
Abstract
The solubility of hydrogen in Mg-Fe olivines as a function of temperature and iron concentration was investigated by hydrothermally annealing single crystals of synthetic forsterite and San Carlos olivine. Experiments were performed at temperatures between 1,273 and 1,573 K on samples with compositions between Fa0 and Fa16.9 under a confining pressure of 300 MPa in a gas-medium apparatus with oxygen fugacity, \( f_{{{\text{O}}_{{\text{2}}} }} \), buffered by the Ni:NiO solid-state reaction and silica activity, \( a_{{{\text{SiO}}_{2} }} \), buffered by the presence of enstatite. Hydroxyl concentrations were determined from infrared spectra obtained from polished thin sections in crack-free regions ≤50 µm in diameter. Hydroxyl solubility increases systematically with increasing temperature and with increasing iron content. Combined with published results on the dependence of hydroxyl solubility on water fugacity and pressure, the present results can be summarized by the relation \( C_{{{\text{OH}}}} = Af^{1}_{{{\text{H}}_{{\text{2}}} {\text{O}}}} \exp {\left[ { - {\left( {\Delta E^{{\text{o}}}_{{{\left\{ {} \right\}}}} + P\Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} } \right)}/RT} \right]}\exp {\left( {\alpha X_{{{\text{Fa}}}} /RT} \right)} \) with A=90±10 H/106Si/MPa, α=97±4 kJ/mol, \( \Delta E^{{\text{o}}}_{{{\left\{ {} \right\}}}} \)=50±2 kJ/mol, and \( \Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} \)=(10.0±0.1)×10−6 m3/mol. The subscript {} indicates that hydroxyl ions are incorporated primarily as defect pairs, probably of the type \( {\left\{ {{\left( {{\text{OH}}} \right)}^{ \bullet }_{{\text{O}}} - {\text{V}}^{{''}}_{{{\text{Me}}}} - {\left( {{\text{OH}}} \right)}^{ \bullet }_{{\text{O}}} } \right\}}^{{\text{x}}} \equiv {\left( {2{\text{H}}} \right)}^{{\text{x}}}_{{{\text{Me}}}} . \) Under similar thermodynamic conditions, the water content in olivine in the martian mantle and in olivine from gabbros may be as much as 5 to 25 times larger than in the less iron-rich olivine dominant in Earth’s mantle.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Structurally bound hydrogen ions (i.e., protons), observed as OH stretching bands in infrared spectra, affect the kinetic properties of nominally anhydrous silicate minerals even at the parts per million level. For instance, the high-temperature creep strength of olivine single crystals as well as olivine aggregates decreases markedly when water is added (Chopra and Paterson 1984; Mackwell et al. 1985; Karato et al. 1986). Recent laboratory experiments have quantified the water-weakening effect for dunite, demonstrating that strain rate increases approximately linearly with increasing water content in both the diffusion and the dislocation creep regimes (Kohlstedt et al. 1995; Mei and Kohlstedt 2000a, 2000b; Karato and Jung 2003). Hence, the viscosity of the mantle will vary substantially from water-undersaturated regions, such as beneath mid-ocean ridges, to water-saturated regions, such as in the mantle wedge above a subducting slab.
The diffusivity of hydrogen as a function of temperature and crystal orientation as well as the solubility of hydroxyl (hydrogen) as a function of water fugacity and pressure have been previously investigated to determine the mechanism and thermodynamics of the incorporation of ‘water’ in olivine (Bai and Kohlstedt 1992, 1993; Kohlstedt et al. 1996; Kohlstedt and Mackwell 1998, 1999). In this paper, we extend these studies by reporting experimental results on the solubility of hydrogen ions (protons) in olivine single crystals as a function of iron content and of temperature for samples hydrothermally annealed under water-saturated conditions. These results lead to the theoretical framework necessary for extrapolating laboratory results to the thermodynamic conditions appropriate for the upper mantle.
Experimental procedure
Starting materials
Six gem-quality single crystals of San Carlos olivine with fayalite contents between Fa8.5 and Fa16.9 were selected for this study. The iron content of each sample was determined with a JEOL JXA-8900R electron microprobe operating at an accelerating voltage of 15 kV and a beam current of 20 nA. For comparison, a single crystal of synthetic forsterite (Fa0) was included. This synthetic crystal was grown by the Czochralski method using an Ir crucible. In addition to ~20 wt. ppm of Ir, the primary impurities were several hundred weight ppm of Fe, <10 wt. ppm of Na, and <1 wt. ppm of Ca (Takei and Kobayashi 1974).
Crystals were first aligned using X-ray microdiffraction to identify the (010) plane and then oriented using Laue back reflection X-ray diffraction to distinguish the [100] and [001] directions. Once the three principal axes were identified, samples with dimensions x a ≃4 mm, x b ≃2 mm and x c=0.5–1.5 mm were cut from each crystal with surfaces perpendicular to the principal axes. To minimize the durations of the hydrothermal anneals, the c-direction was kept smallest since hydrogen diffusion is fastest along the [001] direction in olivine (Kohlstedt and Mackwell 1998).
Sample assemblies
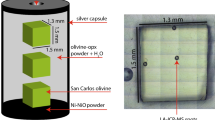
Samples from each of the seven crystals were packed with a mixture of talc and brucite powders inside boxes constructed from Ni foil of 25-µm thickness. Each box contained two or three crystals of the same iron content. The Ni foil kept the powders in contact with the olivine crystals. The talc and brucite powders contributed free water upon their dehydration near 1,073 K. The ratio of talc to brucite was chosen such that the reaction would yield 90% olivine plus 10% enstatite, so that the single crystal samples were buffered at a silica activity set by the presence of enstatite. For each experiment, seven boxes were put into a single nickel capsule 25 mm in length with a 15-mm outer diameter and a 13-mm inner diameter. Six of the boxes contained Fe-bearing olivine and one contained end-member forsterite. The Ni foil and Ni capsule combined with a 0.1-mm-thick layer of NiO powder packed at each end of the capsule maintained the oxygen fugacity at Ni-NiO. Just before welding the cover onto the capsule, a few drops of water were added.
Hydrothermal annealing experiments
Samples were hydrothermally annealed in a gas medium apparatus (Paterson 1990) at 50-K intervals at temperatures ranging from 1,273 to 1,573 K, as summarized in Table 1. The gradient in temperature in the hot zone was ≲ 1 K/mm, and the uncertainty in temperature was less than ±2 K. The confining pressure was maintained at 300±3 MPa, resulting in water fugacities between ~275 and ~310 MPa for temperatures between 1,273 and 1,573 K, respectively (Mackwell and Kohlstedt 1990). To ensure that the concentrations of all of the point defect species (specifically, vacancies and interstitials on the metal, silicon and oxygen sub-lattices) equilibrated to the thermodynamic conditions (P, T, \( f_{{{\text{O}}_{2} }} \), \( a_{{{\text{SiO}}_{{\text{2}}} }} \) and \( f_{{{\text{H}}_{2} {\text{O}}}} \)) imposed during the experiment and that the crystals were fully saturated with water, the duration of the experiments was determined based on the defect diffusivities of Nakamura and Schmalzried (1984), Dimos et al. (1988), and Wanamaker (1994) and the hydrogen diffusivity of Kohlstedt and Mackwell (1998). The annealing experiments lasted from 14 h at 1,273 K to 1 h at 1,573 K. At the end of each hydrothermal annealing experiment, the Ni capsule was filed open to verify that water was present, that is, that water-saturated conditions had been maintained throughout the entire experiment. Samples were subsequently cleaned and doubly polished. After polishing and prior to FTIR analysis, samples were stored in a vacuum oven at ~400 K, 2 kPa for several hours.
Infrared analyses
The water content in each crystal was analyzed with a Nicolet Series II Magna-IR System Fourier transform infrared (FTIR) spectrometer fitted with a Nic-Plan IR microscope and a KBr beam splitter. A beam aperture of 25–50 µm was used for our analyses. Unpolarized IR spectra were collected at a wavenumber resolution of 2/cm, with the incident beam perpendicular to the (010) face while dried air flowed over the sample. The [100] and [001] axes of all of the samples were oriented in the same way with respect to the FTIR specimen stage, as the infrared radiation becomes partially polarized as it passes through the microscope. At least five spectra were collected from each sample with 256 scans per spectrum. Spectra were collected both near the center and near the edge of each sample. Background was removed from FTIR spectra using a spline fit. Concentrations of OH were determined from FTIR spectra using the weighted correction method of Paterson (1982) with integration limits of 3,200 to 3,650/cm and an orientation factor of 1/2 (e.g., Mackwell and Kohlstedt 1990); the resulting concentrations were then increased by a factor of 3.5 following the recent calibration of Bell et al. (2003). As cautioned in Bell et al. (2003), it should be noted that “there is no theoretical basis for a consistent relationship” between OH concentrations determined from polarized and unpolarized infrared spectra [see also Libowitzky and Rossman (1996)]. Hence, the correction factor of 3.5 may be sensitive to the specific sample and measurement conditions (including thickness of the sample and optics of the spectrometer system). However, based on the recent results of Koga et al. (2003), this correction factor appears to work well for the samples and FTIR apparatus used in the present study.
Results
To illustrate the dependence of hydrogen solubility on temperature and iron content, FTIR spectra from samples with four different iron contents annealed at 1,273, 1,373, 1,473 and 1,573 K are compared in Fig. 1; hydrogen contents for all seven samples and all seven annealing temperatures are summarized in Table 1. Hydroxyl concentrations calculated from spectra taken at different locations in a single sample differed by less than 5%. Hydroxyl concentration did not vary significantly between the edge and center of any of the crystals, indicating that the hydrothermally annealed crystals were fully saturated with hydrogen. Hydroxyl concentrations calculated from spectra taken from different samples cut from the same starting crystal also differed by less than 5%.

Unpolarized FTIR spectra illustrating the dependence of OH solubility on T and X Fa. Spectra were obtained from samples of a synthetic forsterite and b–d San Carlos (Fe-bearing) olivine hydrothermally annealed at a confining pressure of 300 MPa at temperatures of 1,273, 1,373, 1,473 and 1,573 K. The wavenumbers of seven and eight absorption peaks are labeled for forsterite and Fe-bearing olivine, respectively
Based on the data in Fig. 1 and Table 1, two trends are clear. First, hydrogen solubility increases systematically with increasing Fa content. Second, hydrogen solubility increases systematically with increasing temperature of the hydrothermal anneal.
Seven distinct peaks appear in the FTIR spectra from hydrothermally annealed samples prepared from the synthetic Fa0 crystal. Eight peaks emerge in the spectra from the iron-bearing samples. The peaks in spectra from the Fa0 samples at wavenumbers <3,600/cm appear to have counterparts in the spectra from the iron-bearing samples. Although the peak at 3,612/cm in the forsterite crystals is absent in the Fe-bearing samples, this peak is present in San Carlos crystals annealed at higher water fugacities (Kohlstedt et al 1996).
The locations of the peaks in the Fe-bearing crystals are independent of Fe content. However, the relatively large peaks present at wavenumbers <3,400/cm [the group II peaks of Bai and Kohlstedt (1993)] in the San Carlos sample with the lowest Fe content (Fa8.5) are significantly smaller in samples with higher Fe contents (Fa12.0 to Fa16.9). The heights of the peaks with wavenumbers >3,400/cm (the group I peaks of Bai and Kohlstedt) all increase with increasing Fe content. The heights of all of the peaks in both the forsterite and the San Carlos crystals increase with increasing temperature.
The systematic increase in solubility of hydroxyl and, therefore, of hydrogen with increasing temperature and increasing Fe content for the San Carlos samples is further illustrated with the Arrhenius plot in Fig. 2. In this figure, values of solubility have been normalized to a water fugacity of 300 MPa based on the observation that hydroxyl (i.e., hydrogen) solubility in olivine is linearly proportional to water fugacity, \( C_{{{\text{OH}}}} \propto f^{{\text{1}}}_{{{\text{H}}_{{\text{2}}} {\text{O}}}} \) (Bai and Kohlstedt 1992; Kohlstedt et al. 1996). At each temperature, the solubility of H in the synthetic Fa0 crystals is lower than anticipated based on extrapolation of the results obtained on the Fe-bearing samples, as discussed in the following section.

Arrhenius plot illustrating the dependence of OH/H solubility normalized to a water fugacity of 300 MPa on temperature for samples of forsterite and Fe-bearing olivine annealed at a confining pressure of 300 MPa. Solid lines result from a non-linear least squares fit of Eq. (7b) to the entire data set of C OH as a simultaneous function of T and X Fa for Fe-bearing olivines. Dashed line labelled ‘linear least-squares fit’ was obtained by fitting the forsterite data alone. Solid line labeled ‘extrapolated’ is the OH solubility predicted for forsterite based on the fitting parameters determined for Fe-bearing olivine. Note that values for C OH reported in this figure were determined from FTIR spectra using the weighted correction method of Paterson (1982) increased by a factor of 3.5 based on the work of Bell et al. (2003)
Finally, electron microprobe analyses revealed (1) no change in Fe:Mg ratio in the Fe-bearing olivine crystals due to cation exchange with the talc + brucite buffer powder and (2) no increase in Ni content of either the Fe-free or the Fe-bearing crystals due to cation exchange with the NiO buffer powder. These results are consistent with published values for cation diffusivity in olivine, which indicate that diffusion distances for cations will be <1 µm under anhydrous conditions (e.g., Chakraborty 1997) and <10 µm under hydrous conditions similar to those used in the present study (Wang et al. 2003).
Discussion
Hydroxyl defects in olivine
The total concentration of hydroxyl (i.e., hydrogen) ions in olivine can be expressed using the Kröger–Vink notation (Kröger and Vink 1956) as the weighted sum of all possible water-derived point defects:
In Eq. (1a), the { } brackets identify defect complexes formed between hydroxyl ions at oxygen sites and point defects such as metal and silicon vacancies. This notation can be simplified by writing, for example, the defect complex \( {\left\{ {{\left( {{\text{OH}}} \right)}^{ \bullet }_{{\text{O}}} - {\text{V}}^{{''}}_{{{\text{Me}}}} } \right\}}^{'} \) as \( {\text{H}}^{'}_{{{\text{Me}}}} \). Hence, Eq. (1a) becomes
To explain the high solubility of hydrogen incorporated into the structure of the nominally anhydrous mineral olivine in high-pressure hydrothermal experiments combined with the linear dependence of water solubility on water fugacity, a major portion of the hydroxyl ions in olivine must form defect complexes (Bai and Kohlstedt 1992; Kohlstedt et al. 1996). Because metal vacancies are one of the majority point defects in olivine under anhydrous conditions, we have argued that the primary water-derived defect complex in olivine is (Kohlstedt and Mackwell 1998, 1999)
This conclusion is consistent with hydroxyl solubility in olivine measured as a function of oxygen fugacity in two-stage hydrothermal annealing experiments (Bai and Kohlstedt 1993). Hence, the total hydroxyl concentration can be approximated from Eq. (1a) as
Thermodynamics of water in olivine
To interpret the solubility results reported in the present paper in terms of point defect thermodynamics, the effect of both temperature and iron content must be taken into account. The reaction equation for the formation of the defect complexes in Eq. (2) can be written as
or equivalently
The law of mass action applied to the reaction described by Eqs. (4a) and (4b) for the introduction of H into olivine yields
Note that in our experiments the activities of enstatite, \( a_{{{\text{MeSiO}}_{{\text{3}}} }} \), and olivine, \( a_{{{\text{Me}}_{{\text{2}}} {\text{SiO}}_{{\text{4}}} }} \), are fixed at unity by the presence of both of these phases throughout the experiments. In Eq. (5), K {} and ΔG {} are the reaction constant and Gibbs energy of formation, respectively, for the reaction given in Eqs. (4a) and (4b) for producing defect complexes, T is absolute temperature, P is pressure, X Fa is the mole fraction of fayalite, and R is the gas constant.
The point defect model derived by Nakamura and Schmalzried (1983) for Fe-bearing olivine provides a useful starting point for analyzing the results of the present experiments. Under anhydrous conditions, the concentration of metal vacancies in olivine increases exponentially with increasing iron content (Nakamura and Schmalzried 1983). A similar result has been reported for (Mg,Co)O in which the concentration of cation vacancies increases exponentially with increasing Co concentration (Schwier et al. 1973; Dieckmann and Schmalzried 1975) and is consistent with the composition dependence of cation interdiffusivities reported for several olivines (Morioka 1989). The former observations have been interpreted as indicating that the change in the Gibbs energy of formation of metal vacancies varies linearly with mole fraction of fayalite and CoO, respectively. Under hydrous conditions, the concentration of defect complexes formed between metal vacancies and hydroxyl ions also increases exponentially with increasing iron content, as illustrated in Fig. 3. Again, phenomenologically this behavior suggests that the Gibbs energy of formation for metal vacancy–hydroxyl ion point defect complexes varies linearly with mole fraction of fayalite. Schmalzried (1995) has discussed this type of behavior for ideal solutions by writing the Gibbs energy of defect formation as a series expansion in terms of composition:

Semi-log plot of OH/H solubility normalized to a water fugacity of 300 MPa versus mole fraction of fayalite for samples hydrothermally annealed at a confining pressure of 300 MPa and four temperatures. Solid lines result from a non-linear least squares fit of Eq. (7b) to the data for C OH as a simultaneous function of T and X Fa for Fe-bearing olivines. Smooth, dashed curves illustrate that data for forsterite are overestimated by extrapolation of the fit obtained for the Fe-bearing olivine samples. Note that values for C OH reported in this figure were determined from FTIR spectra using the weighted correction method of Paterson (1982) increased by a factor of 3.5 based on the work of Bell et al. (2003)
where α i is the coefficient of the i th term in this MacLaurin-type series expansion and α=α1. In Eq. (6), \( \Delta E^{{\text{o}}}_{{{\left\{ {} \right\}}}} \), \( \Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} \), and \( \Delta S^{{\text{o}}}_{{{\left\{ {} \right\}}}} \) are the changes in internal energy, molar volume and entropy due to the incorporation of H as part of a defect complex into olivine. Substitution of Eq. (6) into Eq. (5) then yields an expression for the solubility of hydrogen in olivine,
or, simply,
We used a non-linear least squares fitting program to fit Eq. (7b) to our results in Table 1 for water solubility simultaneously as a function of T and X Fa in order to obtain A, \( \Delta E^{{\text{o}}}_{{{\left\{ {} \right\}}}} \) and α. A value of \( \Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} \)=10.6×10−6 m3/mol was used initially (Kohlstedt et al. 1996). This value for \( \Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} \) was previously determined from the lower-pressure data of Bai and Kohlstedt (1992) obtained at 1,573 K combined with the higher pressure data of Kohlstedt et al. (1996) obtained at 1,273 and 1,373 K. In the present study, the value of \( \Delta E^{{\text{o}}}_{{{\left\{ {} \right\}}}} \) was used to normalize the data of Bai and Kohlstedt (1992) and Kohlstedt et al. (1996) to 1,373 K in order to recalculate \( \Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} \). Iteration of this procedure yields A = 90±10 H/106Si/MPa,\( \Delta E^{{\text{o}}}_{{{\left\{ {} \right\}}}} \)=50±2 kJ/mol, \( \Delta V^{{\text{o}}}_{{{\left\{ {} \right\}}}} \)=(10.0±0.1)×10−6 m3/mol, and α=97±4 kJ/mol. In Fig. 3, best-fit curves are plotted as solid lines through the experimental data for San Carlos olivine, with smooth, dashed curves drawn to connect the results for iron-bearing olivine to those for forsterite.
In both Figs. 2 and 3, the extrapolations of the curves representing Eq. (7b) for the iron-bearing olivines do not intersect the solubility results for forsterite. The extrapolation predicts solubility levels for forsterite that are greater than those observed experimentally. Similar behavior was reported for olivine for the dependence of metal vacancies on oxygen partial pressure (Nakamura and Schmalzried 1983).
Geological implications
Although the variation in composition of olivine in the Earth’s mantle is relatively small with values clustered near Fa10, olivines in the martian mantle (e.g., McGetchin et al. 1981) and in gabbroic intrusions (e.g., Morse 1979) are significantly richer in iron. Based on the results presented above, the solubility of water in these more fayalitic olivines should also be greater. For example, at 1,373 K, extrapolation of Eq. (7b) indicates that the solubility of hydrogen in Fa30 and Fa50 will be more than 5 and 25 times greater, respectively, than the solubility in Fa10 for the same depth (pressure) and water fugacity conditions. Extrapolation of Eq. (7b) describing the solubility of water in olivine to compositions and thermodynamic conditions far from those explored in this investigation must, of course, be conducted with caution. Not only do the errors in C OH introduced by the uncertainty in each of the parameters in Eq. (7b) become significant, but also the mechanism of incorporation of hydrogen into olivine may change to include other OH defects such as those expressed in Eqs. (1a) and (1b), particularly at very high hydrogen concentrations.
Finally, it should be noted that the effect of water on the kinetic properties of the more Fe-rich olivines such as in the martian mantle and in gabbroic rocks has yet to be studied. A direct extrapolation of the results presented here combined with published data quantifying the effect of water on the rheological properties of olivine (Mei and Kohlstedt 2000a, 2000b; Karato and Jung 2003) suggest that, under hydrous conditions, rock viscosity should decrease rapidly with increasing Fe content due to the associated increase in water-derived point defects. In addition, olivine in the martian mantle may provide an even greater reservoir for water than it does in the Earth’s mantle.
References
Bai Q, Kohlstedt DL (1992) Substantial hydrogen solubility in olivine and implications for water storage in the mantle. Nature 357:672–674
Bai Q, Kohlstedt DL (1993) Effects of chemical environment on the solubility and incorporation mechanism for hydrogen in olivine. Phys Chem Min 19:460–471
Bell DR, Rossman GR, Maldener A, Endisch D, Rauch F (2003) Hydroxide in olivine: a quantitative determination of the absolute amount and calibration of the IR spectrum. J Geophys Res 108:2105, 10.1029/2001JB000679
Chakraborty S (1997) Rates and mechanisms of Fe-Mg interdiffusion in olivine at 980°–1300 °C. J Geophys Res 102:12317–12331
Chopra PN, Paterson MS (1984) The role of water in the deformation of dunite. J Geophys Res 89:7861–7876
Dieckmann R, Schmalzried H (1975) Point defects in oxide solid solutions (III): mobilities of cations and vacancies in (Co,Ni)O- and (Co,Mg)O-solid solutions and the calculation of correlation factors. Ber Bunsenges Phys Chem 70:1108–1119
Dimos D, Mackwell SJ, Kohlstedt DL (1988) Transient creep of olivine: point defect relaxation times. Phil Mag A57:779–789
Karato S-I, Jung H (2003) Effects of pressure on high-temperature dislocation creep in olivine. Phil Mag 83:401–414
Karato S-I, Paterson MS, FitzGerald JD (1986) Rheology of synthetic olivine aggregates: influence of grain size and water. J Geophys Res 91:8151–8176
Koga K, Hauri E, Hirschmann M, Bell D (2003) Hydrogen concentration and analyses using SIMS and FTIR: comparison and calibration for nominally anhydrous minerals. Geochem Geophys Geosyst 4:1019, 10.1029/2002GC000378
Kohlstedt DL, Mackwell SJ (1998) Diffusion of hydrogen and intrinsic point defects in olivine. Zeitschr Phys Chem 207:147–162
Kohlstedt DL, Mackwell SJ (1999) Solubility and diffusion of ‘water’ in silicate. In: Wright K, Catlow R (eds) Microscopic properties and processes in minerals. Kluwer, Dordrecht, pp 539–559
Kohlstedt DL, Evans B, Mackwell SJ (1995) Strength of the lithosphere: constraints imposed by laboratory experiments. J Geophys Res 100:17587–17602
Kohlstedt DL, Keppler H, Rubie DC (1996) Solubility of water in the α, β and γ phases of (Mg,Fe)2SiO4. Contrib Mineral Petrol 123:345–357
Kröger FA, Vink HJ (1956) Relation between the concentration of imperfections in crystalline solids. In: Seitz F, Turnball D (eds) Solid state physics, 3. Academic Press, New York, pp 367–435
Libowitzky E, Rossman GR (1996) Principles of quantitative absorbance measurements in anisotropic crystals. Phys Chem Miner 23:319–327
Mackwell SJ, Kohlstedt DL (1990) Diffusion of hydrogen in olivine: implications for water in the mantle. J Geophys Res 95:5079–5088
Mackwell SJ, Kohlstedt DL, Paterson MS (1985) The role of water in the deformation of olivine single crystals. J Geophys Res 90:11319-11333
McGetchin TR, Pepin RO, Phillips RJ (1981) Basaltic volcanism on the terrestrial planets. Pergamon Press, New York, pp 1246–1254
Mei S, Kohlstedt DL (2000a) Influence of water on plastic deformation of olivine aggregates: 1. Diffusion creep regime. J Geophys Res 105:21457–21469
Mei S, Kohlstedt DL (2000b) Influence of water on plastic deformation of olivine aggregates: 2. Dislocation creep regime. J Geophys Res 105:21471–21481
Morioka M (1989) Cationic diffusion in olivine. In: Karato S-I, Toriumi M (eds) Rheology of solids and of the earth. Oxford University Press, Oxford, pp 156–168
Morse SA (1979) Kiglapait geochemistry. I: Systematics, sampling and density. II: Petrography. J Petrol 20:555–624
Nakamura A, Schmalzried H (1983) On the nonstoichiometry and point defects of olivine. Phys Chem Min 10:27–37
Nakamura A, Schmalzried H (1984) On the Fe2+–Mg2+ interdiffusion in olivine. Ber Bunsenges Phys Chem 88:140–145
Paterson MS (1982) The determination of hydroxyl by infrared absorption in quartz, silicate glasses and similar materials. Bull Mineral 105:20–29
Paterson MS (1990) Rock deformation experimentation. In: Duba AG, Durham WB, Handin JW, Wang WF (eds) The brittle–ductile transition in rocks. Geophys Monogr Ser vol 56. AGU, Washington, DC, pp 187–194
Schmalzried H (1995) Chemical kinetics of solids. VCH Verlagsgesellschaft, Weinheim, p 38
Schwier VG, Dieckmann R, Schmalzried H (1973) Punktfehlstellen in Oxidmischphasen (I) Fehlstellenenthermodynamik der Mischphasen (CoxMg1–x)O und (CoxMg1–x)2SiO4. Ber Bunsenges Phys Chem 77:402–408
Takei H, Kobayashi T (1974) Growth and properties of Mg2SiO4 single crystals. J Crystal Growth 23:121–124
Wanamaker BJ (1994) Point defect diffusivities in San Carlos olivine derived from reequilibration of electrical conductivity following changes in oxygen fugacity. Geophys Res Lett 21:21–24
Wang ZY, Hiraga T, Kohlstedt DL (2003) Effect of hydrogen on Fe-Mg interdiffusion in olivine. Appl Phys Lett (in press)
Acknowledgments
Financial support for this research was provided by the National Science Foundation through grant EAR-0106981 and the National Science Foundation of China through grants 10299040-1 and 40174028. The authors thank Shun Karato for providing the synthetic forsterite crystal, Steve Mackwell for invaluable comments and insights, and Mark Zimmerman for critical help with the design of the experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial responsibility: T.L. Grove
Rights and permissions
About this article
Cite this article
Zhao, YH., Ginsberg, S.B. & Kohlstedt, D.L. Solubility of hydrogen in olivine: dependence on temperature and iron content. Contrib Mineral Petrol 147, 155–161 (2004). https://doi.org/10.1007/s00410-003-0524-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00410-003-0524-4