
Abstract
We present first-principles calculations on the structural, mechanical, electronic, thermodynamic, lattice dynamic and magnetic properties of RbN in the CsCl, rocksalt (Rs) and zinc-blende (ZB) structures centred on spin-polarised density functional theory (DFT). It was established that in all the three structures, ferromagnetic (FM) state is more stable than the non-magnetic (NM) state. The results show that RbN exhibits half-metallic characteristics at the equilibrium lattice parameters and have small energy gaps of 1.205, 1.092 and 1.364 eV for CsCl, Rs and ZB structures respectively. We find that only the CsCl and Rs structures are mechanically, lattice dynamically and thermodynamically stable. Furthermore, the structures exhibit equal integer magnetic moment of 2 \({\mu }_\mathrm{B}\) approximately.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Few years back, various researchers carried out rigorous studies on half-metallic (HM) ferromagnetic materials in the area of spintronics due to their wide applications. The dual character which the HM ferromagnets exhibit, either as metallic in one spin direction or as semiconductor (or insulator) in the other spin direction within the Fermi level make them very relevant in electronic industries. In the MC (\(\hbox {M}{=}\hbox {Mg}\), Ca, Sr and Ba) binary compounds, both the magnetic properties and the electronic structures are studied in the zinc-blende (ZB) structure [1]. It was observed that these compounds show the character of HM ferromagnetic materials with HM gaps up to 0.83 eV, and subjecting the lattice constants to strain, the half-metallicities for CaC, SrC and BaC were sustained up to 14, 13 and 9% respectively. The electronic structures, magnetic properties and half-metallicity of \(\hbox {Ti}_{{2}}\hbox {IrZ}\) (\(\hbox {Z} = \hbox {B}\), Al, Ga and In) Heusler alloys were investigated with \(\hbox {AlCu}_{\mathrm {2}}\hbox {Mn-}\) and \(\hbox {CuHg}_{\mathrm {2}}\)Ti-type structures within local density approximation (LDA) and generalised gradient approximation (GGA) for the exchange correlation potential [2]. It was observed that \(\hbox {CuHg}_{\mathrm {2}}\)Ti-type structure in FM state was energetically more favourable than \(\hbox {AlCu}_{\mathrm {2}}\)Mn-type structure in all compounds except \(\hbox {Ti}_{\mathrm {2}}\)IrB which was stable in \(\hbox {AlCu}_{\mathrm {2}}\)Mn-type structure in NM state. Recently, the effect of arsenic (As) composition on structural parameters, phase transition pressure, elastic constants, optical and acoustic phonon frequencies at high symmetry points \({\Gamma }\), X and L, static and electronic dielectric constants and Born effective charge were studied in the ternary alloys \(\hbox {YP}_{{1-}{x}} \hbox {As}_{x}\) [3]. In the rocksalt (Rs) structure, employing first-principles calculations to study the electronic and magnetic properties of MP (\(\hbox {M}=\hbox {K}\), Rb) [4], the results reveal that the compounds are HM ferromagnets by large band gaps of 0.46 and 0.74 eV at the equilibrium lattice constants. Recently, the structural, electronic and magnetic properties of \({\mathrm {Cd}}_{\mathrm {1-}x}\mathrm {V}_{x}\mathrm {Se}\) in the ZB structure were investigated, using the first-principles calculations [5], and the results confirmed that the compound exhibits HM characteristics. Also, the total magnetic moment of \(3\, {\mu _{\mathrm{B}}}\) obtained in the study confirms their HM characteristics. Other theoretical investigations where HM ferromagnetic characteristics were confirmed are for \({\mathrm {Zn}}_{1-x}{\mathrm {Cr}}_{x}\mathrm {Se}\) [6], \({\mathrm {Cd}}_{1-x}{\mathrm {Cr}}_{x}\mathrm {Te}\) [7], \({\mathrm {Al}}_{1-x}{\mathrm {Cr}}_{x}\mathrm {As}\) alloys [8], CrM (\(\hbox {M}=\hbox {P}\), As, Sb, Bi) [9], CrSb [10], XY (\(\hbox {X}=\hbox {V}\), Cr, Mn and \(\hbox {Y}=\hbox {N}\), P, As, Sb, S, Se, Te) [11]. The spin-polarised calculation carried out on the RbN and CsN compounds in CsCl structure, using the first-principles method, show that these materials exhibit HM ferromagnetic property and has a magnetic moment of about \(2 \,\mu \hbox {B}\) [12]. Most semiconductors crystallise in the ZB structure. Hence, if RbN in the ZB structures exhibits HM properties then it can be grown epitaxially. Secondly, the material is unique because of its low magnetic moment. And because they are \(s{-}p\) electrons they have low stray fields, and also their rate of energy loss is minimum. How stable are these properties in other structures, especially in the Rs and ZB structures? Are these materials mechanically and thermodynamically stable? Motivated by these questions, we present a first-principles investigation on the structural, mechanical, electronic, thermodynamic, lattice dynamic and magnetic properties of RbN in CsCl, Rs and ZB structures.

The crystal structures of RbN. (a) CsCl, (b) Rs and (c) ZB.

Total energies as functions of lattice parameters for the CsCl, Rs and Zb structures of RbN.
2 Methods
The structural optimisation and prediction of properties of RbN binary compound are performed by adopting the quantum espresso (QE) code as employed in Giannoziet al [13]. This calculation, based on plane-wave pseudopotential (PWPP) method within the framework of the DFT and exchange correlation was calculated using the GGA formalism [14]. To achieve convergence, a plane wave basis set with kinetic energy cut-off of 55 Ry, charge density cut-off of 220 Ry, convergence threshold of \(1.0\times {10}^{8}\) and lattice parameters of 3.623, 6.101 and \(6.701\,{\AA }\) was applied in CsCl, Rs and ZB structures respectively. The crystal structures used in this calculation are presented in figure 1. The ground-state properties of RbN in CsCl, Rs and ZB structures were obtained by performing the structural optimisation for the lattice parameters through energy minimisation as presented in figure 2.
3 Results and discussions
3.1 Structural and mechanical properties
We performed spin-polarised and spin-unpolarised calculations of the total energy as a function of the lattice constant of RbN in the three structures (CsCl, Rs and ZB). Our results and other available literature results are summarised in table 1. We observed a fair agreement between the present results and the literature report [12]. The elastic constants are functions of the stability and stiffness of RbN as recorded in the present calculation. The mechanical properties of RbN in the CsCl, Rs and ZB structures are computed and presented in table 2.

Electronic band structures of RbN in CsCl structure.

Electronic band structures of RbN in Rs structure.
The mechanical stability of a solid depends on the structural features and the corresponding elastic constants [15]. There are three independent elastic constants for cubic phases, \(C_{11}\), \(C_{12}\) and \(C_{44}\). The mechanical stability is measured by the following conditions: \(C_{11}+2C_{12}>0\), \(C_{11}-C_{12}>0\), \(C_{44}>0\) and \(C_{11}>0\) [16]. The structures that satisfy these conditions are CsCl and Rs, and therefore they are mechanically stable while the ZB structure is not stable mechanically. The bulk modulus B, Young Modulus E and shear modulus G are the parameters that are used to quantify the mechanical properties of solids. They show the extent of resistance of the alloys to volume and shear deformation respectively. The higher their values, the stronger the deformation resistance offered by the alloys. From table 1, the results of the bulk, Young and shear moduli show that the deformation resistance decreases from \(\mathrm {CsCl\, to\, Rs\, to\, ZB}\) structure. An empirical relation to determine the plastic properties of materials is given by the ratio of the bulk modulus to the shear modulus B/G. The critical threshold value of the ratio for delineating ductile from brittle materials is about 1.75 [17]. From our results presented in table 2, it is obvious that the structures are brittle in nature because the B/G ratios are lower than the critical threshold value. The anisotropy factor A is a measure of the degree of anisotropy in solid structures [18]. For anisotropic material, the anisotropy factor \(=1\). If the value of A is zero, the material is said to be isotropic. But when the values are deviating from zero and approaching unity it indicates high elastic anisotropy. The present calculated results of A show that the three structures are isotropic. These results are obtained using the relation, from eq. (1), as recorded in [19].

Electronic band structures of RbN in ZB structure.

The spin-dependent partial density of states of RbN in the three structures, with positive and negative DOS corresponding to the majority and minority states.

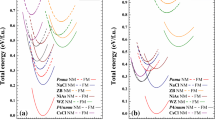
Total energies as functions of lattice constants for the ferromagnetic (FM) and non-magnetic (NM) states of RbN in the CsCl, Rs and ZB structures.
The stability of the material against shear stress can be determined from the value of the Poisson’s ratio of the material. It also discloses the nature of the bonding forces. The value range is of the order \(0<\upsilon <0.5\) [18]. The low value of Poisson’s ratio indicates a large volume compression of the material and when \(\upsilon =0.5\) no volume change occurs. The higher is the Poisson’s ratio, the better is the malleability. From the calculated results of the Poisson’s ratio in table 2, it is clear that the three structures of the RbN are of good plasticity. The microhardness H is another mechanical property that was studied. It was found that the resistance to a localised plastic deformation depends on H. This is given by [19, 20].
The microhardness results obtained for the three structures show that the ZB structure is extremely soft when compared with Mohs scale [21].

Total magnetic moment per formula unit as a function of lattice constants for RbN in the three structures.
3.2 Electronic and magnetic properties
In discussing the origin of half-metallicity in the RbN compound, we present the calculated spin-polarised band structures of RbN in the CsCl, Rs and ZB structures as shown in figures 3–5. As predicted, the minority-spin channel is metallic whereas there is an energy gap around the Fermi level in the majority channel which portrays a semiconductor property. This phenomenon shows that RbN exhibits HM properties in the three structures studied. The calculated spin-resolved total and partial densities of states (PDOS) of RbN at its equilibrium lattice constants are presented in figure 6. From the PDOS of the compound, the plot reveals that the valence bands in the three structures are mainly dominated by the \(\mathrm {N}\) atoms. Here, both the spin-polarised FM phase and spin-unpolarised NM phase were investigated. The calculated results show that the FM states are more stable than the NM states in the three structures studied as presented in figure 7. Hence, RbN exhibits FM character. The ability of these structures of RbN to maintain steady magnetic properties makes them very promising. It is clear from table 3 and figure 8, that the CsCl, Rs and ZB structures exhibit similar features and have approximately the same local magnetic moments of \(2\, \mu _{\mathrm {B}}\) at the equilibrium lattice constant. The total magnetic moment of RbN in the three structures mainly originate from N–p orbitals of the \(\mathrm {N}\) atom and with very small contribution from the \(\mathrm {Rb}-\mathrm {s}\) orbital of the \(\mathrm {Rb}\) atom. Considering the strain of the lattice constants, the half-metallicities for RbN in CsCl, Rs and ZB structures were sustained up to 5.4, 1.8 and 1.5% respectively. The normal electronic configuration of Rb and N are \([\hbox {Kr}]5\hbox {s}^{\mathrm {1}}\) and \([\hbox {He}]2\hbox {s}^{\mathrm {2}}2\hbox {p}^{\mathrm {3}}\). Consequently, the existence of electronic states at the Fermi level in majority-spin state and a band gap at the Fermi level in minority-spin state validate the HM characteristic of the RbN compound in the three structures investigated. It is clear that there is a relatively strong hybridisation between the s-orbital of the Rb and the p-orbital of the N atoms within the Fermi level. This hybridisation results in the splitting of the s–p-orbitals into three states: the bonding states below the Fermi level, the non-bonding states around the Fermi level and the antibonding states above the Fermi level. The s–p hybridisation is the origin of the HM band gap in the RbN compound [22]. The negative sign of the magnetic moment of Rb is due to a negative exchange splitting between spin-up and spin-down electrons in the s-orbital. Therefore, half-metallicity originates from both covalent and s–p hybridisations in the RbN compound as shown in figure 6.

Phonon dispersion curves along symmetry directions of RbN in the CsCl structure.

Phonon dispersion curves along symmetry directions of RbN in the Rs structure.
3.3 Lattice dynamics properties
In this study we calculated the lattice dynamics curves and the corresponding total and partial PDOS employing the GGA-PAW approximation at the equilibrium lattice parameters for the CsCl and Rs structures of RbN, along the high symmetry directions of the Brillouin zone. The present results are illustrated in figures 9 and 10 and the phonon frequencies at high symmetry points of the Brillouin zone were equally investigated. The important features observed in the calculation of the lattice dynamics curves are: Positive frequencies were obtained in the Brillouin zone of the CsCl and Rs structures, while imaginary (negative) frequencies were obtained in the ZB structure of the RbN compound. This indicates that RbN is lattice dynamically stable in the CsCl and Rs structures but is unstable in the ZB structure. The acoustical and optical phonon modes for the two stable structures are alienated by frequency gaps of \(21.62\,{\mathrm {cm}}^{{-1}}\) and \(33.55\,{\mathrm {cm}}^{{-1}}\) for CsCl and Rs structures respectively. At the Brillouin zone of the first \(({\Gamma })\), the following frequencies were obtained: \(156.03\, {\mathrm {cm}}^{{-1}}\) for CsCl and \(167.55\,{\mathrm {cm}}^{{-1}}\) for Rs respectively.
3.4 Thermodynamic properties
The thermodynamic properties of RbN in the three structures have been investigated within a temperature range of 0–800 K. In all the calculations, quasiharmonic approximation (QHA) was applied to compute the Debye temperature \(\varTheta _{\mathrm {D}}\), vibrational entropy S, specific heat capacity at constant volume \(C_{\mathrm{v}}\), free energy \({\Delta }F\) and internal energy \({\Delta }E\) as shown figure 11. From the results, it is observed that the ZB structure is thermodynamically unstable, and so it is not presented in this report. The heat capacity of these two structures increases rapidly as the temperature increases and it approaches the Dulong–Petit limit at very high temperature. It is also observed that at very low temperature, \(C_\mathrm{v}\) is proportional to \(T^{3}\). The entropy and internal energy graphs also show that as the temperature increases the values of the properties increased. But, the free energy values decrease as the temperature increases. The Debye temperature provides information on the strength of covalent bonds in solids, and so higher Debye temperature is an indication of stronger covalent bonds in the compounds [23]. From the present work, shown in figure 6, the CsCl structure of RbN has the strongest covalent bond. The zero-temperature energy of RbN in various structures is presented in table 4.

Variation of thermodynamic properties as a function of temperature for RbN in the CsCl and Rs structures: (a) specific heat capacity, (b) entropy, (c) internal energy and (d) free energy.
4 Conclusion
In the present work, the structural, mechanical, electronic, thermodynamic, lattice dynamic and magnetic properties of RbN in the CsCl, Rs and ZB structures were examined, by performing first-principles calculations. The structural optimisation for the lattice parameter through the energy minimisation was reported in this study. It was observed that the lattice constants increase and the bulk modulus decreases from \(\mathrm {CsCl\, to\, Rs\, to\, ZB}\) respectively. Also, from the investigation of the spin-polarised ferromagnetic (FM) and spin-unpolarised non-magnetic (NM) phases, we found that the FM states are more stable than the NM states, and hence the RbN compound exhibits FM character. Our calculations show that RbN compound in the three structures have HM characteristic at their equilibrium lattice parameters and have energy gaps of 1.205, 1.092 and \(1.364\, \mathrm {eV}\) for CsCl, Rs and ZB structures respectively. It was also observed that the structures exhibit approximately equal integer magnetic moment of \(2\, \mu _{\mathrm {B}}\). We reported that among the three structures investigated, only the CsCl and Rs structures are thermodynamically stable, and their heat capacities approached the Dulong–Petit limit at very high temperature. From the phonon dispersion curves, we reported also that only the CsCl and Rs structures are dynamically stable while imaginary frequencies were observed in the case of ZB structure. It was found that the CsCl and Rs structures satisfied the mechanical stability conditions for cubic phases. From our calculations of the shear and Young moduli, the Poisson’s ratio and the Zener anisotropy, we found that CsCl and Rs structures of the RbN compound are mechanically stable. The B/G ratio shows that the three structures are brittle in nature as the values are lower than the critical threshold value. Also, it was established from table 2 that the structures are isotropic and they are of good plasticity as observed from the Poisson’s ratio calculations. Finally, it was reported that the microhardness H decreases from \(\mathrm {CsCl\, to\, Rs}\, \mathrm {to\, ZB}\) structure.
References
G Y Gao, K L Yao, E Şaşıoğlu, L M Sandratskii, Z L Liu and J L Jiang, Phys. Rev. B 75(17), 174442 (2007)
K H Sadeghi and F Ahmadian, Pramana – J. Phys. 90: 16 (2018)
Y Chaouche and M Souadkia, Pramana – J. Phys. 93: 65 (2019)
Q Gao, L Li, H HXie, G Lei, J B Deng and X R Hu, Mater. Chem. Phys. 163, 460 (2015)
F Ahmadian and N Makaremi, Solid State Commun. 152(17), 1660 (2012)
X F Ge and Y M Zhang, J. Magn. Magn. Mater. 321(3), 198 (2009)
N A Noor, S Ali and A Shaukat, J. Phys. Chem. Solids 72(6), 836 (2011)
Y H Zhao, G P Zhao, Y Liu and B G Liu, Phys. Rev. B 80(22), 224417 (2009)
M Zhang, H Hu, G Liu, Y Cui, Z Liu, J Wang, G Wu, X Zhang, L Yan, H Liu and F Meng, J. Phys.: Condens. Matter 15(29), 5017 (2003)
B G Liu, Phys. Rev. B 67(17), 172411 (2003)
I Galanakis and P Mavropoulos, Phys. Rev. B 67(10), 104417 (2003)
A Lakdja, H Rozale and A Chahed, Comput. Mater. Sci. 67, 287 (2013)
P Giannozzi, S Baroni, N Bonini, M Calandra, R Car, C Cavazzoni, D Ceresoli, G L Chiarotti, M Cococcioni, I Dabo and A Dal Corso, J. Phys.: Condens. Matter 21(39), 395502 (2009)
J P Perdew, K Burke and M Ernzerhof, Phys. Rev. Lett. 77(18), 3865 (1996)
Z Wu, X Hao, X Liu and J Meng, Phys. Rev. B 75(5), 054115 (2007)
G V Sin’ko and N A Smirnov, J. Phys.: Condens. Matter 14(29), 6989 (2002)
S F Pugh, XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science 45(367), 823 (1954)
S Boucetta, J. Mag. Alloys 2(1), 59 (2014)
C H Cheng, C F Yu and W H Chen, J. Alloys Compd 546, 286 (2013)
A El-Adawy and N El-KheshKhany, Solid State Commun. 139(3), 108 (2006)
W D Callister Jr, in: Science and engineering materials – An introduction, 7th edn (John Wiley and Sons, 2006) p. 155
X-P Wei, J-B Deng, Ge-Y Mao, S-B Chu and X-R Hu, Intermetallics 29, 86 (2012)
F Bakare, M I Babalola and B E Iyorzor, Mater. Res. Express 4(11), 116502 (2017)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Iyorzor, B.E., Babalola, I.M. Spin-polarised DFT study of the structural stability and half-metallicity of RbN in the CsCl, rocksalt and zinc-blende structures. Pramana - J Phys 94, 83 (2020). https://doi.org/10.1007/s12043-020-01950-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12043-020-01950-9
Keywords
- Half-metallic ferromagnetism
- spin polarisation
- band structure
- density of state
- lattice vibration curve
- generalised gradient approximation
- quasiharmonic approximation