bulk (B brown lines) and GB systems: a at 0 GPa, and b at 17 GPa with insets (magnified) showing differences between bulk and GB systems near the valence and conduction band edges and the effects of pressure on them. c, d display projected DOS (only for the states showing significant differences at the band edges) for the 35.5° at 0 GPa (also at 17 GPa for O -p states)
Both the top of the valence band and the bottom of the conduction band extend into the gap. A notable feature of the boundary projected density of states (PDOS) is a splitting of interface states from the conduction band, originating from s contributions of Mg and O atoms (Fig. 7). The size of splitting depends on the boundary tilt angle and is as high as 1 eV. Another feature is the appearance of small kinks at the top of the valence band (Fig. 7) arising mainly from occupied O p states. These two types of interfacial states are characteristically different in that the ones from the conduction band edges display one-dimensional pipe like extended features whereas the ones from the valence band edges are rather separated high-density regions (Figs. 8 and S4). For all these states, the electrons remain confined mostly in and around the GB voids. Potential electron trapping below the bulk conduction band due to the presence of the interfacial states was previously predicted for wide gap metal oxides such as MgO and HfO2 (McKenna and Shluger 2008, 2011; Verma and Karki 2010). At the ambient pressure, the DOS magnitudes for these states are higher for the configurations with bigger excess volumes.

Interfacial charge density isosurfaces near the valence (left panel) and conduction (right panel) band edges corresponding to the non-stepped (130)/[001] (35.5°) at zero pressure, viewed along two directions as indicated. Isosurface values are set at 0.001r −3, where r is the Bohr radius. The images display golden Mg spheres, red O spheres, and blue Si spheres. (see S4 for isosurfaces at 17 GPa)
The overall effects of pressure are to broaden the bands and widen the band gap (Fig. 7). For the shallow electronic states below the conduction band in particular, a pressure-induced reduction in the s contribution is compensated by an increase in p states. The DOSs are somewhat pushed toward higher energy (Fig. 7) and the nature of the charge density distributions arising from the interfacial states remains essentially unchanged with pressure (Fig. S4).
Discussion and future work
The energetics based on our first principles simulations of three types of tilt GBs including (0l1)/[100], (1l0)/[001], and (012)/[100] suggests that several configurations are possible in Mg2SiO4 forsterite with comparable formation energies over the pressure regime of 0–17 GPa. Relatively low GB formation enthalpies and small excess volumes predicted for the non-stepped configurations imply that the stepped tilt boundaries with more open space (particularly coherent ones) are less realistic. While several Mg-terminated tilt boundaries were simulated here, an exhaustive search naturally involves many more possible (perhaps complex) configurations arising from consideration of other crystal surfaces as the boundary.
All tilt boundaries studied here share characteristic features such as under-coordinated atoms, distorted bonds, and excess volume, which can affect the physical properties of a polycrystalline phase to a great extent. In particular, the predicted open space and distorted boundary regions can serve as favorable sinks for point defects and impurities (incompatible elements) and can also provide rapid pathways for ionic diffusion that has been inferred in experimental studies (e.g., Kingery 1976; Farver and Yund 1991; van Orman et al. 2003; Hiraga et al. 2003). Incompatible elements such as large Ca tend to favorably segregate to the GBs and enhance the boundary stability (Yan et al. 1998; Hiraga et al. 2004; Hayden and Watson 2007; Wang et al. 2011). Such defects can be accommodated more easily in more open configurations such as some of the stepped boundaries studied here. It is interesting to see how the relative stability of different GBs is affected by the presence of impurities.
Defects segregated to the GBs can combine to form stable defect complexes with/without net charge. The predicted boundary-induced electronic states below the conduction band and at the valence band top are also important. Any excess electrons trapped in the boundary region can attract protons from the bulk to form hydrogen-related defects including molecular species (H2 and H2O), which can fit the interfacial void space. The coordination defects (four and fivefold Mg–O coordination in an otherwise fully octahedral environment) and wide (Mg–O and Si–O) bond distributions at the interface resemble the essence of Mg2SiO4 liquid/glass structure (de Koker et al. 2008). The boundary regions can be considered to behave as an amorphous phase at elevated temperatures, thereby facilitating ionic diffusion and impurity accommodation/localization (Zhang et al. 2009).
In summary, Mg2SiO4 forsterite—a major mantle mineral can exhibit complex boundary structures, which are likely to serve as high-defect and high-mobility regions, and can thus play crucial role in controlling the mantle rheology and distribution of chemical elements. To better understand such grain boundary properties and their implications for the Earth’s mantle requires direct simulations of defect segregation and migration in the grain boundary regions, which are anticipated to happen in the near future.
References
Adjaoud O, Marquardt K, Jahn S (2012) Atomic structures and energies of grain boundaries in Mg2SiO4 forsterite from atomistic modeling. Phys Chem Miner 39:749–760
Burgers JM (1939) Some considerations on the fields of stress connected with dislocations in a regular crystal lattice. Proc K Ned Akad Wet 42, 293, 315, 378
Ceperley DM, Alder BJ (1980) Ground state of the electron gas by a stochastic method. Phys Rev Lett 45:566–569
Cooper RP, Kohlstedt DL (1982) Interfacial energies in the olivine-basalt system. In: Akimoto S, Manghnani MD (eds) Advances in earth and planetary sciences, vol 12: high pressure research in geophysics. Center for Academic Publication, Tokyo, pp 217–228
de Koker NP, Stixrude L, Karki BB (2008) Thermodynamics, structure, dynamics, and freezing of Mg2SiO4 liquid at high pressure. Geochim et Cosmochim Acta 72:1427–1441
de Leeuw NH, Parker SC, Catlow RA, Price GD (2000) Proton containing defects at forsterite 010 tilt grain boundaries and stepped surfaces. Am Mineral 85:1143–1154
Drury MR, Fitz Gerald JD (1996) Grain boundary melt films in an experimentally deformed olivine orthopyroxene rock: implications for melt distribution in upper mantle rocks. Geophys Res Lett 23:701–704
Duffy DM (1986) Grain boundaries in ionic crystals. J Phys C Solid State Phys 19:4393–4412
Duyster J, Stockhert B (2001) Grain boundary energies in olivine derived from natural microstructures. Contrib Mineral Petrol 140:567–576
Farver JR, Yund RA (1991) Measurement of oxygen grain boundary diffusion in natural, fine-grained, quartz aggregates. Geochim Cosmochim Acta 55:1597–1607
Faul UH, Fitz Gerald JD, Farla RJM, Ahlefeldt R, Jackson I (2011) Dislocation creep of fine-grained olivine. J Geophys Res 116:B01203
Gleiter H (1971) The structure and properties of high-angle grain boundaries in metals. Phys Stat Sol (b) 45:9
Harris DJ, Watson GW, Parker SC (1999) Computer simulation of pressure induced structural transitions MgO [001] tilt grain boundaries. Am Mineral 84:138–143
Hartmann K, Wirth R, Heinrich W (2010) Synthetic near ∑5 (210)/[100] grain boundary in YAG fabricated by direct bonding: structure and stability. Phys Chem Miner 37:291–300
Hayden LA, Watson EB (2007) A diffusion mechanism for core-mantle interaction. Nature 450:709–711
Heinemann S, Wirth R, Gottschalk M, Dresen G (2005) Synthetic [100] tilt grain boundaries in forsterite: 9.9 to 21.5. Phys Chem Mineral 32:229–240
Hiraga T, Anderson IM, Kohlstedt DL (2003) Chemistry of grain boundaries in mantle rocks. Am Mineral 88:1015–1019
Hiraga T, Anderson IM, Kohlstedt DL (2004) Grain boundaries as reservoirs of incompatible elements in the Earth’s mantle. Nature 427:699–703
Kingery WD (1974) Plausible concepts necessary and sufficient for interpretation of ceramic grain boundary phenomena: i. grain boundary characteristics, structure, and electrostatic potential. J Am Ceram Soc 57:1–8
Kingery WD (1976) Introduction to Ceramics, 2nd edn. Wiley, New York, pp 250–257
Kresse G, Furthmüller J (1996) Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 6:15–50
Kresse G, Joubert D (1999) From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B 59:1758–1775
Kuru Y, Wohlschlogel M, Welzel U, Mittemeijer EJ (2009) Large excess volume in grain boundaries of stressed, nanocrystalline metallic thin films: its effect on grain-growth kinetics. Appl Phys Lett 95:163112
McKenna KP, Shluger AL (2008) Electron trapping polycrystalline materials with negative electron affinity. Nat Mater 7:859–872
McKenna KP, Shluger AL (2011) Electron and hole trapping in polycrystalline metal oxide materials. Proc. R. Soc. A. doi:10.1098/rspa.2010.0518
Poirier JP (1985) Creep of Crystals: high-temperature deformation processes in metals, ceramics and minerals. Cambridge University Press, Cambridge
Read WT, Shockley W (1950) Dislocation models of crystal grain boundaries. Phys Rev 78:275–289
Rohrer GS (2011) Measuring and interpreting the structure of grain boundary networks. J Am Ceram Soc 94:633–646
Van Orman JA, Fei Y, Hauri EH, Wang J (2003) Diffusion in MgO at high pressures: constraints on deformation mechanisms and chemical transport at the core-mantle boundary. Geophys Res Lett 30:1056. doi:10.1029/2002GL016343
Verma AK, Karki BB (2010) First-principles simulations of MgO tilt grain boundary: structure and vacancy formation at high pressure. Am Mineral 95:1035–1041
Wang Z, Saito M, McKenna KP, Gu L, Tuskimoto S, Shluger AL, Ikuhara Y (2011) Atom-resolved imaging of ordered defect superstructures at individual grain boundaries. Nature 479:380–383
Wirth R (1996) Thin amorphous films (1–2 nm) at olivine grain boundaries in mantle xenoliths from San Carlos. Arizona Contrib Mineral Petrol 124:44–54
Yan Y, Chishol MF, Duscher G, Maiti A, Pennycook SJ, and Pantelides ST (1998) Impurity-induced structural transformation of a MgO grain boundary, Physical Review Letters 81:3675–3678
Zhang H, Srolovitz DJ, Douglas JF, Warren JA (2009) Grain boundaries exhibit the dynamics of glass-forming liquids. Proc Natl Acad Sci 106:7735–7740
Acknowledgments
This work was supported by the National Science Foundation grant (EAR-1014514). High-performance computing resources were provided by Louisiana State University (http://www.hpc.lsu.edu).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ghosh, D.B., Karki, B.B. First principles simulations of the stability and structure of grain boundaries in Mg2SiO4 forsterite. Phys Chem Minerals 41, 163–171 (2014). https://doi.org/10.1007/s00269-013-0633-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-013-0633-1