
Abstract
We determine the valence electron density and the electron band structure of stibnite, bismutinite, guanajuatite and antimonelite using the density functional theory. All the compounds present similar electronic properties and exhibit a quasi-1D character. We perform a detailed analysis of the charge topology, the atomic static charges and volumes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sulphosalts are relatively rare minerals, found in polymetallic hydrothermal ore deposits. Their systematics and cristallochemistry were clarified only recently. Their structure may be described as different combinations of several building units (Nowacki 1971; Makovicky 1981, 1989, 1993). This approach also explains the transferability and the variation of certain properties within this family of minerals. However, despite the many structural and chemical data available, little is known about the physical properties of these minerals (Pauling 1970; Hummel and Armbruster 1987; Ghosal and Sack 1999).
Within the Pb–Bi and Pb–Sb sulphosalts group, the stibnite (Sb2S3) and bismuthinite (Bi2S3) minerals are important end-members. They present solid solutions over a wide range of geologic conditions. The crystal structure is formed of 1-D ribbons of polymerized [A4B6] n . Each ribbon is linked to its four neighbors by weak A–B and B–B bonds. The Se-bearing equivalents, Bi2Se3—guanajuatite and Sb2Se3—antimonselite, are isostructural, and present numerous similar properties.
In this paper, we explore the electronic properties of these four compounds using the density-functional theory. We compute the valence electron density, the electron band structure and the corresponding electronic density-of-states (DOS). We also analyze the charge topology and determine the atomic static (Bader) charges and volumes.
Computational details and crystal structures
All the calculations are performed in the framework of the local density approximation (LDA) of the density functional theory (DFT) (Hohenberg and Kohn 1964; Kohn and Sham 1965). According to the LDA, the exchange-correlation energy, E xc in a point r is equal to the E xc of a homogeneous electron gas that has the same density as the electron gas at point r. We use the ABINIT package (Gonze et al. 2002), which is based on pseudopotentials and planewaves. It relies on the adaptation to a fixed potential of the band-by-band conjugate-gradient method (Payne et al. 1992) and on a potential-based conjugate-gradient algorithm for the determination of the self-consistent potential (Gonze 1996).
As usual with planewave basis sets, the numerical accuracy of the calculation can be systematically improved by increasing the cut-off kinetic energy of the planewaves and the density of the sampling of the Brillouin zone. According to the Bloch’s theorem, for a periodic system, integrals in real space over the infinite system are replaced by integrals over the finite first Brillouin zone in reciprocal space. Such integrals are performed by summing the function values of the integrand (for example: the charge density) at a finite number of points in the Brillouin zone called the k-point mesh. Choosing a sufficiently dense mesh of integration points is crucial for the relevancy of the results and it is therefore one of the major objectives when performing convergence tests. We use the Monkhorst–Pack (Monkhorst and Pack 1976) scheme for generating the grid of special k points. We perform tests where the k-point grid density and the plane-wave cut-off kinetic energy are consecutively and independently increased. The variation of the total energy is further monitored. A difference of 1 mHa (1 Hartree = 27.211 eV) between two successive grids/cut-off energies is considered indicative of sufficient convergence. We finally choose a grid of 4×4×4 k points and a 30 Hartree cut-off for the kinetic energy of the planewaves for all the four materials.
In the pseudopotential approach, the core electrons are replaced by pseudopotentials, while the outer electrons are treated explicitly. We use Troullier–Martins pseudopotentials (Troullier and Martins 1991) for all the four elements. The considered valence electrons for Bi, Sb, S and Se are 6s26p3, 5s25p3, 3s23p4 and 4s24p4, respectively.
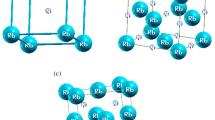
All the calculations are performed on the Pnma orthorhombic experimental structures, illustrated in (Fig. 1). The unit cell parameters and the internal coordinates of the five atoms from the asymmetric unit cell are listed in Tables 1 and 2, respectively. The values, from the ICSD database (2001), were originally reported by Bayliss and Nowacki (1972) for Sb2S3, by Voutsas et al. (1985) for Sb2Se3, by Kanishcheva et al. (1981) for Bi2S3, and by Atabaeva et al. (1973) for Bi2Se3.

3D view of the crystal structure of A2B3 compounds. The ribbons of [A4B6]n are parallel to the b-axis. The larger spheres are for B atoms and the smaller ones for A atoms.
The computation of the electron density and the DOS is done self-consistently. We use the tetrahedron method for the calculation of the DOS (Lehmann and Taut 1972).
Finally, we apply the Bader’s atom-in-molecule method to determine the static atomic charges and volumes (Bader 1979; Bader 1979). According to the Bader approach, the direct space is divided into pieces based on the topological analysis of the electron density. The electronic space of each piece is completely attributed to an atom.
Results and discussions
Valence electron density
The electron density distribution is similar in the four minerals. The electron density maxima are on the order of 1.05–1.20 e−/Å3 for S, 0.80–0.90 e−/Å3 for Se and 0.30–0.40 e−/Å3 for Sb and Bi. We illustrate in Fig. 2 the valence electron density in a cross-section that passes through the innermost atoms (marked by crosses in Fig. 1) of the [A4B6] n ribbons. We observe a strong bonding between the A and B atoms within the ribbons. The inter-ribbons space is depleted of electrons, the inter-ribbons bonding being weaker.

Valence electron density of the a Bi2S3, b Bi2Se3, c Sb2S3 and d Sb2Se3. The cross-section passes through the inner atoms (marked by crosses in Fig. 1) of the [A4B6] n ribbons. The distance between the side ticks is 2 Bohr (1 Bohr=0.52917724 Å).
Electronic band structure and the Density Of States
Based on the previously computed valence electron density, we determine non-self-consistently the electronic band structure. The electron bands along a high-symmetry path through the Brillouin zone and the corresponding electronic-density of states (DOS) are exemplified in Fig. 3 for Sb2S3.

Electron band structure (left) and electronic density of states (right) for Sb2S3. The path through the first Brillouin zones follow the high-symmetry lines and/or its edges: Γ(0 0 0)–X(1/2 0 0)–S(1/2 1/2 0)–Y(0 1/2 0)–Γ–S–R(1/2 1/2 1/2)–T(0 1/2 1/2)–Z(0 0 1/2)–U(1/2 0 1/2)–R–Z–Γ.
The electron band structure and the corresponding DOS are similar for the four minerals. They are all insulators, with 1.55, 1.14, 1.47 and 0.90-eV LDA gaps for Sb2S3, Sb2Se3, Bi2S3 and Bi2Se3, respectively. The computed gaps are most likely underestimated with respect to the measured values, as usual in DFT. For example, optical measurements on thin films of Bi2S3 show a 1.58-eV gap (Mahmoud et al. 1997).
We distinguish three groups of electronic bands, separated by gaps. The energy limits of the three groups, and the gaps between them, are synthesized in Table 3. Figure 4 shows the electron density corresponding to each of the three groups of electronic bands in a section passing through the middle of the ribbons in Sb2S3. The electronic charge of each group stems from complicated schemes of hybridization between the s and p electrons present in the systems.

Partial valence electron density corresponding to the three groups of electronic bands for Sb2S3; top lowest-energy group, middle mid-energy group, bottom: highest-energy group. The cross section passes through the inner atoms (marked by crosses in Fig. 1) of the [Sb4S6] n ribbons. In the top image, the electronic clouds are positioned around the S atoms. The distance between the side ticks is 2 Bohr (1 Bohr=0.52917724 Å).
In order to infer the participation of the different electronic states to the groups of bands, we compute the angular-momentum projection of the DOS inside a sphere centered on some atom. The sphere volume corresponds to the Bader atomic volume (see below). The resulting projected atomic DOS is shown in Fig. 5. The lowest-energy group (8 bands with 16 electrons) corresponds to hybrids of the Sb s and S s electrons. The mid-energy group (12 bands with 24 electrons) corresponds to combined Sb s and S p states, while the charge of the highest-energy group (36 bands with 72 electrons) stems essentially from hybrids of Sb p and S p electrons.

Angular-momentum projected atomic DOS for Sb2S3. Vertical axis is energy in eV, horizontal axis the DOS in e−/eV
Within each group, the bands are highly dispersive along the direction (b*=[010]) reciprocal to the ribbons direction in the real space (b=[010]). The bands are less dispersive along the other directions. Due to the complementarity between the reciprocal space and the direct space, highly dispersive electronic bands in the reciprocal space correspond to highly delocalized electronic states in the real space, while less dispersive (flat) bands correspond to localized electronic states. In the studied A2B3 minerals, the dispersive character of the bands is highly dependent on the orientation of the path. The dispersion diagrams shown in Fig. 3 reveal the existence of electrons confined within the [A4B6] n ribbons: the electronic states are localized within these ribbons, but delocalized along them. This image and the presence of abrupt peaks (van Hove singularities) in the DOS (Fig. 3) underline the low-dimensionality of these systems that behave as quasi-1D ones.
Topology analysis
Table 4 lists the atomic charges and the atomic volumes for the four minerals. The absolute values of the Bader atomic charges are larger in sulfides than in selenides, as a consequence of the higher electronegativity of S than Se. The charges are remarkably similar in the two sulphides than in the two selenides.
The atomic volumes behave differently. The volumes of the anions are relatively constant from one phase to another, while the volumes of the cations change more. Se atoms are larger than S atoms and Bi than Sb. We also observe that the inner atoms (closer to the ribbons center) have smaller volumes than the outer atoms (closer to the ribbons margins).
We also analyze the electron density corresponding to the three groups of bands, within the same approach. In the case of Sb2S3, the topological analysis shows that for the lowest-in-energy group of electronic bands there are 0.36–0.39 electrons around each Sb atom and 1.09 electrons around each S atom. For the mid-energy group, there are 1.44–1.47 electrons around each Sb atom and 1.02–1.04 around each S atom, while in the case of the highest-in-energy group, there are 2.12–2.13 electrons around each Sb atom and 4.55–4.67 electrons around each S atom.
Conclusions
We use the local-density approximation of the density-functional theory to investigate the electron density, the electron band structure and the corresponding density-of-states of four A2B3 minerals: Bi2S3—bismuthinite, Bi2Se3—guanajuatite, Sb2S3—stibnite and Sb2Se3—antimonselite, whose structure is formed by the packing of [A4B6] n ribbons. The chemical bonds are strong within the [A4B6] n ribbons and much weaker between them, as the electron density is concentrated within these ribbons. The electron band structure reveals a quasi-1-dimensional character, the electrons being localized along the ribbons. This may generate interesting behavior under pressure where an insulator-to-metal transition might be expected at large compression. There are three groups of electronic bands corresponding, in increasing order of energy, to hybrids of A s and B s electrons, A s and B p electrons and A p and B p electrons. The topological analysis of the charge shows atomic static charges on the order of +0.9–1.1 for the A atoms and −0.6 to −0.8 for the B atoms.
References
Nowacki W (1971) Crystal chemistry of sulphosalts. Soc Mining Geol Jpn, spec issue 2:3-9
Makovicky E (1981) The building principles and classification of bismuth-lead sulphosalts and related compounds. Fortsch Miner 59:137-190
Makovicky E (1989) Modular classification of sulphosalts—current states. Definition and application of homologous series. N Jb Miner Abh 160:269-297
Makovicky E (1993) Rod-based sulphosalt structures derived from the SnS and PbS archetypes. Eur J Miner 5:545-591
Pauling L (1970) Crystallography and chemical bonding of sulfide minerals. Miner Soc Am Spec Pap 3:125-131
Hummel W, Armbruster T (1987) Tl1+, Pb2+ and Bi3+ bonding and ordering in sulphides and sulphosalts. Schweiz Miner Petrogr Mitt 67:213-218
Ghosal S, Sack RO (1999) Bi–Sb energetics in sulfosalts and sulfides. Miner Mag 63:723-733
Hohenberg P and Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:B864-B871
Kohn W and Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140:A1133-A1138
Gonze X, Beuken JM, Caracas R, Detraux F, Fuchs M, Rignanese GM, Sindic L, Verstraete M, Zerah G, Jollet F, Torrent M, Roy A, Mikami M, Ghosez P, Raty JY and Allan DC (2002) First-principles computation of material properties : the ABINIT software project. Comp Mater Sci 25:478-492
Payne MC, Teter MP, Allan DC, Arias TA and Joannopoulos JD (1992) Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev Mod Phys 64:1045-1097
Gonze X (1996) Towards a potential-based conjugate gradient algorithm for order-N self-consistent total energy calculations. Phys Rev B 54:4383-4386
Monkhorst HJ and Pack JD (1976) Special points for Brillouin-zone integrations. Phys Rev B 13:5188-5192
Troullier N and Martins JL (1991) Efficient pseudopotentials for plane-wave calculations. Phys Rev B 43:1993-2006
ICSD–Inorganic Crystal Structure Database (2001) http://www.fiz-informationsdienste.de/en/DB/icsd/.
Bayliss P and Nowacki W (1972) Refinement of the crystal structure of stibnite, Sb2S3. Z Kristall 135:308–315
Voutsas GP, Papazoglou AG and Rentzeperis PJ (1985) Z Kristall 171:261-268
Kanishcheva AS, Mikhailov YN and Trippel AF (1981) Izv Akad Nauk SSSR. Neorganicheskie Mater. 17:1972-1975
Atabaeva EY, Mashkov SA and Popova SV (1973) Kristallografiya 18:173-174
Lehmann G and Taut M (1972) On the numerical calculation of the density of states and related properties. Phys Status Solidi B 54:469
Bader RFW, Anderson SG and Duke AJ (1979) Quantum topology of molecular charge distributions. 1. J Amer Chem Soc 101:1389-1395
Bader RFW, Nguyen-Dang TT and Tal Y (1979) Quantum topology of molecular charge distributions. II. Molecular structure and its change. J Chem Phys 70:4316-4329
Mahmoud S, Eid AH and Omar H (1997) Optical characteristics of bismuth sulfide (Bi2S3) thin films. Fizika A 6:111-120
Acknowledgements
X.G. acknowledges financial support from the Belgian FNRS.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Caracas, R., Gonze, X. First-principles study of the electronic properties of A2B3 minerals, with A=Bi,Sb and B=S,Se. Phys Chem Minerals 32, 295–300 (2005). https://doi.org/10.1007/s00269-005-0470-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-005-0470-y