
Abstract
Emerging evidence suggests that an increased density of pre-treatment CD8+ tumor-infiltrating lymphocytes (TILs) is associated with good response to chemoradiotherapy (CRT) in patients with locally advanced rectal cancer. However, the significance of T-cell complexity in the clinical setting remains unknown. High-throughput T-cell receptor (TCR) β sequencing was applied to quantify the TCR repertoire of pre-treatment biopsies from 67 patients with advanced rectal cancer receiving preoperative CRT. Diversity index was used to represent the complexity of the TCR repertoire in a tumor. Pre-treatment CD8+ TIL densities were assessed by immunohistochemistry. Changes in TCR repertoire before and after CRT were also analysed in 23 patients. Diversity indices were significantly higher for good responders than for non-responders (P = 0.031). The multivariate analysis revealed that both CD8+ TIL density and TCR diversity index were independently associated with good response to CRT (P < 0.001 and P = 0.049, respectively). Patients who were high for both CD8+ TIL density and TCR diversity (double-high) had markedly better responses to CRT than double-low patients (84.2% vs 16.7%, P < 0.0001). Larger changes in TCR repertoires before and after CRT were correlated with better recurrence-free survival (P = 0.027). The complexity and dynamic change in the TCR repertoire might serve as a useful indicator of response to CRT in combination with CD8+ TIL density in patients with rectal cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neoadjuvant chemoradiotherapy (CRT) followed by total mesorectal excision is the standard of care for patients with locally advanced rectal cancer. Whereas some patients respond completely to CRT and can avoid surgery [1], others respond poorly. Predicting responses to CRT would enable a more tailored treatment strategy: when a poor response to CRT is anticipated, CRT can be omitted to minimize any potential morbidity or detrimental functional consequences associated with CRT [2]. Multiple studies have previously identified potential biomarkers that can effectively distinguish between good and poor responders, including the expression levels of ectopic p53 protein [3], Ki67 and thymidylate synthase [4], and several gene sets [5] and microRNA sets [6].
T cell-mediated adaptive immune response plays an important role in cancer progression [7]. A higher density of CD8+ tumour-infiltrating lymphocytes (TILs) has been reported to be associated with better prognosis in patients with various types of cancer, including colorectal cancer [8]. Whereas various clinical and pathological variables are linked with response to radiotherapy, recent studies have elucidated that the clinical efficacy of radiation is attributed to CD8+ T-cell infiltration and function [9]. We and other groups have previously shown that CD8+ TIL abundance in pre-treatment biopsies is associated with better responses to CRT in patients with rectal cancer [10,11,12,13,14]. Furthermore, recent next-generation sequencing studies have unveiled that the complexity of the T-cell receptor (TCR) repertoire could serve as a biomarker for prognosis and response to therapy in specific types of cancer [15,16,17,18]. For example, a less-diverse intra-tumoral TCR repertoire is associated with a better response to anti-PD-1 therapy in metastatic melanoma [19], whereas a more diverse TCR repertoire is associated with sensitivity to anti-PD-1 therapy in metastatic Merkel cell carcinoma [17] and sensitivity to chemotherapy in diffuse large B-cell lymphoma [16]. However, no study has evaluated the impact of the TCR repertoire on the response to CRT in rectal cancer.
In the present study, we aimed to quantify the complexity of the TCR repertoire in the tumors of patients with advanced rectal cancer before they commenced CRT, and to investigate its correlation with treatment response. We also sought to examine how the TCR repertoires change between pre- and post-CRT samples. We show that response to CRT is associated with not only the CD8+ TIL density but also the complexity of the TCR repertoire in pre-treatment samples. In addition, we show that larger variations in the TCR repertoire are associated with a favourable long-term outcome. Our data provide new insight into the impact of pre-treatment TCR complexity and TCR repertoire shifts over time on CRT response and recurrence in patients with rectal cancer.
Materials and methods
Patients
All patients underwent oral 5-fluorouracil-based CRT with a total dose of 45.0 or 50.4 Gy, followed by total mesorectal excision within a median interval of 48 days (interquartile range 43–55 days) from the completion of CRT. In our institution, neoadjuvant CRT is indicated for patients with clinical stage II–III tumors when the inferior border of the tumor is located below the peritoneal reflection. The current study was approved by the institutional review board of the Cancer Institute Hospital (Tokyo, Japan; approval number “2013-1003”) and was conducted in compliance with the Declaration of Helsinki. Signed informed consent was obtained from all participants.
Tumor samples
Tumor samples used in the current study were obtained through pre-treatment endoscopic biopsies or following surgical excision [10]. Of the 107 biopsy samples with previous whole-exome sequencing and/or expression microarray data, TCR repertoire sequencing was performed in 69 samples for which > 800 ng RNA was available. Two samples were excluded due to a failure in library construction. Finally, 67 pre-treatment samples were analyzed (Supplementary Table 1). Supplementary Table 2 presents the clinical characteristics. Post-treatment tumor samples were collected from surgical specimens to assess the dynamic changes in TCR repertoire after CRT; specimens were only available for 23 patients because of the difficulties associated with obtaining sufficient amount of tissues from surgical specimens after CRT, especially in cases when only scar or necrotic ulcerative tissues are available after tumor shrinkage. Biopsies and post-treatment tumor samples were stored at − 150 °C. The procedures for DNA and RNA preparation, whole-exome sequencing, expression microarrays, and microsatellite instability (MSI) testing were described previously [10].
Tumor regression grade
The response to preoperative CRT was assessed on surgically resected specimens according to Dworak’s criteria [20] for tumor regression grade (TRG): TRG 1, dominant tumor mass with obvious fibrosis; TRG 2, dominantly fibrotic changes with few tumor cells; TRG 3, very few (difficult to find microscopically) tumor cells in the fibrotic tissue; and TRG 4, no viable tumor cells. Specimens graded as TRG 4 and TRG 3 were grouped as “good responders”, whereas those graded as TRG 1 were “non-responders” [21]. TRG 2 specimens were categorised as “partial responders” and were excluded from exome sequencing/microarray analysis [10].
T-cell receptor sequencing and repertoire analysis
The complimentary determining region 3 (CDR3) of the TCR β chain was amplified using the iRepertoire multiplex primer set (HTBI-M; iRepertoire, Huntsville, AL, USA) following the manufacturer’s instructions [22]. The Qiagen OneStep RT-PCR kit (Qiagen, Valencia, CA, USA) was used for reverse transcription. Amplification and library preparation were performed with the iR-Processor and iRock 2.0 (iRepertoire, Huntsville, AL, USA). Paired-end sequencing was performed on purified libraries using an Illumina MiSeq v2 500-cycle Reagent Kit (Illumina, San Diego, CA, USA). The web tools provided by iRepertoire (https://www.irepertoire.com) were used to perform basic informatics analyses, such as barcode demultiplexing and filtering, V(D)J alignment, and CDR identification, and to calculate diversity indices [22]. The Morisita–Horn index was used to assess the extent of overlap in the TCR repertoires between samples; the range is zero (no overlap) to one (perfect overlap) [23].
Neoantigen prediction
Using our previous whole-exome sequencing data [10], we performed HLA genotyping and neoantigen predictions using Optitype [24] and NetMHCpanv2.8 software [25]. We considered as neoantigens all 8 to 11-mer peptides harbouring each substituted amino acid with binding affinities of < 50 nM to HLA class I alleles.
Immunohistochemistry
CD8+ TIL density was quantified using the Aperio ImageScope system (Leica Biosystems, Newcastle, UK), as described previously [10, 26]. Among 67 samples with TCR repertoire sequencing data, two samples could not be evaluated due to an insufficient number of cancer cells.
Statistical and bioinformatic analyses
Continuous variables were compared using the Mann–Whitney U test, and categorical variables were compared using Fisher’s exact test or χ2 test. Paired continuous variables were compared using Wilcoxon matched-pairs signed rank test. Spearman correlation was used to determine the relationship between two sets of data. Receiver operating characteristic (ROC) curve analysis with the Youden index was used to determine the optimal cut-off value. The Kaplan–Meier method with log-rank test was used for survival analyses. Recurrence-free survival (RFS) was defined as the time from surgery to any recurrence. Statistical analyses were performed with GraphPad Prism 7 software (GraphPad, San Diego, CA). Univariate and multivariate analyses were performed using the logistic regression model with JMP software V10.0.2 (SAS Institute, USA) to evaluate predictors of CRT response. The Morisita–Horn index was computed using the divo package in R (v3.6.3; R Foundation for Statistical Computing, Vienna, Austria). Differentially expressed genes in the microarray between groups were obtained using SAGx package in R/Bioconductor. Gene ontology (GO) analysis was performed using the online platform Metascape [27], where differentially expressed genes are assigned to a set of predefined terms (Kyoto Encyclopedia of Genes and Genomes Pathway, GO Biological Processes, Reactome Gene Sets, Canonical Pathways, and CORUM). For estimation of term similarity, the arrangement calculator Cohen kappa coefficient was used; a kappa value > 0.3 was set as a threshold for selecting the terms for clustering. Two-sided P values < 0.05 were considered statistically significant.
Results
Patient characteristics
The clinical characteristics of the 67 patients included in this study are summarized in Supplementary Table 2. Forty (60.0%) patients were male, and 39 (58.2%) patients were clinical stage III. Thirty-two (47.8%) patients were good responders (TRG 3/4), and 35 (52.2%) patients were non-responders (TRG 1).
Relationship between TCR repertoire and response to CRT
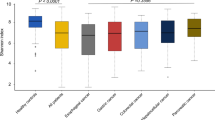
To assess the complexity of the TCR repertoire in a primary tumor before CRT, we isolated RNAs from pre-treatment biopsy specimens and performed TCRβ sequencing. We obtained a total of 646,753 ± 155,125 sequence reads (average ± SD) mapped to the V, D, J, and C segments, and identified 1,734 ± 1,212 unique CDR3 clonotypes that are important for the recognition of a unique antigen on an HLA molecule. The frequencies of the top 10 clonotypes ranged from 1.9% to 89.7% (54.5 ± 23.8%), indicating high variability in TCR diversity within a tumor between patients (Fig. 1a). We used the diversity index to evaluate TCR complexity in a tumor sample, with higher values indicative of higher complexity. The diversity index was significantly higher for good responders than non-responders (P = 0.0309, Fig. 1b). The TCR diversity index was also significantly higher for ypN-negative patients than for ypN-positive patients, and it tended to be higher for ypT0-2 patients than for ypT3-4 patients (P = 0.0451, P = 0.0673, respectively; Fig. 1c). RFS was not significantly different between high and low TCR diversity index groups [Fig. 1d; median follow-up, 74 months after surgery (interquartile range, 67–85 months)].

Response to preoperative chemoradiotherapy (CRT) and survival according to T-cell receptor (TCR) repertoire diversity. a Clonotypes for TCR repertoire of pre-treatment biopsy samples. Frequencies of top 10 clonotypes in a sample are presented as a colour map after sorting by diversity index. Diversity index is shown as a heatmap below the colour map. Tumor regression grade (TRG), microsatellite instability (MSI) status and mutational status of POLE exonuclease (exo) domain for each sample are shown. b TCR repertoire diversity index and tumor regression grade (TRG). The median value is represented by a horizontal line. Data were analysed using a Mann–Whitney U test. c TCR repertoire diversity index and post-CRT tumor and lymph node stage (left; ypT, and right; ypN). The median value is represented by a horizontal line. Data were analysed using Mann–Whitney U-tests. (d) Kaplan–Meier curves for recurrence-free survival by TCR repertoire diversity index. Groups were compared with the log-rank test
No positive correlation of TCR repertoire diversity with mutational/neoantigen load
Given the positive link between mutational load and sensitivity to CRT [10], we suspected that there might also be a possible correlation between mutational load and TCR complexity. We identified four hypermutators (Fig. 1a): two had POLE proof-reading domain mutations; and two—without POLE mutations—showed MSI-H accompanied by germline mutations in MSH2. One tumor with a POLE mutation also had an MSI-H phenotype. With the exclusion of four hypermutators, we observed no correlation for the number of single nucleotide variants (SNVs)/indels or the number of neoantigens (Fig. 2a, b; P = 0.1810 and P = 0.6762).[10] Correlations were also not significant when the hypermutator samples were included (SNVs/indels; P = 0.1144, and neoantigens; P = 0.4523). The TCR diversity index for the hypermutators was high (≥ 4.9) in 3 tumors but low (= 4.2) in 1 tumor with MSI-H. CD8+ TIL density was high (≥ 73 cells/mm2) for all 4 hypermutators.

Relationship between T-cell receptor (TCR) repertoire diversity and mutational/ neoantigen load. a Diversity index and number of single nucleotide variations (SNVs)/insertions and deletions (indels). b Diversity index and number of neoantigens predicted from SNVs. Spearman’s correlation test was used to compute rho and P value
Relationship between TCR repertoire diversity and CD8+ TIL density
Consistent with previous reports [10], we observed a positive association for CD8+ TIL density with good response (TRG 3/4) to CRT (Fig. 3a; P = 0.0010). Although not significant, there was a high trend for an association between diversity index and CD8+ TIL density (Fig. 3b; P = 0.0970). Using univariate logistic regression analyses, we found that patients with a higher density of CD8+ TILs and with more complex T-cell repertoires in the tumor had a higher frequency of CRT-sensitive (TRG 3/4) tumors (Table 1; P < 0.001 and P = 0.009). Importantly, these correlations were independent of various clinical parameters, as revealed by the multivariate test (Table 1; P < 0.001 and P = 0.049). To determine whether the diversity index could be used to refine patient subgroups based on CD8+ TIL density, we divided patients into four groups according to a combination of (1) CD8 + TIL density (TIL-high; TILhi, and TIL-low; TILlo) and (2) TCR diversity index (DI-high; DIhi, and DI-low; DIlo). Whereas the TILhi/DIhi group had the highest frequency of responders (TRG 3/4; 84.2%), the lowest frequency was observed in the TILlo/DIlo group (16.7%). The TILhi/DIlo and TILlo/DIhi groups exhibited an intermediate frequency of responders (TRG 3/4; 52.9% and 36.4%, respectively) (Fig. 3c). A significant difference in the frequency of responders (TRG 3/4) was detected between TILhi/DIhi and TILlo/DIlo groups (P < 0.0001 by Fisher exact test). These findings suggest that T-cell complexity is associated with good response in combination with CD8+ TIL density.

CD8+ tumor-infiltrating lymphocyte (TIL) density and T-cell receptor (TCR) diversity in response to preoperative chemoradiotherapy (CRT). a CD8+ TIL density and tumor regression grade (TRG). Horizontal line indicates median value. P value was computed by Mann–Whitney U test. b CD8+ TIL density and TCR diversity. Rho and P value were computed by Spearman’s correlation test. c Combinatory status of CD8+ TIL density and TCR diversity and TRG. P value was computed by χ2 test and Fisher’s exact test
Differences in gene expression between TILhi/DIhi and TILlo/DIlo groups
To gain biological insight into the link between TCR repertoire status and CRT response, we assessed differentially expressed genes between TILhi/DIhi and TILlo/DIlo groups by exploring transcriptome data. Among the differentially expressed genes (q < 0.1; between the groups), 108 and 743 genes were upregulated in TILhi/DIhi and TILlo/DIlo tumors, respectively (Fig. 4a, Supplementary Table 3). Whereas upregulated genes in the TILhi/DIhi group included gene ontological (GO) annotations of “leucocyte activation”, “immune-response activating cell surface receptor signaling pathway”, and “cytokine signalling pathway”, genes enriched in the TILlo/DIlo group were associated with “ribosome biogenesis and assembly” (P value cut-off = 0.001). In a network analysis, we observed intra- and inter-cluster similarities in the clusters enriched in the TILhi/DIhi group; the genes expressed in the TILlo/DIlo group showed no cluster similarity (Fig. 4b). These findings imply the relevance of immunological processes in CRT sensitivity for TILhi/DIhi tumors.

Differentially expressed genes between double-high and double-low tumors for CD8+ TIL density and TCR diversity. a Heatmap of differentially expressed genes. Expression levels of genes with q-value < 0.1 are shown (red; overexpressed, and green; underexpressed). Q-value was computed by Benjamini and Hochberg’s method. b Network of enriched clusters in the TILhi/DIhi group compared with the TILlo/DIlo group. Each node represents an enriched term, and node size is proportional to the number of input genes that fall into that term. The thickness of the node connection represents the term similarity, as calculated by Cohen kappa coefficient
Similarity of TCR repertoire before and after CRT
Recent findings have indicated that exposing tumor cells to radiation evokes neoantigen production and activation of immunological processes against tumor cells [28]. To explore the effect of CRT on the TCR repertoire, we measured the T-cell complexity in both pre- and post-CRT samples obtained from biopsy and surgical specimens (n = 23 patients). We found that the diversity index significantly increased after CRT (data not shown, P = 0.0085). When we assessed this change in the diversity index according to CRT response, we found that the diversity index significantly increased after CRT in non-responders (TRG 1 patients; P = 0.0074) but was not significantly altered in good responders (TRG 3/4 patients; P = 0.6875) (Fig. 5a). Neither an increase nor decrease in the diversity index during CRT significantly affected patient RFS (Fig. 5b). The similarity in the TCR repertoire before and after CRT was then assessed using the Morisita–Horn index. We found that the Morisita–Horn index was not significantly different between good responders (TRG 3/4) and non-responders (TRG 1) (P = 0.6828) but tended to be higher in patients with recurrence than in those without (P = 0.0810, Fig. 5c). Of note, patients with a higher Morisita–Horn index exhibited shorter RFS [Fig. 5d; P = 0.0270, median follow-up of 84 months after surgery (interquartile range 66–94 months)], which may suggest that the extent of alterations to the TCR repertoire during CRT is an important prognostic factor.

Alteration of T-cell receptor (TCR) repertoire before and after CRT. a Change in TCR diversity index before and after CRT in good responders (left; TRG 3/4) and non-responders (right; TRG 1). P value was computed by Wilcoxon matched-pairs signed rank test. b Kaplan–Meier curve analysis for recurrence-free survival between increased and decreased TCR repertoire diversity. P value was computed by the log-rank test. c Morisita–Horn index and recurrence. Horizontal line indicates median value. P value was computed by Mann–Whitney U-test. d Kaplan–Meier curve analysis for recurrence-free survival between high and low Morisita–Horn indices. P value was computed by the log-rank test
Discussion
We applied high-throughput next-generation sequencing to characterize the TCR repertoire in pre-treatment biopsy specimens, and showed that TCR diversity in a pre-treatment sample was positively correlated with good response to CRT. This suggests that a diverse TCR repertoire before CRT provides adequate immune surveillance and, hence, ensures broad tumor antigen control. Whereas the importance of TCR complexity in tumor tissues has been previously noted in predicting patient responses to chemotherapy and immunotherapy [16, 17], few studies have evaluated the impact of TCR complexity on the response to radiotherapy, including CRT. One previous study described a lack of correlation between TCR diversity and survival in head and neck squamous cell carcinoma treated with CRT [29]. To our knowledge, the present work is the first to show the relevance of TCR complexity on CRT response.
CD8+ TIL density and TCR diversity index were independently associated with response to CRT, indicating that not only density but also complexity of TIL coordinately impact on CRT responses in rectal cancer. Noteworthy, improved RFS was associated with CD8+ TIL density in a previous study [10] but not with TCR diversity in the current study. There could be two reasons for this difference: First, CD8+ TIL density better reflects the number of T cells with effector activity than does the diversity index, which does not distinguish between CD8 and CD4. Second, CD8+ TIL density reflects not only the number of unique clonotypes but also the number of expanded clones; i.e., the extent of T-cell migration and expansion. Comparatively, the TCR diversity index is based on the number of unique clonotypes, and therefore, only captures the extent of T-cell migration. Neither CD8+ TIL density nor TCR diversity index are a perfect index to reflect the extent of CD8+ TIL, particularly in terms of recognizing tumor-associated antigens. Therefore, it is important to note that the TCR diversity is useful as a biomarker along with CD8+ TIL density, with the combination offering a more precise way to identify good responders than using CD8+ TIL density alone.
Functional characterization of the immune microenvironment in TILhi/DIhi tumors using microarray data revealed a higher expression of genes involved in T-cell activation: e.g., IL2RB, CD3D, TCIRG1 [30], LCP2 [31], RASGRP1 [32]. A previous study of rectal cancer similarly showed enrichment of immune activation-related pathway markers in CRT responders [33]. LCP2 is an adapter protein that acts as a substrate in the TCR-activated protein tyrosine kinase pathway, and is important for T-cell activation [31]. TCIRG1 is a membranous protein expressed in T-cells and is essential for T-cell activation [30]. RasGRP1 is responsible for Ras activation in T-cells, and mediates the activation of conventional αβ T-cells [32].
The TCR repertoire becomes more diverse after CRT as compared with baseline readings. This change is in accordance with previous observations showing that radiation enhances TCR repertoire diversity in intra-tumoral T-cells in mouse models of metastatic melanoma [34]. The increase in TCR diversity is perhaps caused by an elevation in the expression of tumor-associated antigens after irradiation, which can develop additional tumor-reactive clones [18]. Importantly—yet possibly derived from different sampling regions—we found a dissimilarity in the clonotypes between pre- and post-CRT to be associated with improved RFS but not with TRG. This finding suggests that the newly emerged T-cell clones, which can recognize a new set of tumor antigens, can endow the host with long-lasting immunity, and, therefore, effectively suppress tumor recurrence. Because the sample number is limited, a future study with larger sample size is needed to confirm the impact of changes in the TCR repertoires before and after CRT on prognostication and perhaps to rule out any positional bias associated with sampling region.
There were several limitations in this study, most of which were predominantly due to the small sample size and the retrospective study design. Partial responders were excluded from whole-exome sequencing, expression microarray, and TCRβ sequencing analyses [10], which may have caused a bias in our study population. We do not have data regarding the impact of TCR diversity on CRT response according to T-cell type (e.g., CD8 + T-cells, CD4 + T-cells, CD4 + CD25 + regulatory T-cells). Furthermore, we restricted our evaluation of TCR diversity to the TCRβ CDR3 region: this might have led to an underestimation of diversity, because a functional TCR is composed of α and β chains.
In conclusion, to our knowledge, this is the first study exploring the TCR repertoire in patients with rectal cancer who were treated with CRT. Our data suggest that the pre-treatment complexity of the TCR repertoire is associated with CRT responses in combination with CD8+ T-cell density, and that a lower overlap of the TCR repertoire before and after CRT is associated with lower rates of recurrence. These results should be validated in a larger cohort study.
References
van der Valk MJM, Hilling DE, Bastiaannet E, Meershoek-Klein Kranenbarg E, Beets GL et al (2018) Long-term outcomes of clinical complete responders after neoadjuvant treatment for rectal cancer in the International Watch & Wait Database (IWWD): an international multicentre registry study. Lancet 391:2537–2545
Honda M, Akiyoshi T, Noma H, Ogura A, Nagasaki T, Konishi T et al (2016) Patient-centered outcomes to decide treatment strategy for patients with low rectal cancer. J Surg Oncol 114:630–636
Spitz FR, Giacco GG, Hess K, Larry L, Rich TA, Janjan N et al (1997) p53 immunohistochemical staining predicts residual disease after chemoradiation in patients with high-risk rectal cancer. Clin Cancer Res 3:1685–1690
Kikuchi M, Mikami T, Sato T, Tokuyama W, Araki K, Watanabe M et al (2009) High Ki67, bax, and thymidylate synthase expression well correlates with response to chemoradiation therapy in locally advanced rectal cancers: proposal of a logistic model for prediction. Br J Cancer 101:116–123
Watanabe T, Kobunai T, Akiyoshi T, Matsuda K, Ishihara S, Nozawa K (2014) Prediction of response to preoperative chemoradiotherapy in rectal cancer by using reverse transcriptase polymerase chain reaction analysis of four genes. Dis Colon Rectum 57:23–31
Campayo M, Navarro A, Benitez JC, Santasusagna S, Ferrer C, Monzo M et al (2018) miR-21, miR-99b and miR-375 combination as predictive response signature for preoperative chemoradiotherapy in rectal cancer. PLoS ONE 13:e0206542
van der Leun AM, Thommen DS, Schumacher TN (2020) CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 20:218–232
Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C et al (2018) International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391:2128–2139
Weichselbaum RR, Liang H, Deng L, Fu YX (2017) Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol 14:365–379
Akiyoshi T, Tanaka N, Kiyotani K, Gotoh O, Yamamoto N, Oba K et al (2019) Immunogenomic profiles associated with response to neoadjuvant chemoradiotherapy in patients with rectal cancer. Br J Sur 106:1381–1392
Anitei MG, Zeitoun G, Mlecnik B, Marliot F, Haicheur N, Todosi AM et al (2014) Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin Cancer Res 20:1891–1899
Yasuda K, Nirei T, Sunami E, Nagawa H, Kitayama J (2011) Density of CD4(+) and CD8(+) T lymphocytes in biopsy samples can be a predictor of pathological response to chemoradiotherapy (CRT) for rectal cancer. Radiat Oncol 6:49
Teng F, Meng X, Kong L, Mu D, Zhu H, Liu S et al (2015) Tumor-infiltrating lymphocytes, forkhead box P3, programmed death ligand-1, and cytotoxic T lymphocyte-associated antigen-4 expressions before and after neoadjuvant chemoradiation in rectal cancer. Transl Res 166:721–732
Shinto E, Omata J, Sikina A, Sekizawa A, Kajiwara Y, Hayashi K et al (2020) Predictive immunohistochemical features for tumour response to chemoradiotherapy in rectal cancer. BJS Open 4:301–309
Cui JH, Lin KR, Yuan SH, Jin YB, Chen XP, Su XK et al (2018) TCR Repertoire as a novel indicator for immune monitoring and prognosis assessment of patients with cervical cancer. Front Immunol 9:2729
Keane C, Gould C, Jones K, Hamm D, Talaulikar D, Ellis J et al (2017) The T-cell receptor repertoire influences the tumor microenvironment and is associated with survival in aggressive B-cell lymphoma. Clin Cancer Res 23:1820–1828
Spassova I, Ugurel S, Terheyden P, Sucker A, Hassel JC, Ritter C et al (2020) Predominance of central memory T cells with high T-cell receptor repertoire diversity is associated with response to PD-1/PD-L1 inhibition in merkel cell carcinoma. Clin Cancer Res 26:2257–2267
Liu YY, Yang QF, Yang JS, Cao RB, Liang JY, Liu YT et al (2019) Characteristics and prognostic significance of profiling the peripheral blood T-cell receptor repertoire in patients with advanced lung cancer. Int J Cancer 145:1423–1431
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L et al (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571
Dworak O, Keilholz L, Hoffmann A (1997) Pathological features of rectal cancer after preoperative radiochemotherapy. Int J Colorectal Dis 12:19–23
Ryan R, Gibbons D, Hyland JM, Treanor D, White A, Mulcahy HE et al (2005) Pathological response following long-course neoadjuvant chemoradiotherapy for locally advanced rectal cancer. Histopathology 47:141–146
Wang C, Sanders CM, Yang Q, Schroeder HW Jr, Wang E, Babrzadeh F et al (2010) High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proc Natl Acad Sci USA 107:1518–1523
Rempala GA, Seweryn M (2013) Methods for diversity and overlap analysis in T-cell receptor populations. J Math Biol 67:1339–1368
Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O (2014) OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics 30:3310–3316
Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O et al (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13
Ogura A, Akiyoshi T, Yamamoto N, Kawachi H, Ishikawa Y, Mori S et al (2018) Pattern of programmed cell death-ligand 1 expression and CD8-positive T-cell infiltration before and after chemoradiotherapy in rectal cancer. Eur J Cancer 91:11–20
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O et al (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature Commun 10:1523
Lhuillier C, Rudqvist NP, Elemento O, Formenti SC, Demaria S (2019) Radiation therapy and anti-tumor immunity: exposing immunogenic mutations to the immune system. Genome Med 11:40
Saloura V, Fatima A, Zewde M, Kiyotani K, Brisson R, Park JH et al (2017) Characterization of the T-Cell receptor repertoire and immune microenvironment in patients with locoregionally advanced squamous cell carcinoma of the head and neck. Clin Cancer Res 23:4897–4907
Heinemann T, Bulwin GC, Randall J, Schnieders B, Sandhoff K, Volk HD et al (1999) Genomic organization of the gene coding for TIRC7, a novel membrane protein essential for T cell activation. Genomics 57:398–406
Koretzky GA, Abtahian F, Silverman MA (2006) SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat Rev Immunol 6:67–78
Gorentla BK, Zhong XP (2012) T cell receptor signal transduction in T lymphocytes. J Clin Cell Immunol Suppl 12:5
Ji D, Yi H, Zhang D, Zhan T, Li Z, Li M et al (2018) Somatic mutations and immune alternation in rectal cancer following neoadjuvant chemoradiotherapy. Cancer Immunol Res 6:1401–1416
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E et al (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520:373–377
Acknowledgements
The authors thank Yuki Ota, Sayuri Amino, Kumiko Sakurai and Noriko Yaguchi for technical assistance; Minako Hoshida for administrative assistance; and Rebecca Jackson for her editorial support. We thank all members of Colorectal Surgery Division, Cancer Institute Hospital, Japanese Foundation for Cancer Research for assistance of sample collection.
Funding
This study was supported in part by JSPS KAKENHI Grant Number 18K08664 (to T. A.), 18K08635 (to M.U), 20K09022 (to Y.F), Grant from Foundation for Promotion of Cancer Research in Japan (to T. A.).
Author information
Authors and Affiliations
Contributions
T.A. and S.M. designed the study and wrote the manuscript. T.A., N.Y., M.U., and Y.F. contributed to data collection. T.A., N.T., K.K., O.G., and S.M. contributed to the bioinformatics analysis. All authors contributed to the analysis and interpretation of data. All authors critically reviewed the manuscript and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
This study was approved by the Institutional Review Boards of the Japanese Foundation for Cancer Research (Tokyo, Japan) and performed in accordance with the Declaration of Helsinki. The reference number was 2013–1003.
Informed consent
Written informed consent was obtained in accordance with local institutional ethical guidelines.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Akiyoshi, T., Gotoh, O., Tanaka, N. et al. T-cell complexity and density are associated with sensitivity to neoadjuvant chemoradiotherapy in patients with rectal cancer. Cancer Immunol Immunother 70, 509–518 (2021). https://doi.org/10.1007/s00262-020-02705-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02705-6