
Abstract
The kinetics of hydrogen diffusion in enstatite was studied by hydrogen–deuterium exchange experiments in the range of 1–5,000 bar and 700–850°C using synthetic single crystals of pure and Cr-doped enstatites. The OH- and OD-content in the samples was quantified after each thermal treatment with Fourier transformed infrared spectroscopy. H–D-exchange rates were measured parallel to the three crystallographic axes. In addition, in order to visualize diffusion profiles, OH and OD were mapped for some samples, utilizing synchrotron IR micro-spectroscopy. Hydrogen self-diffusivities derived from D/H exchange experiments at one atmosphere are very similar to the chemical diffusivity of hydrogen in natural Fe-bearing orthopyroxene, which was reported previously (Stalder and Skogby 2003) to exhibit a small, but significant anisotropy (D[001] > D[100] > D[010]). Activation energies are estimated to be 211 (±31) kJ/mol for diffusion parallel [100] and 185 (±28) kJ/mol for diffusion parallel [010]. Lattice diffusion of hydrogen is decelerated by more than one order of magnitude when Cr is dissolved in enstatite. In comparison to the chemical composition, pressure seems to have only a minor influence on hydrogen diffusion. Compared to other minerals in the Earth’s upper mantle, enstatite exhibits the highest activation energy for hydrogen diffusion, suggesting faster diffusion than in other mafic minerals at mantle temperatures, but slower diffusion at crustal conditions. Thus under upper mantle conditions, physical properties that are expected to be influenced by hydrogen mobility, such as electrical conductivity, may in enstatite be more intensely affected by the presence of hydrogen than in other upper mantle minerals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During the past 20 years numerous studies have shown that considerable amounts of hydrogen can be incorporated by defects in the crystal lattice of nominally anhydrous minerals of the Earth’s upper mantle (e.g., Kitamura et al. 1987; Skogby et al. 1990; Bell and Rossman 1992; Bai and Kohlstedt 1992; Bell et al. 1995; Ingrin and Skogby 2000; Rauch and Keppler 2002; Stalder 2004; Zhao et al. 2004). On the one hand, the solubility and high mobility of hydrogen in mantle minerals has far reaching consequences for physical properties such as rheology (Chopra and Paterson 1984; Mackwell et al. 1985; Hirth and Kohlstedt 1996; Mei and Kohlstedt 2000) and electrical conductivity (Karato 1990; Wang et al. 1999). In addition, incorporated hydrogen plays an important role during partial melting, as it can be released as water and reduce mantle solidus temperatures. Even if pyroxenes only account for approximately 30 wt% of the Earth’s upper mantle, their generally higher OH-contents as compared to olivine (Ingrin and Skogby 2000) make them major hosts for water in the upper mantle.
OH-concentration of natural orthopyroxenes found at the Earth surface varies strongly with geological setting (Skogby et al. 1990). It has been argued that samples from xenoliths from the upper mantle might have undergone partial water loss during ascent (Mackwell and Kohlstedt 1990; Ingrin and Skogby 2000), as diffusion experiments on single crystals of mantle minerals have demonstrated a high diffusivity of hydrogen (olivine: Mackwell and Kohlstedt 1990; Kohlstedt and Mackwell 1998; 1999; Demouchy and Mackwell 2003; orthopyroxene: Stalder and Skogby 2003; Carpenter 2003; clinopyroxene: Ingrin et al. 1995; Hercule and Ingrin 1999; Carpenter et al. 2000). High H-mobility in natural (i.e. Fe-bearing) orthopyroxene from the Earth`s mantle is—similar to olivine—probably achieved by electron-hole diffusion coupled to a counter flux of protons, which is rate-limited by proton self-diffusion (Mackwell and Kohlstedt 1990).
In this study the kinetics of hydrogen-deuterium exchange in enstatite was studied experimentally in the range of 1–5,000 bar and 700–850°C using synthetic single crystals of pure and Cr-doped enstatites. H–D-exchange rates were measured with Fourier transformed infrared spectroscopy parallel to the three crystallographic axes. After verification that H–D-exchange was fully reversible, diffusion coefficients were determined in both time dependent exchange experiments and from diffusion profiles. In order to visualize diffusion profiles, in some samples OH and OD was mapped with a spatial resolution of 10–20 μm utilizing the synchrotron IR-beamline at BESSY II in Berlin-Adlershof.
Experimental
Enstatite synthesis and preparation
Single crystals of pure enstatite and Cr-doped enstatite were synthesized from specpure oxides at 25 kbar in a piston–cylinder apparatus under H2O- or D2O-saturated (D2O from Sigma-Aldrich, 99.9% purity) conditions (Stalder 2002), leading to four different starting materials. Samples of each charge were analysed with a JEOL JXA 8900 RL electron microprobe using an accelerating voltage of 15 kV, a sample current of 15 nA and a beam focus diameter of 5 μm. For Cr the analysis lasted 30 s on the peak and 15 s on the background, for Si and Mg the analysis lasted 15 s on the peak and 5 s on the background. Pure enstatites were stoichiometric within analytical error (Stalder 2004). No other elements than Mg, Si and O were detected. Three crystals of Cr-doped OD-bearing enstatites contained 0.64 (±0.19) wt% Cr2O3. Similar Cr2O3-contents (0.58 ± 0.09 wt%) were determined in two crystals of Cr-doped OH-bearing enstatite. No other elements than Mg, Si, Cr and O were detected. The oxidation state of Cr was not determined directly, but on the basis of crystal chemical considerations and the greenish colour (Stalder 2004) chromium is expected to be incorporated as VICr3+.
Crystals of each charge were selected by handpicking, embedded in a thermoplastic resin and oriented parallel (100) or (010) using a microscope with polarised transmitted light. All crystals were ground and polished on both sides and the thickness of each section was measured with an accuracy of ± 2 μm using a mechanical micrometer. After preparation for diffusion, experiments specimens were between 50 and 290 μm thick. The area of the polished faces was in the range of 0.2–2 mm2.
Hydrogen isotope exchange experiments
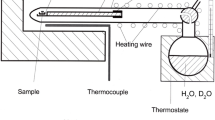
Kinetic studies at one atmosphere were performed by stepwise thermal treatment in a vertical furnace purged with a constant H2-gas flow of 60 ml/min. Temperatures were measured with a Pt100–Pt90Rh10–thermocouple located approximately 5 mm above the sample. The temperature was calibrated against the melting point of Au and is estimated to be correct within ± 2°C. For H–D-exchange experiments samples were inserted in an Au-container (5.0 mm in diameter) which was placed in the central zone of the furnace. The furnace was then flushed with hydrogen and subsequently heated to the desired temperature at a rate of 20°C/min. For cooling the heating power was switched off, resulting in an initial cooling rate of 12°C/min. As a finite time is required to reach the nominal temperature and to quench the sample, additional H–D-exchange may occur during the temperature ramps. In order to account for heating and cooling, a run time correction was applied following the equation (Ingrin et al. 1995):
The activation energy was estimated from Stalder and Skogby (2003) to be approximately 200 kJ/mol. The resulting error in time is estimated to be ±2 min.
High-pressure D–H-exchange experiments were performed in an internally heated pressure vessel (IHPV) at 2,000 and 5,000 bar. Oriented crystals initially containing OD were sealed together with H2O in an Au-capsule with 5.0(4.6) mm outer (inner) diameter. Analogously, crystals initially containing OH were equilibrated with D2O (Sigma-Aldrich, 99.9% purity). Quartz was added to each charge in excess to the silica solubility in the fluid expected for the given P–T conditions (Manning 1994), in order to prevent hydrothermal alteration of orthopyroxene to olivine. IHPV-runs were initially pressurised to approximately half of the final pressure and then heated to the final temperature causing a simultaneous increase in pressure; the final pressure was held constant within ± 50 bar. A heating rate of 100°C/min up to 760°C and 20°C/min between 760 and 800°C was applied, leading to a short overstepping (up to about 5°C for 1 min). Temperatures were controlled within ± 1°C using a Pt100–Pt90Rh10–thermocouple placed next to the sample. Experiments were finished by switching off the heating power resulting in an initial cooling rate of approximately 100°C/min. In order to facilitate the retrieval of the crystal, the capsule was carefully heated over a flame until it was sufficiently inflated. Then, the capsule was cleaned, weighed, pierced, dried and weighed again, in order to check for fluid loss during the experiment. In all runs reported here, no significant weight loss could be observed. All experimental run conditions are summarized in Table 1.
Infrared spectroscopy
Polarised IR-spectra of each sample was recorded parallel to the crystallographic c-axis (Fig. 1) in transmitted light using a Bruker IFS 88 FTIR-spectrometer coupled to a Bruker microscope IR scope II at the University of Hannover. Measurements were performed using a globar light source, a KBr beamsplitter and a wire-grid polarizer. For each spectrum 64 scans in the range between 0 and 6,000 cm−1 were acquired with a spectral resolution of 2 cm−1. Backgrounds in the spectra were fitted using a polynomial function between 4,000 and 3,500 cm−1, 2,950 and 2,650 cm−1, and 2,200 and 2,100 cm−1. Total absorption was obtained by integration over the wavenumber range from 3,000 to 3,600 cm−1 for OH and 2,200 to 2,700 cm−1 for OD. During analysis of samples #192–195 significant interferences of the OD-absorption band close to 2,300 cm−1 with the CO2-stretching modes occurred due to variations in atmospheric CO2 during the IR session. Therefore, in these samples for quantification of H/D exchange only the integral absorption of the most pronounced OH-absorption band (range of 3,300–3,420 cm−1) and the corresponding OD-absorption band (2,450–2,550 cm−1) was considered.

IR-spectra (E//c) showing the OH- and OD-absorption bands in pure enstatite #111 (a) and Cr-doped enstatite #113 (b). The three different spectra compare the sample after synthesis under D2O-saturated conditions (I, thick black curves), after the first thermal treatment in H2 at ambient pressure (II, thin black curves), and after a second long term thermal treatment in H2 (III, thick grey curves). In the case of the Cr-doped sample, the grey curve show the spectrum of a Cr-doped enstatite with comparable amounts of Cr as #113 synthesized under H2O-saturated conditions, because the second exchange experiment with of #113 failed (see text)
Crystals equilibrated at high pressure in the IHPV sometimes exhibited a very fine-grained overgrowth, which probably consists of quartz and was precipitated from the fluid phase during quenching. Attempts to remove this overgrowth in an ultrasonic bath were not successful. Spectra of crystals from IHPV runs containing H2O exhibited a step-like increased background in the OH-absorption region (Fig. 2), whereas those from runs containing D2O showed a similar feature in the OD-absorption region. This supported the assumption that the additional absorption feature was produced by OH- or OD-containing quench deposits on the crystal surface. As an approximation, background correction was carried out by adjusting a linear baseline between 2,940 and 3,600 cm−1 (Fig. 2).

IR-spectrum of an originally OD-bearing enstatite equilibrated with H2O at 2 kbar (sample thickness 290 μm). The step in the background at approximately 2,900 and 3,700 cm−1 is probably due to OH-containing precipitates on the surface of the crystal. Since polynomial fitting of the stepped background did not furnish unequivocal results, a linear baseline correction was applied (dotted line)
In order to visualize the two-dimensional distribution of OH and OD, some samples were mapped utilizing the synchrotron IR-beamline at BESSY II in Berlin-Adlershof, Germany, using a Nicolet 870 FTIR spectrometer equipped with a KBr-beamsplitter, a Continuμm microscope and a MCT-detector. Spectra were recorded using a rectangular aperture with a spatial resolution of 10 × 10 to 20 × 20 μm. No polarizer was applied. For each spectrum 8–32 scans in the range between 2,000 and 4,000 cm−1 were acquired with a spectral resolution of 2 cm−1. Between IR-analyses an automatic X-Y-stage moved the sample with increments of 10–20 μm. The higher beam brilliance and intensity of synchroton IR-spectroscopy in comparison to usual FTIR-spectroscopy was insofar advantageous. Smaller beam diameters and shorter counting times generated sufficiently high signal-to-noise ratio absorption signals in the OH- and OD-vibration regions, and hence enabled mapping with high spatial resolution. Spectra were automatically processed yielding peak height, peak area, peak height ratio or ratio of two chosen peak areas, and the resulting data were encoded by colour. After data reduction a smoothing function averaging over the eight nearest neighbour points was applied (Fig. 3). Between 378 and 1,225 spectra were recorded to create one map. The time to create a map of 300 × 300 μm (consisting of 400 spectra à 16 scans) took several hours. Two maps revealing the OH and OD distribution within the sample are shown in Fig. 3. Both sample #97 (Fig. 3a) and #191 (Fig. 3b) exhibit higher OH/OD-ratios towards the rim. In case of sample #191 the diffusion rim appears to be only slightly broader towards the (001) face than towards the (100) face, suggesting that diffusion in the Cr-doped enstatite is nearly isotropic. In the pure enstatite (sample #97), however, the diffusion rim developed along [001] is significantly enlarged compared to that along [100], suggesting a distinct hydrogen diffusion anisotropy.

Maps showing the spatial distribution of the OH/OD absorbance ratio for a pure enstatite sample #97b after annealing in H2O (2 kbar, 800°C, 3 h) and b Cr-doped enstatite sample #191 after annealing in H2 (1 bar, 800°C, 24 h). OH/OD-absorbances are discriminated by colours as indicated on the right hand side. These ratios were determined by dividing peak areas in the ranges 3,330–3,400 cm−1 and 2,480–2,520 cm−1 (a), and 3,435–3,460 cm−1 and 2,540–2,570 cm−1 (b). Concentration ratios OH/OD may be calculated by scaling with εOH/εOD (cf. Fig. 4). The solid black line highlights the boarder of the crystal; spectra taken outside or on the edge of the sample lead to random values. Note the horizontal scratch in the upper half of a, causing locally poor quality spectra resulting in deviating values for total absorbance
Analytical solutions of diffusion problems
Because the thickness of the samples perpendicular to the polished surfaces was much smaller than parallel to them, diffusion can be described using the solution of Fick’s second law for one-dimensional diffusion in a solid medium with a homogeneous initial concentration bounded by two parallel planes (Carslaw and Jaeger 1959; Ingrin et al. 1995). This approach is justified only if the diffusion from the sides is not significantly faster than from the basal planes, as supported by the data presented in Table 1. In H–D-exchange experiments only one species (i.e. OH or OD) was initially present. Then the average OH- and OD-concentration across the crystal plate follows the equation
for the removed species and
for the introduced species, where c av is the average concentration across the whole thickness of the crystal plate, c 0 the initial concentration of the diffusing species, D the diffusion coefficient, t the time and 2L the thickness of the sample.
As orientation of crystals parallel to (001) was not successful (the preferred cleavage parallel (110) split the crystal into many fragments as soon as the thickness became smaller than 350 μm), diffusion parameters along [001] were calculated from diffusion profiles. For some crystals diffusivities along [100] were also calculated from measured diffusion profiles (Table 1). At a given time the concentration as a function of the location x along a profile can be expressed as
where erfc is the complementary error function, c(x) the at position x and X the crystal length along the profile. As diffusion along the measured profile is superimposed with the diffusion through the basal planes, another fitting parameter a 0 is introduced, taking into account that the concentration at the diffusion plateau is not 0. Along the profile the contribution of diffusion from the basal planes is assumed to be constant. In first approximation the two diffusion processes—on the one hand through the basal planes, and on the other hand from the side—can be treated as two independent processes, because the diffusion coefficient obtained by fitting the profile is mainly determined by the length over which the concentration changes.
As the diffusion experiments were carried out at much lower pressures (and lower hydrogen fugacities) than the syntheses, defects in the synthetic crystals were not in equilibrium with the thermodynamic conditions prevailing during the H/D exchange experiments. Therefore, a decrease in bulk (OH + OD)-content in the sample is expected to be superimposed to the D/H exchange. However, the kinetics of dehydration is much slower than hydrogen self-diffusion (Stalder and Skogby 2003) and it is only expected to change the total (OH + OD) significantly close to the surface.
Results
Quantification of species concentrations and reversibility of H–D-exchange
Integral absorbances (A OH and A OD) of the OH and the OD bands are plotted for Cr-free and Cr-bearing enstatite in Fig. 4. For pure enstatite two parallel trends are apparent (Fig. 4a). The first trend is defined by sample #99 (black circles), which originally contained OH and was equilibrated with D2O. #110 (open triangles), #111 (open circles) and #112 (open squares) were produced by D2O-saturated syntheses and then annealed in H2-atmosphere at ambient pressure. Absorbances of all samples synthesized under D2O-saturated conditions (before and after partial H–D-exchange) plot on a straight line which intersects the ordinate at a value close to the integral absorbance of enstatites synthesized under H2O-saturated conditions and vice versa. It is therefore concluded that the molar content of OH of the enstatite crystals synthesized under H2O-saturated conditions is similar to the OD-content in crystals synthesized under D2O-saturated conditions. As all these data follow a trend with a slope of −1.8, the ratio of the extinction coefficients for OH and OD ε*OH/ε*OD = 1.8 can be derived. Knowledge of this ratio is necessary for calculating diffusion coefficients from concentration profiles measured across the crystals.

a Integral absorbances of OH- vs. OD-absorption bands (A OH and A OD ) for pure enstatites (normalized to 1 cm sample thickness). Samples #99 (black circles), #110 (open triangles), #111 (open circles), #112 (open squares), #97a (open diamond) and #97b (black squares), see text and Table 1. All data follow a trend which represents the ratio of the integral molar extinction coefficients. b Integral absorbances of OH- vs. OD-absorption bands for Cr-doped enstatites. Samples #113 (open circles), #114 (black circles), #115 (grey circles). See text
Reversibility of H–D-exchange was tested using sample #97 (open diamond). Complete H–D-exchange was achieved within 24 h by annealing the sample in D2O atmosphere at 800°C, 2,000 bar (black square on the OD-axis). Then the sample was stepwise re-equilibrated with H2O at same P-T conditions (black squares along the broken line). The data of the hydrothermally treated samples form a second trend parallel to the first one, but shifted towards lower absorbances (Fig. 4a). We attribute this shift to partial dehydration during the initial long term hydrothermal treatment in D2O. Using the ε*OH/ε*OD ratio of 1.8 the loss of the total (OH + OD) is calculated to be about 20%. Based on this value a chemical diffusivity of hydrogen of \( \log \ifmmode\expandafter\tilde\else\expandafter\sim \fi{D}_{{\text{H}}} [010] = - {\text{14}}{\text{.1 (}} \pm {\text{0}}{\text{.3)}} \)m2/s is estimated by applying Eq. 2a. The value is two orders of magnitude lower than diffusion coefficients derived from hydrogen H/D exchange at the same conditions (Table 1) implying that short term D/H exchange experiments are not significantly influenced by desorption kinetics in our study.The plot of integral absorbances of OH- vs. OD-absorption bands of Cr-bearing enstatites is shown in Fig. 4b. One problem was that cracks evolved during H/D exchange experiments at 1 atmosphere. After the first exchange step the abundance of cracks was very low, and the exchange rate can be still interpreted in terms of one-dimensional diffusion (#113, #191). However, subsequent annealing experiments resulted in significant losses in (OH + OD). In contrast, D–H-exchange experiments at high pressure were less affected by crack formation (#114, #115). But in these experiments background correction of the IR spectra was more difficult due to formation of fine-grained overgrowth on the crystal surface, probably caused by precipitation from the fluid phase. In order to establish trends for ε*OH/ε*OD we used crystals #113 (open circles), #114 (black circles) and #115 (grey circles) which were synthesized in the same batch under D2O-saturated conditions and later equilibrated with H2 (#113) or H2O (#114 and #115), respectively. Because complete D-H-exchange could not be achieved, two OH-bearing enstatites (black triangles) with similar Cr content were synthesized under the same condition as the OD-bearing crystals (but with excess of H2O instead of D2O) and used to calculate the ratio of the integral molar extinction coefficients. Combining these data yields roughly the same slope for A OH/t vs. A OD/t and hence a ratio of ε*OH/ε*OD as found for Cr-free samples. Thus we infer that the ε*OH/ε*OD ratio of 1.8 is typical for enstatite and used this value for evaluating relative concentrations of H and D in the diffusion samples.
Kinetics of H–D-exchange
Diffusion results are summarized in Table 1. In pure enstatite, H/D exchange is anisotropic, whereby D[001] is approximately one order of magnitude faster than D[010]. In Fig. 5a the time dependent OD → OH exchange in sample #97 is displayed. Figure 5b shows a diffusion profile parallel [100] and [001], respectively, confirming the distinct diffusion anisotropy shown in Fig. 3a. In contrast, diffusion anisotropy in Cr-doped enstatite appears to be much weaker, as illustrated by the diffusion profiles measured on #191 along [100] and [001] (Fig. 6). This result is consistent with the OH/OD distribution map (Fig. 3b), which does not reveal a significant anisotropy in diffusivity within error.

Results of D-H exchange with sample #97 at 800°C and 2 kbar. a Variation of integral intensities with time in the central area of the (010) section. b OD-diffusion profiles along [100] and along [001] after the last hydrothermal equilibration step. Note the different diffusivities along both crystallographic axes. Thick black curves correspond to the diffusion coefficient and thin grey curves correspond to the upper and lower limit of the given error

OH-diffusion profiles along [100] a and [001] b for sample #191 after annealing in H2 (see also Fig. 3b). Thick black curves correspond to the diffusion coefficient and thin grey curves correspond to the upper and lower limit of the given error
The effect of pressure on H-mobility does not follow a clear trend. In Cr-free samples H-mobility is enhanced by about half an order of magnitude between 1 and 2,000 bar, but no clear trend is observed between 2,000 and 5,000 bar. H-mobility in Cr-doped enstatites is also slightly higher at 5,000 bar than at ambient pressure. The diffusion anisotropy at 2,000 bar (Fig. 5) is slightly less pronounced, but in the same order as at 1 bar. An increase of diffusivity with pressure cannot be explained with the concept of activation volume. A possible explanation could be pressure-induced formation or incorporation of defects. However, the observed effects may be influenced by different prevailing oxygen fugacity in experiments at ambient and at elevated pressure as well.
The effect of doping is much more pronounced than that of pressure. H-mobility in Cr-bearing enstatites is in general, nearly two orders of magnitude lower than in pure enstatite. The diffusion anisotropy in the Cr-bearing crystals is less pronounced (Fig. 6), and cannot be resolved within error of the diffusion profile measurements.
Diffusion coefficients from different temperatures were fitted using a least squares regression to an Arrhenius equation
where D 0 is the diffusivity at infinite temperature and E a is the activation energy. Results are shown in Fig. 7. Values for D 0 and E a of hydrogen self-diffusion in pure enstatite along [100], [010] and [001] are listed in Table 2. The high error for the diffusion parameters parallel [001] is due to very imprecise data at 700°C.

Arrhenius plot for H-self-diffusion in synthetic enstatite along [100] (black circles), [010] (black squares) and [001] (black triangles). Diffusivities for Cr-bearing samples are shown with grey symbols. The grey array indicates the range of chemical diffusivities of hydrogen in natural upper mantle orthopyroxenes (Stalder and Skogby 2003; Carpenter 2003), determined from hydrogen extraction experiments. Metal vacancy diffusivities (dashed line) are from Stalder and Skogby (2003). These values were calculated from dehydrogenation experiments taken into account that \(\mathop D\limits^\sim \approx 3D_{V_{{\text{Me}}} } \)(Kohlstedt and Mackwell 1998)
Discussion
Hydrogen diffusion in natural mantle-derived orthopyroxenes and in synthetic enstatite
As noted above dehydration kinetics are slow in our experiments compared to D/H interdiffusion. Hence, in first approximation we can consider the D/H exchange as a process in a quasi-homogeneous medium. Thus, the diffusion coefficients derived from isotope exchange experiments are expected to be close to the self-diffusivity of hydrogen. These values have to be clearly distinguished from chemical diffusion coefficients which are determined from experiments in which concentration gradients (or more precise gradients in the chemical potential of hydrogen) occur.
Chemical diffusion of hydrogen has previously been studied on natural mantle-derived orthopyroxenes from Kilbourne Hole/New Mexico and Hoher Hagen/Germany (Stalder and Skogby 2003) and San Carlos/Arizona (Carpenter 2003) using hydrogen extraction experiments. The mantle-derived samples were Fe-bearing with Fe/(Fe + Mg) between 0.08 and 0.10. The major finding of both studies was that diffusion is anisotropic in the order D[001] > D[100] > D[010]. Activation energies between 127 and 213 kJ/mol (with an average of 175 kJ/mol) were determined for different samples and orientations. Diffusion along the fastest diffusion direction (i.e. [001]) was one order of magnitude faster than along the slowest diffusion direction (i.e. [010]). In addition, Carpenter (2003) studied hydrogen self-diffusion on orthopyroxene from San Carlos at 1 GPa. Diffusivities were nearly identical to those derived from hydrogen extraction experiments and it was concluded that chemical diffusion of hydrogen (as protons) is coupled to a counter-flux of polarons (electron holes). Such a mechanism has been proposed earlier to explain rapid dehydrogenation of other Fe-bearing mafic silicates (e.g., olivine: Mackwell and Kohlstedt 1990; clinopyroxene: Hercule and Ingrin 1999) with an iron content exceeding a critical value (approximately Fe/(Fe + Mg) > 0.08). Extraction or incorporation of hydrogen in Fe-bearing minerals can be achieved by the reaction:
Here we use the notation of Kröger and Vink (1956) to characterize point defects with respect to species (or elements), charge and crystallographic site. Positive, neutral and negative charge relative to the regular structural sites is indicated by superscripts dot, x and prime, respectively. h • refers to a polaron (an electron hole localized on an iron atom occupying an octahedrally coordinated M-site). The chemical diffusion of hydrogen depends on self-diffusion of hydrogen and on diffusion of polarons by
Since D h >> D H (Mackwell and Kohlstedt 1998, 1999) the equation simplifies to
In other words, due to the accelerating effect of rapidly moving polarons the chemical diffusion of hydrogen is enhanced by a factor of two compared to the self-diffusion of hydrogen. If protons are localized on metal vacancies, reaction 5 modifies to
Again, the incorporation and extraction of hydrogen is controlled by the interdiffusion of protons and polarons, but the mobility of protons may be reduced by interaction with M vacancies, \( V^{{\prime\prime}}_{{\text{M}}} . \) Hence, in this case, the self-diffusion of hydrogen will decrease with increasing concentration of M vacancies. In absence of heterovalent elements, such as iron, hydrogen can be incorporated in pyroxene only by substitution of regular elements, either Mg or Si, because incorporation of hydrogen as molecular H2O is less likely in the densely packed pyroxene structure. In pure enstatite the most prominent hydrogen derived point defect is a metal vacancy VM1″ on the M1-position charge balanced by two interstitial protons H •i :
Reaction 8 or similar ones were suggested to control the dehydrogenation of iron-poor pyroxene and olivine (Mackwell and Kohlstedt 1990; Hercule and Ingrin 1999). The extraction of hydrogen involves parallel diffusion of M vacancies and protons to the surface of the crystal and annihilation of M vacancies at the surface by dissociation reactions of pyroxene or by supply of MgO from an external source. Diffusion mechanisms in which the main cationic constituents of the orthopyroxene lattice are involved, i.e. Si-diffusion (Mackwell 1991), Mg–Fe-interdiffusion (Ganguly and Tazzoli 1994) or Mg-self-diffusion (Schwandt et al. 1998), are more than seven orders of magnitude slower than hydrogen self-diffusion in the temperature range investigated here, and hence are not very likely to control the hydrogen extraction kinetics. If controlled by reaction 8, fluxes of M vacancies and protons are coupled by charge balance and the chemical diffusion of hydrogen is given as (Kohlstedt and Mackwell 1998; 1999)
The diffusion of M vacancies is much slower than self-diffusion of hydrogen \( {\text{(}}D_{{V_{{{\text{Mg}}}} }} {\text{ << }}D_{{\text{H}}} {\text{)}} \) resulting in
The Mg-vacancy diffusivity calculated from dehydration kinetics of pure synthetic enstatite (Stalder and Skogby 2003) is in the studied temperature range about 2 log units slower than hydrogen self-diffusion and matches the hydrogen self-diffusion determined in Cr-doped enstatite along [010] within error (Fig. 7). Using the chemical diffusivity of hydrogen from #97 (Table 1) the diffusivity of Mg-vacancies at 800°C and 2 kbar estimated by Eq. 9a is log D V [010] = −14.6 (±0.3) m2/s, which is in excellent agreement with the data derived from the experiments of Stalder and Skogby (2003).
The slow self-diffusion of hydrogen in Cr-doped enstatite implies that another defect than in pure enstatite controls the mobility of hydrogen. A probable incorporation mechanism for hydrogen is a coupled substitution of 2Mg2+ by Cr3+ + H+ (Stalder 2004). Thus, in Cr-doped enstatite the protons are probably connected to Cr3+ by strong local Coulomb-forces generating very immobile defect associates, e.g., {Cr• M+VM″ + H •i } or, if protons are localised on M-positions, {Cr• M+ H ′M }. In both cases H/D exchange at low concentration levels of Cr-defects is rate-limited by the mobility of the Cr-defect rather than by H self-diffusion. In this context we can also explain the formation of cracks: the extraction of hydrogen requires for the conservation of local charge neutrality the movement of Cr3+ or O2−, causing distortion and stress in the crystal lattice. A possible defect associate formed after extraction of hydrogen could be {2Cr• M+VM″}.
The fact that the chemical composition influences hydrogen diffusion is also evident when chemical diffusivities of hydrogen in natural mantle orthopyroxenes are compared to hydrogen self-diffusivities in pure synthetic enstatite from this study (Fig. 7). According to Eq 6a, the chemical diffusivity of hydrogen in Fe-bearing samples is rate-limited by hydrogen self-diffusion, but enhanced by a factor of two by polarons. Considering this, the hydrogen self-diffusion in natural (i.e., Fe-, Al-, and Cr-bearing) orthopyroxenes is about one order of magnitude slower than in pure enstatite (Fig. 7), illustrating the negative effect of trivalent cations on hydrogen diffusion.
Comparison to other mantle minerals
In comparison to other nominally anhydrous mantle minerals, the activation energy for hydrogen self-diffusion in orthopyroxene exhibits the highest values, clustering around 200 kJ/mol. Activation energies reported for hydrogen self-diffusion for other mantle minerals, as olivine (Kohlstedt and Mackwell 1998, 110 kJ/mol //[001], 145 kJ/mol //[100], 180 kJ/mol //[010]), diopside (Hercule and Ingrin 1999, 149 kJ/mol //[001] and //[100]*, 143 kJ/mol //[010]; Carpenter et al. 2000, 153 kJ/mol //[001]*, 181 kJ/mol //[100],) and garnet (Blanchard and Ingrin 2004a, 140 kJ/mol, Kurka et al. 2005, 102 kJ/mol) are lower. Dehydration studies on Fe2+-poor crystals, where hydrogen diffusion kinetics are prevailingly controlled by metal-vacancy diffusion, revealed that activation energies are generally higher than that for hydrogen self-diffusion. Orthopyroxene (Stalder and Skogby 2003) and garnet (Wang et al. 1996; Blanchard and Ingrin 2004b; Kurka et al. 2005), clustering around 300 kJ/mol, show significantly higher activation energies than olivine (Demouchy and Mackwell 2003, 211 kJ/mol) and clinopyroxene (Ingrin et al. 1995, 136 kJ/mol). In summary, H-mobility in enstatite at 800°C is slightly slower than in olivine and clinopyroxene, but extrapolation to mantle temperatures suggest higher H-diffusivities for orthopyroxene than for other nominally anhydrous minerals in the upper mantle. Physical properties influenced by hydrogen mobility (as electrical conductivity) are therefore suggested to be more intensely affected by the presence of hydrogen in orthopyroxene than in other upper mantle minerals under mantle conditions.
Petrological application
The data for Cr-free samples imply that H-diffusivities are fast enough to allow considerable H-exchange between a mantle xenolith consisting of mm-sized crystals and its host magma upon ascent. However, Cr decelerates hydrogen mobility considerably and, if it is assumed that Cr does not affect the activation energy for H-diffusion, orthopyroxenes with a high Cr/Fe-ratio are suggested to be equilibrated with the host magma less readily.
Trivalent cations such as Al3+ and Cr3+ are abundant in natural samples and, as demonstrated for Cr in this study, may reduce the diffusivity of hydrogen significantly. On the other hand, it is not very likely that hydrogen self-diffusion is a simple function of Fe-content, as extraction experiments on a (010)-section of a pure synthetic ferrosilite yields nearly identical diffusivities (Stalder, unpublished data) as hydrogen self-diffusion in enstatite. However, as natural samples contain both Fe and trivalent cations, probably the ratio of Fe/(Cr + Al) controls hydrogen diffusion.
References
Bai Q, Kohlstedt DL (1992) Substantial hydrogen solubility in olivine and implications for water storage in the mantle. Nature 357:672–674
Bell DR, Rossman GR (1992) Water in the Earth’s mantle: the role of nominally anhydrous minerals. Science 255:1391–1397
Bell DR, Ihinger PD, Rossman GR (1995) Quantitative analysis of trace OH in garnet and pyroxenes. Am Miner 80:465–474
Blanchard M, Ingrin J (2004a) Kinetics of deuteration in pyrope. Eur J Miner 16:567–576
Blanchard M, Ingrin J (2004b) Hydrogen diffusion in Dora Maira pyrope. Phys Chem Miner 31:593–605
Carpenter SJ, (2003) The kinetics of hydrogen diffusion in single crystals orthopyroxene. PhD thesis, Pennsylvania State University, 249 pp
Carpenter S, Mackwell S, Dyar D (2000) Hydrogen in diopside: diffusion profiles. Am Miner 85:480–487
Carslaw HS, Jaeger JC (1959) Conduction of heat in solids, 510 pp, Clarendon Press, Oxford
Chopra PN, Paterson MS (1984) The role of water in the deformation of dunite. J Geophys Res 89:7861–7876
Demouchy S, Mackwell S (2003): water diffusion in synthetic iron-free forsterite. Phys Chem Miner 30:486–494
Ganguly J, Tazzoli V (1994) Fe2+–Mg interdiffusion in orthopyroxene: retrieval from the data on intercrystalline exchange reaction. Am Miner 79:930–937
Hercule S, Ingrin J (1999) Hydrogen in diopside: diffusion, kinetics of extraction-incorporation, and solubility. Am Miner 84:1577–1587
Hirth G, Kohlstedt DL (1996) Water in the oceanic upper mantle: implications for rheology, melt extraction and the evolution of the lithosphere. Earth Planet Sci Lett 144:93–108
Ingrin J, Hercule S, Charton T (1995) Diffusion of hydrogen in diopside: results of dehydration experiments. J Geophys Res 100:15489–15499
Ingrin J, Skogby H (2000) Hydrogen in nominally anhydrous upper-mantle minerals: concentration levels and implications. Eur J Miner 12:543–570
Karato S (1990) The role of hydrogen in the electrical conductivity of the upper mantle. Nature 347:272–273
Kitamura M, Kondoh S, Morimoto N, Miller GH, Rossman GR, Putnis A (1987) Planar OH-bearing defects in mantle olivine. Nature 328:143–145
Kohlstedt DL, Mackwell SJ (1998) Diffusion of hydrogen and intrinsic point defects in olivine. Z Phys Chem 207:147–162
Kohlstedt DL, Mackwell SJ (1999) Solubility and diffusion of ‘water’ in silicate minerals. In: Catlow R (ed.) Microscopic properties and processes in minerals. Kluwer, Dordrecht, pp 539–559
Kröger FA, Vink HJ (1956) Relations between the concentration of imperfection in crystalline solids. In: Seitz, F & Turnball, D (Hrsg.) Solid state physics 3, Academic, London, pp 367–435
Kurka A, Blanchard M, Ingrin J (2005) Kinetics of hydrogen extraction and deuteration in grossular. Mineral Mag 69:359–371
Mackwell SJ (1991) High-temperature rheology of enstatite: implications for creep in the mantle. Geophys Res Lett 18:2027–2030
Mackwell SJ, Kohlstedt DL (1990) Diffusion of hydrogen in olivine: implications for water in the mantle. J Geophys Res 95:5079–5088
Mackwell SJ, Kohlstedt DL, Paterson MS (1985) The role of water in the deformation of olivine single crystals. J Geophys Res 90:11319–11333
Manning CE (1994) The solubility of quartz in H2O in the lower crust and upper mantle. Geochim Cosmochim Acta 58:4831–4839
Mei S, Kohlstedt DL (2000) Influence of water on plastic deformation of olivine aggregates 1. Diffusion creep regime. J Geophys Res 105:21457–21469
Rauch M, Keppler H (2002) Water solubility in orthopyroxene. Contrib Mineral Petrol 143:525–536
Schwandt CS, Cygan RT, Westrich HR (1998) Magnesium self-diffusion in orthoenstatite. Contrib Mineral Petrol 130:390–396
Skogby H, Bell DR, Rossman GR (1990) Hydroxide in pyroxene: variations in the natural environment. Am Miner 75:764–774
Stalder R (2002) Synthesis of enstatite at high pressure. Eur J Miner 14:637–640
Stalder R (2004) Influence of Fe, Cr and Al on hydrogen incorporation in orthopyroxene. Eur J Miner 16:703–711
Stalder R, Skogby H (2003) Hydrogen diffusion in natural and synthetic orthopyroxene. Phys Chem Miner 30:12–19
Wang L, Zhang Y, Essene E (1996) Diffusion of the hydrous component in pyrope. Am Miner 81:706–718
Wang ZC, Ji SC, Dresen G (1999) Hydrogen-enhanced electrical conductivity of diopside crystals. Geophys Res Lett 26:799–802
Zhao YH, Ginsberg SB, Kohlstedt DL (2004) Solubility of hydrogen in olivine: dependence on temperature and iron content. Contrib Mineral Petrol 147:155–161
Acknowledgments
This project was kindly supported by the Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BMBF 05 ES3XBA/5). We thank Andreas Kronz (University of Göttingen) for microprobe analyses, and Stephen Elphick and Hans Keppler are thanked for their constructive reviews.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stalder, R., Behrens, H. D/H exchange in pure and Cr-doped enstatite: implications for hydrogen diffusivity. Phys Chem Minerals 33, 601–611 (2006). https://doi.org/10.1007/s00269-006-0112-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-006-0112-z