
Abstract
Immunotherapy targeting the PD-L1/PD-1 pathway using antibodies is effective in the clinical treatment of a multitude of cancers. This makes research of the regulatory mechanisms of PD-1 expression in cancer cells intriguing. PD-L1 expression can be categorized into inducible expression, attributed to extrinsic factors in the microenvironment, and constitutive expression, attributed to intrinsic cancer-driving gene alteration. The mechanisms of PD-L1 expression in cancer cells operate at multiple levels, including gene amplification, chromatin modification, transcription, posttranscription, translation and posttranslation. Moreover, some open questions in this field that need to be answered in future research are proposed. Studies of regulatory mechanisms of PD-L1 expression pave the way for the application of more effective approaches in the future of cancer immunotherapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Regulatory mechanisms of PD-L1 expression in cancer cells are of interest since PD-1 is a critical immune checkpoint pathway in antitumor immunity. Cancer cells exploit PD-L1 to subvert T cell-mediated immunosurveillance. PD-L1 expressed by cancer cells can engage the PD-1 expressed by cancer-specific CTL, inducing apoptosis or functional exhaustion of the CTLs [1]. Based on the mechanism of PD-L1/PD-1 signaling, immunotherapy using antibodies against PD-L1 (atezolizumab, avelumab and durvalumab) or PD-1 (nivolumab and pembrolizumab) achieves great success in the clinical treatment of cancer. At present, this antibody therapy has been approved for multiple cancers, including Hodgkin’s lymphoma, melanoma, NSCLC, renal cell carcinoma (RCC), bladder cancer, HNSCC, Merkel cell carcinoma, HCC and gastric cancer. In spite of effectiveness of the immune checkpoint blockade in the treatment of cancers, the responses to checkpoint blockade are not universal. Indeed, there are particular tumors and tumor subtypes that are less sensitive to the checkpoint blockade. Several predictive biomarkers for the effective response of antibody therapy targeting PD-L1/PD-1 signaling are identified, including TIL infiltrate and localization at the leading edge [2], TIL activation status [3], and mutational burden of the tumor itself [4], as well as the PD-L1 expression in cancer cells. For example, in reference to NSCLC, the overall survival of patients treated with atezolizumab is improved by 4.2 months compared with patients treated with docetaxel. The improvement of the overall survival of all patients with PD-L1 expression is 5.4 months [5]. The effectiveness of blocking the PD-L1/PD-1 signaling pathway in cancer treatment and the correlation of PD-L1 expression with the differential responses to the checkpoint blockade therapy highlight the importance of studying the regulatory mechanisms of PD-L1 expression in cancer cells.
Studies about the regulation of PD-L1 expression are accumulating. In this paper, the expression of PD-L1 is divided into inducible expression and constitutive expression according to causes of PD-L1 expression in cancer cells. Then, the molecular mechanisms whereby expression of PD-L1 in cancer cells is regulated are discussed. Finally, several questions are posed to be answered by further research in this field.
Inducible and constitutive expression of PD-L1 in cancer cells
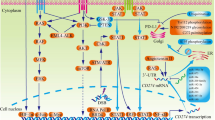
Both extrinsic and intrinsic factors are responsible for the expression of PD-L1 in cancer cells. Correspondingly, the expression of PD-L1 is classified into inducible expression, which is ascribed to extrinsic stimuli, and constitutive expression, which is ascribed to intrinsic genetic alteration of the malignant cells (Fig. 1). Among a number of extrinsic factors, IFN-γ is the most potent inducer of PD-L1 expression, acting mainly via the JAK/STAT1/interferon regulatory factor (IRF) 1 pathway in multiple types of cancers, such as melanoma [6], NSCLC [7], HCC [8], HNSCC [9], gastric cancer [10] and myeloma [11]. TNF-α induces the expression of PD-L1 mainly through NF-κB signaling in breast cancer [12], prostate cancer and colon cancer [13]. Epidermal growth factor (EGF) induces expression of PD-L1 via the JAK2/STAT1 pathway in HNSCC [9] or via both the PI3K/AKT and JAK/STAT1 pathways in NSCLC [14]. IL-17 causes PD-L1 expression through NF-κB and ERK1/2 signaling in prostate and colon cancers [13]. IL-27 induces PD-L1 expression in ovarian cancer through activation of STAT3 [15]. IL-4 stimulates the expression of PD-L1 in RCC [16]. In addition to cytokines, 17α-estradiol induces expression of PD-L1 at the posttranscriptional level via the PI3K/AKT signaling pathway in endometrial and breast cancer cells [17]. In addition, the ligands of TLR also cause the inducible expression of PD-L1. For instance, poly(I:C) triggers TLR3 signaling to induce PD-L1 expression in neuroblastoma cells [18], and LPS induces PD-L1 expression through TLR4 signaling in bladder cancer cells [19]. Furthermore, some chemotherapeutic agents, such as paclitaxel, also can induce the expression of PD-L1 in cancer cells through NF-kB signaling [20].

Inducible expression and constitutive expression of PD-L1 in cancer cells. a Inducible expression: PD-L1 expression is induced by 17α-estradiol via the PI3K/AKT pathway. EGF induces PD-L1 expression through the PI3K/AKT/mTOR pathway and the MEK/ERK pathway. IL-17 stimulates the MEK/ERK and NF-κB pathways to elevate expression of PD-L1. TNF-α, LPS, poly(I:C) and paclitaxel activate the NF-κB pathway to enhance expression of PD-L1. IFN-γ induces expression of PD-L1 through the JAK/STAT1/IRF1 pathway. IL-27 induces PD-L1 expression via the JAK/STAT3 pathway. b Constitutive expression: PTEN loss leads to activation of the PI3K/AKT/mTOR pathway, resulting in expression of PD-L1. RAS mutation, EGFR mutation and EML4-ALK translocation induce activation of the PI3K/AKT/mTOR and MEK/ERK pathways, resulting in expression of PD-L1. MYC directly promotes expression of PD-L1
Alteration of oncogenes or tumor suppressor genes can lead to constitutive expression of PD-L1 in cancer cells. For example, MYC protein, an oncogenic transcription factor that is overexpressed in many cancers, binds to the promoter of the PD-L1 gene to cause elevated expression of PD-L1 in T cell acute lymphoblastic leukemia (T-ALL) [21]. The mutation of the RAS oncogene is cancer-driving and increases cancer cell-intrinsic PD-L1 expression through MEK/ERK signaling [22, 23]. Activating mutations of the EGFR gene have been identified as oncogenic drivers in NSCLC. Chromosomal rearrangement involving the anaplastic lymphoma kinase (ALK) and echinoderm microtubule-associated protein–like 4 (EML4) genes is also a cancer-driving event in NSCLC. Both EML4-ALK and mutant EGFR upregulate PD-L1 by activating the PI3K-AKT and MEK–ERK signaling pathways [24, 25]. Phosphatase and tensin homolog (PTEN) is a critical tumor suppressor and inhibitor of PI3K/AKT signaling. Loss of its function is causally linked to increased expression of PD-L1 in glioma [26] and lung cancer [27].
Regardless of the causes of PD-L1 expression in cancer cells, the regulatory mechanisms operating from DNA to protein are similar for the inducible and constitutive expression of PD-L1. In the following, the multiple layers of regulatory mechanisms of PD-L1 expression are reviewed (Fig. 2).

Multiple players regulate PD-L1 expression in cancer cells. a Amplification of the PD-L1 gene elevates the expression of PD-L1. b Chromatin modification regulates the expression of PD-L1. c Transcription factors promote the expression of PD-L1. d PD-L1 3′UTR and microRNAs decrease the stability of PD-L1 mRNA. e MiR-513 and the AKT/mTOR pathway regulate the expression of PD-L1 at the translational level. f Ubiquitination and glycosylation control the stability of the PD-L1 protein. g CMTM4/6 maintains the stability of the PD-L1 protein in the lysosome pathway for degradation of proteins. h Sigma1 maintains the stability of the PD-L1 protein by preventing its autophagic degradation
Regulation of PD-L1 expression by genomic amplification
The PD-L1 gene is located on chromosome 9p24.1. The amplification of this region is identified in nodular sclerosing Hodgkin lymphoma, a subtype of classical Hodgkin lymphoma, with a frequency of 38% and in mediastinal large B-cell lymphoma with a frequency of 63%. The amplification of 9p24.1 tightly correlates to increased expression of PD-L1 in malignant cells [28]. In addition to lymphomas, amplification of the PD-L1 gene has also been detected in solid cancers, such as in 15% of EBV-positive gastric adenocarcinomas [29], 19% of squamous cell carcinomas of the oral cavity [30], 1.9% of small cell lung cancers [31] and 5.3% of NSCLCs [32]. PD-L1 expression is correspondingly elevated in these amplified cases. Moreover, the JAK2 gene is also located in the 9p24.1 region. The amplification of the JAK2 gene enhances the expression of PD-L1 through JAK-STAT signaling in NSCLC [32]. Future research should investigate whether amplification of 9p24.1 occurs in other types of cancers. The subset of cases with amplification of the PD-L1 gene may be particularly susceptible to immune checkpoint blockade therapy.
Regulation of PD-L1 expression by chromatin modification
Histone acetylation in the promoter region of the PD-L1 gene is required for PD-L1 expression. Woods et al. reported that histone deacetylase (HDAC) inhibitor treatment resulted in upregulation of histone acetylation at approximately 455 base pairs upstream of the first exon of the PD-L1 gene. Consequently, HDAC inhibitors enhanced the expression of PD-L1 in both human melanoma cell lines and a murine melanoma model [33]. HDAC inhibitors are FDA-approved for the treatment of cutaneous T-cell lymphoma and myeloma in which setting they are highly efficacious [34, 35]. The antitumor activity of HDAC inhibitors in pre-clinical models is also dependent upon functional interactions with the host immune system [36]. Thus, a combinatorial therapy with HDAC inhibitors and the checkpoint blockade may be a synergistic therapeutic strategy to treat cancer [33]. By contrast, Lienlaf et al. reported that knockdown of HDAC6 resulted in decreased expression of PD-L1 induced by IL-6 in melanomas. Mechanistically, HDAC6 recruited STAT3 to the PD-L1 promoter and activated STAT3 signaling. Additionally, selective HDAC6 inhibitors impaired tumor growth, suggesting HDAC6 as a potential target for tumor immunotherapy [37]. The BET family of proteins is a kind of histone acetylation reader that directly binds to acetylated lysine on histone tails to promote gene transcription. One member of the bromodomain and extra terminal domain (BET) family is bromodomain-containing protein (BRD) 4, which binds to the acetylated histone H3K27Ac in the promoter region of the PD-L1 gene and in a distal super enhancer. BRD4 knockdown or pharmacologic inhibition reduces both constitutive and inducible PD-L1 expression in ovarian cancer cells, murine pancreatic ductal adenocarcinoma cells and lymphoma cells. Consistently, the BET inhibitor JQ1 significantly suppresses PD-L1 expression in vitro and in vivo, increasing the activity of antitumor cytotoxic T cells. These findings support a small-molecule approach to blocking PD-L1 signaling [38, 39]. In addition to histone acetylation, histone methylation controls expression of PD-L1. H3K4me3 marks are enriched in the PD-L1 promoter region around − 1000 to + 2000 bp in both human and mouse pancreatic cancer cells. Myeloid/lymphoid or mixed-lineage leukemia (MLL) 1, one of the H3K4 methylation-specific HMTases, directly binds to the proximal PD-L1 promoter region to catalyze H3K4me3 and activate PD-L1 transcription [40]. However, it is difficult for the MLL1-H3K4me3 axis to be a therapeutic target because H3K4me3 is associated with most transcriptionally active genes in human cells. Therefore, the clinical application of chromatin modifications should be considered with caution because they may influence the expression of many genes rather than PD-L1 only.
Transcriptional regulation of PD-L1 expression
Quite a few transcription factors involved in PD-L1 expression have been identified. Transcription factor AP-1 promotes the expression of PD-L1 in Hodgkin lymphomas by binding to an enhancer region, which is located on intron 1 of the PD-L1 gene approximately 5 kb downstream from the transcription start site [41].
A second transcription factor is IRF1, which is required for expression of PD-L1, particularly for IFN-γ-induced expression in cancer cells [7]. A shRNA assay of interferon receptor pathway was used to systemically screen 33 known interferon receptor pathway signaling molecules in order to identify critical genes responsible for PD-L1 expression induced by IFN-γ. The results show that the JAK1/JAK2-STAT1/STAT2/STAT3-IRF1 axis primarily regulates PD-L1 expression, with IRF1 binding to the PD-L1 promoter [6]. Additionally, two IRF1 binding sites have been identified in the PD-L1 promoter region between − 320 and − 202 bp from the translational start site, designated IRF-1α and IRF-1β [7]. These two IRF1 binding sites may have different affinities for engagement of IRF1 since the expression of PD-L1 in EBV-positive gastric cancer cells is primarily mediated by the IRF-1α site [10].
The expression of PD-L1 in cancer cells is dependent on transcription factor NF-κB as well. There are NF-κB binding sites in the promoter region of the PD-L1 gene [42,43,44]. Recently, an enhancer 140 kb downstream of the PD-L1 gene bound by NF-κB was identified by analysis of expressed enhancers using TCGA RNA-seq data. This finding led to the proposal of a NF-κB-mediated enhancer-promoter interaction model of PD-L1 activation. However, the working rationale of this model remains to be validated experimentally [45].
STAT3 is also involved in expression of PD-L1, but its direct engagement with the PD-L1 promoter is controversial. On the one hand, STAT3 has been found to bind to the PD-L1 gene promoter to increase expression of PD-L1 in chimeric nucleophosmin (NPM)/ALK-carrying T cell lymphoma [46], ALK-negative large-cell lymphoma [47] and pulmonary adenocarcinomas with EML4-ALK translocation [48]. On the other hand, the binding of STAT3 at the PD-L1 promoter has not been detected in melanoma cells [6].
Oncogene MYC codes for a transcription factor that can activate or suppress the expression of its target genes [49]. There are discrepancies in the reports about the role of MYC in the regulation of expression of PD-L1 in cancer cells. Casey et al. used a Tet-off MYC transgenic mouse model of T-ALL or HCC to demonstrate that MYC increases PD-L1 expression by binding to the promoter of the PD-L1 gene. They came to a similar conclusion for human T-ALL cell lines, the HCC cell line HepG2, the melanoma cell line SKMEL28, the NSCLC cell line H1299, and even in primary human T-ALL samples [21]. In contrast, Hogg et al. found that MYC did not affect PD-L1 expression in murine Eµ-MYC lymphoma cells and the breast cancer cell line AT3. Furthermore, they found that MYC suppressed PD-L1 expression with or without exposure to IFN-γ in a Tet-off murine PDAC model or liver cancer model [39]. In line with Hogg et al. Durand-Panteix et al. reported that MYC negatively regulated PD-L1 expression in EBV-immortalized B cells. In addition, they found that MYC not only suppressed the mRNA expression of PD-L1 at the transcription level but also inhibited the secretory lysosomal pathway and PD-L1 membrane expression by decreasing actin polymerization [50]. These discrepancies may be due to the cell-dependent and context-dependent signatures of MYC action [49].
Since hypoxia is one characteristic of the cancer microenvironment, it makes sense that hypoxia-inducible factors (HIF) are implicated in the regulation of PD-L1 expression in cancer cells. HIF1α increases the expression of PD-L1 by binding directly to the PD-L1 proximal promoter in myeloid-derived suppressor cells [51] or pulmonary adenocarcinomas with the EML4-ALK translocation under hypoxic conditions [48]. Under the condition of normoxia, HIF2α regulates PD-L1 expression in clear cell renal cell carcinoma [52].
Posttranscriptional regulation of PD-L1 expression
After transcription, there are some mechanisms to control the stability of mRNA. The 3′-untranslated region (UTR) of the PD-L1 transcripts plays a negative regulatory role in mRNA stability. This insight comes from the observation that genomic structural variations leading to truncation of the 3′UTR of PD-L1 transcripts correlate with elevated expression of PD-L1 mRNA in multiple cancer types, including adult T-cell leukemia/lymphoma, diffuse large B-cell lymphoma and stomach adenocarcinoma. The underlying mechanism may be the impairment of a number of cis-acting elements located in the 3′UTR region and involved in mRNA decay, including an AU-rich element and potential microRNA-binding sites [53]. Elucidation of the underlying mechanisms could provide potential approaches to manipulate the expression of PD-L1 in the majority of cancers without genomic structural variation.
Stability of mRNA is also affected by microRNAs. The regulation of PD-L1 expression by microRNA is reviewed in detail by Wang et al. [54]. The microRNAs that target PD-L1 mRNA to degrade it are listed. MiR-15a, miR-15b, miR-16 and miR-193a-3p target PD-L1 3′UTR to decrease PD-L1 expression in malignant pleural mesothelioma cells [55]. MiR-106b-5p decreases expression of PD-L1 mRNA in pancreatic cancer cells [56]. MicroRNA miR-142-5p suppresses PD-L1 mRNA expression by binding to the PD-L1 3′UTR in mouse pancreatic cancer cells [57]. Direct binding between miR-17-5p and the 3′UTR of PD-L1 mRNA has been demonstrated to result in posttranscriptional downregulation of PD-L1 expression in melanoma cells [58]. MiR-152 directly binds to the PD-L1 3′UTR in gastric cancer cells and inhibits PD-L1 expression [59]. MiR-424 inhibits PD-L1 expression through direct binding to the 3′UTR of PD-L1 in ovarian cancers [60]. MiR-34a inhibits PD-L1 mRNA expression in acute myeloid leukemia through binding to the 3′UTR of PD-L1 [61]. These microRNAs targeting PD-L1 have usually been identified separately in different cancer types. Future research should determine whether they have synergy in the inhibition of PD-L1 expression.
Regulation of PD-L1 translation
MicroRNA not only contributes to degradation of the target mRNA but also inhibits the translation of the mRNA. MiR-513 targeting the 3′UTR of PD-L1 mRNA suppresses PD-L1 translation in cholangiocytes [62]. In addition to regulation of translation of PD-L1 by microRNA, mTOR activation controls synthesis of the PD-L1 protein in cancer cells. The activation of the PI3K–Akt–mTOR–S6K1 pathway due to PTEN loss in glioma cells leads to an increase in the level of the PD-L1 protein. The regulation of this pathway is considered to occur at the translational level because mTOR activation leads to recruitment of PD-L1 transcripts to translationally active polysomes [23]. Additionally, the constitutive expression of PD-L1 ascribed to alteration of oncogenes, such as EGFR, KRAS, and ALK, and the inducible expression of PD-L1 induced by IFN-γ or EGF, depends on the activation of AKT/mTOR pathway in NSCLC cells. Activation of mTOR increases the level of the PD-L1 protein rather than the PD-L1 mRNA [63]. The activation of mTOR usually initiates translation of the mRNA that harbors a 5′TOP motif. However, there is no canonical 5′ terminal oligopyrimidine (TOP) sequence in the PD-L1 5′UTR. A putative regulatory sequence (5′-GCCGCGCTTCTGTCCGCC-3′) may be an alternative to the canonical 5′ TOP motif, but the specific regulatory properties of this sequence remain to be determined [23].
Regulation of PD-L1 protein stability
Recent studies reveal that PD-L1 protein modifications, such as ubiquitination, deubiquitination, and glycosylation, and PD-L1-binding proteins impact the stability of the PD-L1 protein in cancer cells. A protein is usually labeled with a specific polyubiquitin chain before it undergoes degradation through the proteasome or lysosome pathway [64]. As far as the proteasome pathway is concerned, three enzymes, including E1 activating enzyme, E2 conjugating enzyme and E3 ligase, mediate the ubiquitination process, with specificity of the E3 ligase in recognition of target proteins. Several E3 ligases targeting the PD-L1 protein have been identified. The first is Cullin 3SPOP, which ubiquitinates the PD-L1 protein and leads to its subsequent degradation in prostate cancer cells. Furthermore, Cyclin D-CDK4 kinase mediates phosphorylation of SPOP and thereby promotes SPOP degradation by APC/CCdh1, so this cell cycle kinase indirectly controls the stability of the PD-L1 protein [65]. This finding implies that CDK4 kinase may be a therapeutic target for blockade of the PD-L1/PD-1 immune checkpoint in the cancer therapy. The second E3 ligase targeting the PD-L1 protein is β-TrCP, which ubiquitinates the PD-L1 protein phosphorylated by glycogen synthase kinase 3b (GSK3b) in basal-like breast cancer cells. Moreover, the phosphorylation of the PD-L1 protein is controlled by its glycosylation because glycosylated PD-L1 cannot interact with GSK3b. Thus, the glycosylation indirectly prevents ubiquitination and degradation of PD-L1 proteins. In fact, the majority of PD-L1 in cancer cells is glycosylated and cannot be degraded by the proteasome [66, 67]. Another E3 ligase is STUB1, which causes destabilization of PD-L1. It remains to be investigated whether STUB1 directly ubiquitinates the PD-L1 protein or indirectly affects the expression of PD-L1 [68]. Although the regulation of the abovementioned E3 ligases in the expression of PD-L1 has been validated experimentally, the deeper molecular mechanisms, such as how the E3 ligase ubiquitinates the PD-L1 protein, are still unclear.
On the other hand, deubiquitination is the reversed process of ubiquitination, usually resulting in stabilization of the target proteins. So far, only one deubiquitinase, COP9 signalosome 5 (CSN5), has been identified for stabilization of the PD-L1 protein in breast cancer cells. The expression of CSN5 is controlled by NF-κB signaling, which is activated by the proinflammatory cytokine TNF-α, which is derived from tumor-associated macrophages in breast cancer [12].
In addition to protein modification, PD-L1-binding proteins also affect PD-L1 protein stability. CKLF-like MARVEL transmembrane domain containing protein (CMTM) 6, a transmembrane protein of previously unknown function, increases PD-L1 protein expression without affecting PD-L1 transcription in a variety of cancer cell lines, including breast, lung, pancreas, thyroid, and colorectal cancers; melanoma; and chronic myeloid leukemia, as well as in primary dendritic cells. Mechanically, CMTM6 forms a molecular partner of both constitutive PD-L1 and IFN-γ-induced PD-L1 at the plasma membrane and in recycling endosomes, preventing ubiquitination and subsequent lysosomal degradation of PD-L1. In addition, CMTM4, a close family member of CMTM6, has a similar function to CMTM6 [68, 69]. Another PD-L1-binding protein is Sigma1, a unique ligand-regulated integral membrane scaffolding protein that contributes to cellular protein and lipid homeostasis. PD-L1 protein levels in breast cancer cells and prostate cancer cells are suppressed by small molecule inhibition of Sigma1. The mechanism is that the Sigma1 inhibitor induces degradation of PD-L1 via selective autophagy [70].
Although several mechanisms controlling the stability of the PD-L1 protein have been revealed in cancer cells, there remains a point of confusion that should be elucidated as soon as possible: it seems that the degradation of the PD-L1 protein involves both proteasome and lysosome pathways because both the proteasome inhibitor MG132 [65] and the lysosome inhibitor chloroquine [63] elevate the expression of the PD-L1 protein. So far, it is not known which degradation pathway is dominant in cancer cells or whether the degradation pathway is dependent on cancer type.
Concluding remarks
Over the past years, compelling data have been provided to show the landscape of the regulation of PD-L1 expression in cancer cells. Based on the molecular mechanisms underlying PD-L1 expression, some potential therapeutic approaches have emerged. For instance, inhibition of BRD4 may serve as a strategy of immunotherapy for PD-L1-expressing cancers since BRD4 is required for both inducible and constitutive expression of PD-L1, and the BRD4 inhibitor OTX015 has been used to treat hematological malignancies in clinical trials [71, 72]. Another potential therapeutic target is the mTOR pathway because this pathway not only enhances PD-L1 expression, but also promotes proliferation and metastasis of cancers, and mTOR inhibitors have been explored in clinical trials [73, 74]. Besides, the mechanisms underlying intrinsic resistance to the checkpoint blockade and /or relapse following initial responses to the therapy are disclosed through the studies of the regulation of PD-L1 expression. For instance, loss-of-function mutation of JAK1/JAK2 is responsible for absence of PD-L1 expression in cancer cells, resulting in primary and acquired resistance to PD-1 blockade therapy [75, 76]. Indeed, the IFN-γ signaling pathway is one of the most prominent pathways recognized to drive PD-L1 induction in the tumor immune microenvironment. Inactivating mutations in the IFNGR/JAK/STAT pathways are frequently observed in patients relapsing from checkpoint blockade [77,78,79].
There are still many open questions that need to be addressed in future research. First, many transcription factors are involved in the regulation of transcription of the PD-L1 gene. Usually these transcription factors bind to enhancer and promoter regions, often cooperatively, to regulate gene expression. The transcription factors regulating PD-L1 expression would be highly tumor type dependent. Thus, the major transcription factors responsible for PD-L1 expression in certain type of cancer should be identified. Second, several mechanisms controlling the stability of the PD-L1 protein have been revealed, such as ubiquitination, glycosylation and CMTM4/6. The relationship between them should be further analyzed by future research to provide a complete understanding of the regulatory mechanisms of the stability of the PD-L1 protein. Third, the regulatory mechanisms of PD-L1 expression have been discovered in different types of cancer cells, operating at multiple levels, such as transcription, posttranscription, translation and posttranslation. However, it is not known which one is dominant in a given type of cancer. The cancer-type dominant or individual dominant regulatory mechanisms of PD-L1 expression should be determined so that more precise measures can be applied to block the PD-L1/PD-1 pathway in cancer immunotherapy.
Collectively, although antibody therapy targeting the PD-L1/PD-1 pathway is effective, there is resistance or no response to the therapy. Therefore, further intensive research focusing on regulatory mechanisms of PD-L1 expression may provide more options and more effective approaches for cancer immunotherapy targeting the PD-L1/PD-1 pathway.
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- BET:
-
Bromodomain and extra terminal domain
- BRD:
-
Bromodomain-containing protein
- CMTM:
-
CKLF-like MARVEL transmembrane domain containing protein
- CSN5:
-
COP9 signalosome 5
- EGF:
-
Epidermal growth factor
- EML:
-
Echinoderm microtubule-associated protein–like
- GSK3b:
-
Glycogen synthase kinase 3b
- HDAC:
-
Histone deacetylase
- HIF:
-
Hypoxia-inducible factors
- IRF:
-
Interferon regulatory factor
- MLL:
-
Myeloid/lymphoid or mixed-lineage leukemia
- NPM:
-
Chimeric nucleophosmin
- PTEN:
-
Phosphatase and tensin homolog
- TOP:
-
Terminal oligopyrimidine
- UTR:
-
Untranslated region
References
Chen L, Han X (2015) Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Investig 125:3384–3391. https://doi.org/10.1172/jci80011
Diem S, Hasan Ali O, Ackermann CJ, Bomze D, Koelzer VH, Jochum W, Speiser DE, Mertz KD, Flatz L (2018) Tumor infiltrating lymphocytes in lymph node metastases of stage III melanoma correspond to response and survival in nine patients treated with ipilimumab at the time of stage IV disease. Cancer Immunol Immunother CII. 67: 39–45. https://doi.org/10.1007/s00262-017-2061-4
Kansy BA, Concha-Benavente F, Srivastava RM et al (2017) PD-1 status in CD8(+) T cells associates with survival and anti-PD-1 therapeutic outcomes in head and neck cancer. Cancer Res 77:6353–6364. https://doi.org/10.1158/0008-5472.CAN-16-3167
Hellmann MD, Callahan MK, Awad MM et al (2018) Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 33:853–861e4. https://doi.org/10.1016/j.ccell.2018.04.001
Rittmeyer A, Barlesi F, Waterkamp D et al (2017) Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389:255–265. https://doi.org/10.1016/s0140-6736(16)32517-x
Garcia-Diaz A, Shin DS, Moreno BH et al (2017) Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep 19:1189–1201. https://doi.org/10.1016/j.celrep.2017.04.031
Lee SJ, Jang BC, Lee SW et al (2006) Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Lett. 580:755–762. https://doi.org/10.1016/j.febslet.2005.12.093
Li N, Wang J, Zhang N et al (2018) Cross-talk between TNF-alpha and IFN-gamma signaling in induction of B7-H1 expression in hepatocellular carcinoma cells. Cancer Immunol Immunother CII. 67: 271 – 83. https://doi.org/10.1007/s00262-017-2086-8
Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, Freeman GJ, Ferris RL (2016) Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck cancer. Cancer Res 76:1031–1043. https://doi.org/10.1158/0008-5472.CAN-15-2001
Moon JW, Kong S-K, Kim BS et al (2017) IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci Rep. https://doi.org/10.1038/s41598-017-18132-0
Liu J, Hamrouni A, Wolowiec D, Coiteux V, Kuliczkowski K, Hetuin D, Saudemont A, Quesnel B (2007) Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN- and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 110:296–304. https://doi.org/10.1182/blood-2006-10-051482
Lim S-O, Li C-W, Xia W et al (2016) Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. https://doi.org/10.1016/j.ccell.2016.10.010
Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, You Z (2017) Inflammatory cytokines IL-17 and TNF-alpha up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett 184:7–14. https://doi.org/10.1016/j.imlet.2017.02.006
Okita R, Maeda A, Shimizu K, Nojima Y, Saisho S, Nakata M (2017) PD-L1 overexpression is partially regulated by EGFR/HER2 signaling and associated with poor prognosis in patients with non-small-cell lung cancer. Cancer Immunol Immunother CII. 66: 865 – 76. https://doi.org/10.1007/s00262-017-1986-y
Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, Petretto A, Fabbi M, Ferrini S (2015) IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 6:43267–43280. https://doi.org/10.18632/oncotarget.6530
Quandt D, Jasinski-Bergner S, Muller U, Schulze B, Seliger B (2014) Synergistic effects of IL-4 and TNFalpha on the induction of B7-H1 in renal cell carcinoma cells inhibiting allogeneic T cell proliferation. J Transl Med 12:151. https://doi.org/10.1186/1479-5876-12-151
Yang L, Huang F, Mei J, Wang X, Zhang Q, Wang H, Xi M, You Z (2017) Posttranscriptional control of PD-L1 expression by 17beta-estradiol via PI3K/Akt signaling pathway in ERalpha-positive cancer cell lines. Int J Gynecol Cancer 27:196–205. https://doi.org/10.1097/IGC.0000000000000875
Boes M, Meyer-Wentrup F (2015) TLR3 triggering regulates PD-L1 (CD274) expression in human neuroblastoma cells. Cancer Lett 361:49–56. https://doi.org/10.1016/j.canlet.2015.02.027
Qian Y, Deng J, Geng L et al (2008) TLR4 signaling induces B7-H1 expression through MAPK pathways in bladder cancer cells. Cancer Investig 26:816–821. https://doi.org/10.1080/07357900801941852
Peng J, Hamanishi J, Matsumura N et al (2015) Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-kappaB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res 75:5034–5045. https://doi.org/10.1158/0008-5472.CAN-14-3098
Casey SC, Tong L, Li Y et al (2016) MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352:227–231. https://doi.org/10.1126/science.aac9935
Coelho MA, de Carne Trecesson S, Rana S et al (2017) Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47:1083–1099 e6. https://doi.org/10.1016/j.immuni.2017.11.016
Sumimoto H, Takano A, Teramoto K, Daigo Y (2016) RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PloS One 11:e0166626. https://doi.org/10.1371/journal.pone.0166626
Akbay EA, Koyama S, Carretero J et al (2013) Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 3:1355–1363. https://doi.org/10.1158/2159-8290.CD-13-0310
Ota K, Azuma K, Kawahara A et al (2015) Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res 21:4014–4021. https://doi.org/10.1158/1078-0432.CCR-15-0016
Parsa AT, Waldron JS, Panner A et al (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13:84–88. https://doi.org/10.1038/nm1517
Xu C, Fillmore CM, Koyama S et al (2014) Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25:590–604. https://doi.org/10.1016/j.ccr.2014.03.033
Green MR, Monti S, Rodig SJ et al (2010) Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116:3268–3277. https://doi.org/10.1182/blood-2010-05-282780
Cancer Genome Atlas Research N (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513:202–209. https://doi.org/10.1038/nature13480
Straub M, Drecoll E, Pfarr N et al (2016) CD274/PD-L1 gene amplification and PD-L1 protein expression are common events in squamous cell carcinoma of the oral cavity. Oncotarget 7:12024–12034. https://doi.org/10.18632/oncotarget.7593
George J, Saito M, Tsuta K et al (2017) Genomic amplification of CD274 (PD-L1) in small-cell lung cancer. Clin Cancer Res. 23: 1220–1226. https://doi.org/10.1158/1078-0432.CCR-16-1069
Ikeda S, Okamoto T, Okano S et al (2016) PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol 11:62–71. https://doi.org/10.1016/j.jtho.2015.09.010
Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J (2015) HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol Res 3:1375–1385. https://doi.org/10.1158/2326-6066.cir-15-0077-t
Ellis L, Pan Y, Smyth GK et al (2008) Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin Cancer Res 14:4500–4510. https://doi.org/10.1158/1078-0432.CCR-07-4262
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12:1247–1252. https://doi.org/10.1634/theoncologist.12-10-1247
West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW (2013) An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Cancer Res 73:7265–7276. https://doi.org/10.1158/0008-5472.CAN-13-0890
Lienlaf M, Perez-Villarroel P, Knox T et al (2016) Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol 10:735–750. https://doi.org/10.1016/j.molonc.2015.12.012
Zhu H, Bengsch F, Svoronos N et al (2016) BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep 16:2829–2837. https://doi.org/10.1016/j.celrep.2016.08.032
Hogg SJ, Vervoort SJ, Deswal S et al (2017) BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep 18:2162–2174. https://doi.org/10.1016/j.celrep.2017.02.011
Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, Oberlies NH, Pearce C, Liu K (2017) The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst. https://doi.org/10.1093/jnci/djw283
Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, Neuberg D, Shipp MA (2012) Constitutive AP-1 activity and EBV infection induce PD-L1 in hodgkin lymphomas and posttransplant lymphoproliferative disorders: implications for targeted therapy. Clin Cancer Res 18:1611–1618. https://doi.org/10.1158/1078-0432.ccr-11-1942
Chen Y, Zhang J, Guo G et al (2009) Induced B7-H1 expression on human renal tubular epithelial cells by the sublytic terminal complement complex C5b-9. Mol Immunol 46:375–383. https://doi.org/10.1016/j.molimm.2008.10.026
Viola JPB, Huang G, Wen Q, Zhao Y, Gao Q, Bai Y (2013) NF-κB plays a key role in inducing CD274 expression in human monocytes after lipopolysaccharide treatment. PloS One 8:e61602. https://doi.org/10.1371/journal.pone.0061602
Maeda T, Hiraki M, Jin C et al (2018) MUC1-C induces PD-L1 and immune evasion in triple-negative breast cancer. Cancer Res 78:205–215. https://doi.org/10.1158/0008-5472.CAN-17-1636
Chen H, Li C, Peng X, Zhou Z, Weinstein JN, Cancer Genome Atlas Research N, Liang H (2018) A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell 173:386 –386 99 e12. https://doi.org/10.1016/j.cell.2018.03.027
Marzec M, Zhang Q, Goradia A et al (2008) Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci. 105:20852–20857. https://doi.org/10.1073/pnas.0810958105
Atsaves V, Tsesmetzis N, Chioureas D et al (2017) PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 31:1633–1637. https://doi.org/10.1038/leu.2017.103
Koh J, Jang JY, Keam B et al (2016) EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1alpha and STAT3. Oncoimmunology 5:e1108514. https://doi.org/10.1080/2162402X.2015.1108514
Kress TR, Sabo A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15:593–607. https://doi.org/10.1038/nrc3984
Durand-Panteix S, Farhat M, Youlyouz-Marfak I, Rouaud P, Ouk-Martin C, David A, Faumont N, Feuillard J, Jayat-Vignoles C (2012) B7-H1, which represses EBV-immortalized B cell killing by autologous T and NK cells, is oppositely regulated by c-Myc and EBV latency III program at both mRNA and secretory lysosome levels. J Immunol 189:181–90. https://doi.org/10.4049/jimmunol.1102277
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211:781–790. https://doi.org/10.1084/jem.20131916
Ruf M, Moch H, Schraml P (2016) PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer J international du cancer 139:396–403. https://doi.org/10.1002/ijc.30077
Kataoka K, Shiraishi Y, Takeda Y et al (2016) Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature 534:402–406. https://doi.org/10.1038/nature18294
Wang Q, Lin W, Tang X, Li S, Guo L, Lin Y, Kwok H (2017) The roles of microRNAs in regulating the expression of PD-1/PD-L1 immune checkpoint. Int J Mol Sci 18:2540. https://doi.org/10.3390/ijms18122540
Kao SC, Cheng YY, Williams M et al (2017) Tumor suppressor microRNAs contribute to the regulation of PD-L1 expression in malignant pleural mesothelioma. J Thoracic Oncol 12:1421–1433. https://doi.org/10.1016/j.jtho.2017.05.024
Cioffi M, Trabulo SM, Vallespinos M et al (2017) The miR-25-93-106b cluster regulates tumor metastasis and immune evasion via modulation of CXCL12 and PD-L1. Oncotarget 8:21609–21625. https://doi.org/10.18632/oncotarget.15450
Jia L, Xi Q, Wang H et al (2017) miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem Biophys Res Commun 488:425–431. https://doi.org/10.1016/j.bbrc.2017.05.074
Audrito V, Serra S, Stingi A et al (2017) PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 8:15894–15911. https://doi.org/10.18632/oncotarget.15213
Wang Y, Wang D, Xie G, Yin Y, Zhao E, Tao K, Li R (2017) MicroRNA-152 regulates immune response via targeting B7-H1 in gastric carcinoma. Oncotarget 8:28125–28134. https://doi.org/10.18632/oncotarget.15924
Xu S, Tao Z, Hai B et al (2016) miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun 7:11406. https://doi.org/10.1038/ncomms11406
Wang X, Li J, Dong K et al (2015) Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal 27:443–452. https://doi.org/10.1016/j.cellsig.2014.12.003
Gong AY, Zhou R, Hu G et al (2009) MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J Immunol 182:1325–1333
Lastwika KJ, Wilson W IIIrd, Li QK et al (2016) Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res 76:227–238. https://doi.org/10.1158/0008-5472.CAN-14-3362
Clague MJ, Urbe S (2010) Ubiquitin: same molecule, different degradation pathways. Cell 143:682–685. https://doi.org/10.1016/j.cell.2010.11.012
Zhang J, Bu X, Wang H et al (2017) Cyclin D-CDK4 kinase destabilizes PD-L1 via Cul3(SPOP) to control cancer immune surveillance. Nature. https://doi.org/10.1038/nature25015
Li C-W, Lim S-O, Xia W et al (2016) Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 7:12632. https://doi.org/10.1038/ncomms12632
Jiao S, Xia W, Yamaguchi H et al (2017) PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 23: 3711–3720. https://doi.org/10.1158/1078-0432.CCR-16-3215
Mezzadra R, Sun C, Jae LT et al (2017) Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549:106–110. https://doi.org/10.1038/nature23669
Burr ML, Sparbier CE, Chan YC et al (2017) CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549:101–105. https://doi.org/10.1038/nature23643
Maher CM, Thomas JD, Haas DA, Longen CG, Oyer HM, Tong JY, Kim FJ (2018) Small-molecule sigma1 modulator induces autophagic degradation of PD-L1. Mol Cancer Res MCR 16:243–255. https://doi.org/10.1158/1541-7786.MCR-17-0166
Berthon C, Raffoux E, Thomas X et al (2016) Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol 3:e186–e195. https://doi.org/10.1016/s2352-3026(15)00247-1
Amorim S, Stathis A, Gleeson M et al (2016) Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol 3:e196–e204. https://doi.org/10.1016/s2352-3026(16)00021-1
Choueiri TK, Escudier B, Powles T et al (2016) Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol 17:917–927. https://doi.org/10.1016/s1470-2045(16)30107-3
Powles T, Lackner MR, Oudard S et al (2016) Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol 34:1660–1668. https://doi.org/10.1200/JCO.2015.64.8808
Shin DS, Zaretsky JM, Escuin-Ordinas H et al (2017) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 7:188–201. https://doi.org/10.1158/2159-8290.CD-16-1223
Zaretsky JM, Garcia-Diaz A, Shin DS et al (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819–829. https://doi.org/10.1056/NEJMoa1604958
Gao J, Shi LZ, Zhao H et al (2016) Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 167:397–404 e9. https://doi.org/10.1016/j.cell.2016.08.069
Kearney CJ, Vervoort SJ, Hogg SJ et al (2018) Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol. https://doi.org/10.1126/sciimmunol.aar3451
Manguso RT, Pope HW, Zimmer MD et al (2017) In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547:413–418. https://doi.org/10.1038/nature23270
Acknowledgements
I thank Dr. Xiaoyan Wang, Huaiyu Zhou and Jianing Wang in Shandong University School of Basic Medical Sciences for their support and encouragement.
Funding
This work was supported by the National Nature Science Foundation of China [Grant numbers 81372264 and 81672806] and the Key Research and Development Program of Shandong Province [Grant number 2017GSF218066].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Rights and permissions
About this article

Cite this article
Shi, Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother 67, 1481–1489 (2018). https://doi.org/10.1007/s00262-018-2226-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-018-2226-9