
Abstract
Targeted therapies were rationally designed to inhibit molecular pathways in tumor cells critically involved in growth and survival; however, many drugs used in targeted therapies may affect the immune system. In addition, selected conventional chemotherapeutic agents have also been reported to be endowed with direct or indirect effects on immunity, for instance via immunogenic death of tumors. Thus, cancer therapies may have off-target effects, some of which are directed to the immune system. Here, we will review some of these effects in specific therapeutic approaches. We will examine the modulation of the immune contexture in human sarcoma and melanoma induced by anti-angiogenic therapies and by BRAF inhibitors, respectively. We will then discuss how the anti-tumor agent trabectedin is selectively cytotoxic to cells of the monocytic-macrophage lineage and how these immune-related effects can be part of the response to treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Considering the intense interplay of the different cells harbored in the tumor micro-environment, it is not surprising that effects delivered to tumor cells may impact on normal host cells, especially those of the immune system. In the latest years, cancer treatment has gained new impetus due to the availability of new, more selective drugs, specifically designed to target pathways driving the survival and progression of cancer cells. In the scenario of these targeted therapies, the host immune system is emerging as a key component in the response to treatment. Data obtained both in humans and in a preclinical setting with dedicated mouse models, strongly demonstrate that these drugs possess immune-modulating activities [1]. These by-stander immune-related effects stem from the capacity of targeted drugs to directly affect signaling pathways regulating the functional activities or the differentiation/maturation programs of immune cells and/or from their ability to modulate the immune-related features of tumor cells. Imatinib, a drug inhibiting the c-kit tyrosine kinase receptor, is a paradigm for this double activity. As shown in animal models and in humans, Imatinib directly induces the host DCs to promote NK activation, and this immunological effect was associated with prolonged disease-free survival in Imatinib-treated GIST patients [2]. Simultaneously, Imatinib strongly reduces the release of the immunosuppressive enzyme indoleamine 2,3-dioxygenase by tumor cells [3] and Imatinib-sensitive, but not Imatinib-resistant GIST, drive intratumoral macrophage polarization [4].
The active interplay between the immune system and anti-cancer therapies occurs also for conventional chemotherapy. Established evidence demonstrates that some chemotherapeutic treatments may induce immunogenic cell death in cancer cells. The signals released by dying tumor cells activate immune effectors and have an impact on the anti-tumor immune response and long-term success of anti-cancer therapies. Indeed, the anti-tumor activity of some drugs is greatly reduced in immune deficient mice, demonstrating that immune cells are required for a successful therapeutic outcome [5]. Furthermore, some anti-tumor agents, like gemcitabine, doxorubicin and 5-fluoruracile, kill MDSC or block their immunosuppressive function, diminishing the tumor-mediated immunesuppression and likely favoring the setting of a more active anti-tumor response [6, 7].
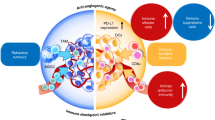
Interaction of anti-cancer therapies with host defense mechanisms occurs at different levels and with multiple mechanisms (Fig. 1) whose dissection is an area of great interest with important implications for the future design of more effective combination therapies. This review will focus on selected anti-tumor strategies pointing specifically to immunity.

Immunomodulating activities of BRAF inhibitors. In melanoma cells, MAPK signaling sustains the production of immunosuppressive and pro-inflammatory cytokines that promote the accumulation of immature myeloid suppressive cells (MDSC), immature dendritic cells (iDC), regulatory T cells, (Treg) and activated fibroblast stromal cells (TAF) by local and systemic mechanisms. Treatment with BRAF inhibitors (BRAFi) leads to a local re-shaping of the infiltrating lymphocytes with enrichment in CD4+ CD8+ activated T cells, while systemically it limits the frequency of MDCS and boosts the presence of Th1-related factors in the plasma of treated patients
Anti-angiogenic drugs are active modulators of the immune contexture in cancer patients
Targeted agents include several drugs with anti-angiogenic properties such as the tyrosin kinase inhibitors sunitinib, pazopanib or axitinib, and they all inhibit the signaling activities of VEGFR, PDGFR, c-KIT, although with different affinity [8]. Neo-angiogenic processes are key events in tumor development and progression. The angiogenic switch occurring at the tumor site results in the formation of new, highly abnormal blood vessels displaying a heterogeneous distribution, irregular blood flow and increased permeability [9, 10]. In addition, ‘endothelial cell anergy’ induced by pro-angiogenic factors, strongly limits the leukocyte–endothelial interaction and the subsequent extravasation of effector cells into tumor sites [11]. As a result, the tumor microenvironment displays poor effector T-cell infiltration and is characterized by hypoxia and acidity, conditions known to foster immunosuppressive cells, including cells of the myeloid lineage and regulatory T cells [12]. Certainly, tumor cells directly drive the cellular events supporting neo-angiogenesis and the expression of pro-angiogenic factors is controlled by oncogene activation. However, considerable evidence has now emerged for the key role played by the resident or newly recruited tumor—infiltrating myeloid cells in the phenomenon of tumor neo-angiogenesis [13].
We explored the presence and the localization of cells expressing myeloid markers in the inflammatory infiltrate of metastatic alveolar soft part sarcoma (ASPS) [14]. In the metastatic form, ASPS expresses an array of angiogenesis-related molecules and this tumor is characterized by a peculiar tumor-associated vasculature [15]. We found that myeloid cells expressing CD14 and CD163 markers constitute the prominent cells in the inflammatory infiltrate. In the ASPS environment, CD14+ CD163+ cells are structurally organized in two distinct localizations. CD14+ CD163+ cells form a network surrounding the endothelial cells or, as single cells, they are interspersed in tumor nests, keeping deep contact with tumor cells. In the perivascular region, CD163+ cells are aligned to VEGFR2+ CD31+ cells of endothelial nature. Of note, this same distribution of immunoreactivity is also typical of the M-CSF receptor, the major regulator of survival, proliferation and functional differentiation of macrophages. Our observations established the presence of M2-like, CD163+ CD14+ macrophages in the tumor microenvironment of naive ASPS. These myeloid cells are active inflammatory components that may promote VEGF-mediated vasculogenesis and, although not physically part of the vasculature, they are thought to provide trophic support to the characteristic ASPS vascular network. These immunophenotypic ASPS signatures, together with the known positivity of ASPS cells for the expression of pro-angiogenic factors [15, 16] provide the rationale for the usage of different anti-angiogenic targeted therapies for this sarcoma. Indeed, bevacizumab, sunitinib and more recently cediranib have been reported to induce durable responses in metastatic ASPS patients [16–19]. Molecular analysis of ASPS after cediranib treatment showed a strong modulation of transcripts related to angiogenesis/vasculogenesis. Of note, genes encoding for markers of inflammatory myeloid cells were also affected, thus indicating the tumor-infiltrating myeloid cells as potential targets of cediranib and their numeric or functional modulation as part of the response to treatment.
In a soft tissue sarcoma of different histology, namely in malignant and dedifferentiated solitary fibrous tumors (M/DSFT), we observed that another anti-angiogenic therapy based on sunitinib malate treatment also induced a profound remodeling in the myeloid infiltrate. At the tumor site, this myeloid shift favored the acquisition of a new immune contexture displaying features of the adaptive immune response enriched with a strong T-cell infiltration (Tazzari M, personal communication).
Several pro-angiogenic factors, in addition to exert their activity on endothelial cells, also possess immunosuppressive functions. VEGF plays key regulatory roles on the adaptive and innate immunity directly inhibiting DC maturation and fostering the accumulation of immature, tolerogenic DCs at the tumor site [20, 21]. Moreover, VEGF promotes the systemic accumulation of MDSC. Since immature DC and MDSC are strong inducers of regulatory T cells (Treg), VEGF is also indirectly involved in boosting Tregs; more recent evidence also indicates that VEGF directly induces Treg proliferation in a VEGFR2-dependent manner in tumor-bearing mice and in metastatic colorectal cancer patients [22]. Thus, drugs inhibiting VEGF-mediated signaling affect the balance of these various cell subsets and impact on the anti-tumor immune response [23]. Sunitinib and Bevacizumab are first-line standard of care in the treatment of renal cancer patients [24]; several data showed that the frequency of circulating Tregs and the different subsets of MDSC, including monocytic MDSC (CD11b+ CD14+ DRneg/low), MDSC defined as CD33+DR− and as CD15+ CD14−, are down-modulated in the blood of renal cancer patients receiving sunitinib treatment [25–28]. Furthermore, in the subset of patients experiencing tumor regression, sunitinib induced the reacquisition of a normal frequency of CD1c/BDCA-1+ myeloid DC [29]. Normalization in the levels of immune-suppressive cells, both Tregs and MDSC, paralleled a regained Th1 function by peripheral CD3+ T cells [26]. Of note, we recently confirmed that the down-modulation of Treg and monocytic MDSC also occurs in the blood of patients with solitary fibrous tumors treated with sunitinib, and by ex vivo analysis we showed that the modulation of these suppressive cells correlates with a regained capacity of T cells to produce Th1-related cytokines (Tazzari M, personal communications).
BRAF inhibitors and immunity: an on-going cross-talk
Melanoma is an immunogenic tumor for which lymphocytic infiltration, defined as brisk, non brisk or absent, has long been known to have prognostic significance [30, 31]. Recently, new studies on a large series of melanoma cases strongly indicated that tumor grade and distribution of lymphocyte infiltration predicted survival, independently of age, sex, tumor site and stage [32]. Furthermore, independent lines of evidence also confirmed that melanoma in its metastatic form is highly suppressive and that several and multi-levels mechanisms of immune evasion are actively operated by melanoma cells [33]. From in vitro and in vivo studies using targeted specific drugs, the emerging concept is that the immunosuppressive ability of melanoma cells is dependent on gene and signaling alterations that drive their transformation [34]. In melanoma, the release of immunosuppressive cytokines, such as IL-6, VEGF, IL-10 and of pro-inflammatory cytokines such as IL-1α and IL-1β, known to aberrantly stimulate stromal cells at the tumor site, is driven by the activated MAPK signaling and abrogated by the treatment of melanoma cells with BRAF or MEK inhibitors or by silencing of the mutated BRAF (V600E) [35–37]. These data, together with the observation that mutated Ras is crucial in sustaining CXCL8 secretion [38], provide a strong rationale for considering the drugs targeting these signaling pathways as endowed with strong immunomodulating capacity. Thus, smoldering of an immunosuppressive tumor microenvironment and reactivation of the host immune system are likely taking part in the response to treatment. Indeed, in patients treated with the BRAF inhibitors vemurafenib or dabrafenib, several immunological effects have been described as correlated/associated with clinical responses. Tumors surgically removed after short term treatment with vemurafenib, displayed enhanced infiltration with activated CD4+ and CD8+ lymphocytes [39]. Increased intratumoral CD8+ cells correlated with the dimensional response to therapy; moreover, post-treatment biopsies displayed an increased degree of necrosis [39]. Of note, T cells infiltrating tumors post BRAF treatment displayed an increased clonality, thus suggesting the expansion of tumor-specific T-cell clones [40].
All together these results indicate that BRAF treatment has unleashed or has newly promoted a T-cell-mediated response to autologous melanoma in treated patients, suggesting that a relieve in the local and/or systemic immunosuppression might have occurred upon drug treatment.
Indeed, in BRAF inhibitor-treated patients, enhanced T-cell infiltration correlated at the tumor site with a decrease in the local production of IL-6 and CXCL8 [36].
At systemic level, we have shown that melanoma patients display enhanced frequency of monocytic MDSC defined as CD11b+ CD14+ DRlow/neg; immunological monitoring of immune cells in the blood of patients at different time points during treatments indicates that vemurafenib reversed MDSC accumulation and decreased immune suppression in patients with advanced melanoma [41, 42]. In agreement with this finding, the profile of chemokines and cytokines in the sera of melanoma patients before and early during treatment with dabrafenib and vemurafenib, indicates that BRAF inhibition leads to a significant decrease in the serum levels of the pro-inflammatory, suppressive CXCL8, while it induced a boost of the Th1-related factors IFNγ, CCL4 and TNFα. Furthermore, these systemic changes correlated with the modulation occurring at the tumor site: the decrease in CXCL8 levels was associated with reduction of the proliferation marker Ki67 in melanoma cells and with an increase in tumor-infiltrating cytotoxic T cells in the corresponding tumor biopsies [43].
On the other hand, the ability of cancer therapies to modulate tumor–host interactions, raises additional crucial questions on the role of immune-related factors in directing the resistance to treatment. In this perspective, we recently found that cytokine/chemokine secretion is altered in BRAF-induced-resistant cell lines as compared to their BRAF-susceptible pairs, and that these altered profiles paralleled those found in the sera of melanoma patients under treatment with Vemurafenib. Our data indicate, in patient settings, the relevance of CCL2, a chemokine previously found to be crucially involved in the host response to BRAF treatment in animal models [44].
Therapeutic effects on monocytes/macrophages: the case of the marine-derived compound trabectedin
It is now established that Tumor-Associated Macrophages (TAM) and related myeloid cells infiltrating the tumor micro-environment promote tumor progression and are associated with poor patient prognosis. In fact, in most established tumors, incoming monocytes are conditioned by the tumor micro-environment and acquire an M2-like functional polarization, displaying a number of pro-tumoral functions, e.g., increase of tumor cell proliferation and survival, tumor dissemination, promotion of angiogenesis and matrix remodeling [45–48]. Strategies to deplete TAM or to inhibit their recruitment in tumors have been successful in experimental settings and are now considered in oncology as promising therapeutic approaches. Indeed, a number of recent studies have demonstrated that inhibitors of the M-CSF receptor are effective in inhibiting macrophage recruitment and/or pro-tumoral differentiation [49, 50]. We recently reported that monocytes and macrophages are susceptible to the cytotoxic effect of the anti-tumor agent trabectedin, a compound originally extracted from a marine organism, the Tunicate Ecteinascidia, and now synthetically produced [51].
Trabectedin is the first marine anti-tumor agent to have reached the market. It is registered in Europe and in several other countries, for second-line treatment of soft tissue sarcoma and for ovarian cancer, in combination with liposomal doxorubicin [52, 53]. Trabectedin binds the minor groove of DNA and blocks cell cycle and proliferation in tumor cells. Other recognized effects on cancer cells are its interference on DNA repair mechanisms and on selected transcription factors.
By treating non-activated resting leukocytes, we demonstrated that trabectedin induces apoptosis selectively on monocytes and macrophages, but not in neutrophils and lymphocytes. We further demonstrated that the drug rapidly triggers the activation of caspase 8 downstream of TRAIL receptors; among leukocyte subsets only monocytes/macrophages express appreciable levels of signaling TRAIL-R, while neutrophils and T lymphocytes preferentially express the non-signaling decoy receptor. When used in vivo in different mouse tumor models, trabectedin was effective in significantly decreasing the number of blood monocytes, spleen and tumor macrophages, but had no effect on neutrophils and lymphocytes [51].
We then asked the question whether the macrophage-depleting effect of trabectedin was relevant for its anti-tumor efficacy. Treatment of mice bearing a trabectedin-resistant tumor cell line resulted in slowed tumor growth, in spite of confirmed resistance of cancer cells to the drug. The hypothesis was that by targeting tumor macrophages trabectedin inhibited the pro-tumoral effects of TAM. In line with this interpretation, the adoptive transfer of macrophages to treated mice significantly reinstated tumor growth. Therefore, macrophage targeting in vivo is a key component of the anti-tumor activity of trabectedin. Effects other than macrophage depletion may also account for its efficacy. Pathological examination of tumor sections revealed that in treated tumors the vessel network, the angiogenic factor VEGF and the chemokine CCL2 were significantly down-modulated. Thus, in addition to direct cytotoxic activity on mononuclear phagocytes, trabectedin may reduce the recruitment of circulating monocytes into tumors and may affect angiogenesis [51].
Patients with soft tissue sarcoma receiving trabectedin as single treatment were studied for blood monocyte counts: a decrease in monocytes occurred within few days after injection of trabectedin in most patients. Furthermore, in tumor sections collected before and after neo-adjuvant therapy, a dramatic decrease of macrophage infiltration and reduction of the vessel network were observed, confirming also in cancer patients that this compound is able to target both the neoplastic compartment and the tumor micro-environment (Fig. 2).

Multiple effects of the anti-tumor agent trabectedin on the tumor micro-environment. In addition to blocking tumor cell proliferation, the anti-tumor agent trabectedin is selectively cytotoxic to cells of the monocyte-macrophage lineage, including Tumor-Associated Macrophages (TAM). Trabectedin also inhibits the production of selected inflammatory mediators such as the chemokines CCL2 and CXCL8, the cytokine IL-6 and the angiogenic factor VEGF. These mechanisms of action impact on the pro-tumoral role of TAM and on the cancer-related inflammation, thus augmenting its anti-tumor activity
Trabectedin is currently used in a limited number of tumors and as second line of treatment; these findings open interesting perspectives for the rational exploitation of this peculiar property in cancer therapy in a wider range of tumors.
Concluding notes
Immune implication of novel-targeted therapies in cancer is extending very far away from what could be expected on the basis of our current knowledge of the molecular events regulating cancer. It is now clear that the immunological status/response of cancer patients is strongly relevant, not only in those patients receiving immune-based therapies. The immune system is indeed potentially affecting the clinical efficacy of treatments previously considered devoid of any immunological implication. This evidence is reinforcing the notion that a broad immunological monitoring of treated cancer patients is a worthwhile effort potentially providing clues and rationale for discontinuing or further sustaining a given treatment, thus possibly ensuring a better standard of care. Moreover, the new accumulating knowledge on the immunological relevance of these new drugs may also provide rationale for the design of novel, previously unforeseen combinations of drug-based therapies with immune-related approaches.
Abbreviations
- ASPS:
-
Alveolar soft part sarcoma
- DC:
-
Dendritic cells
- GIST:
-
Gastrointestinal stromal tumor
- M/DSFT:
-
Malignant and dedifferentiated solitary fibrous tumors
- M-CSF:
-
Monocyte-colony stimulating factor
- MDSC:
-
Myeloid-derived suppressive cells
- PDGFR:
-
Platelet-derived growth factor receptor
- TAM:
-
Tumor-associated macrophages
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- Treg:
-
Regulatory T cells
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G (2013) Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity 39:74–88
Menard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C, Nonn C, Chaput N, Taieb J, Delahaye NF, Flament C, Emile JF, Le Cesne A, Zitvogel L (2009) Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res 69:3563–3569
Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, Taguchi T, Yuan J, Wolchok JD, Allison JP, DeMatteo RP (2011) Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med 17:1094–1100
Cavnar MJ, Zeng S, Kim TS, Sorenson EC, Ocuin LM, Balachandran VP, Seifert AM, Greer JB, Popow R, Crawley MH, Cohen NA, Green BL, Rossi F, Besmer P, Antonescu CR, DeMatteo RP (2013) KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med 210:2873–2886
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G (2012) The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov 11:215–233
Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rebe C, Ghiringhelli F (2010) 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 70:3052–3061
Annels NE, Shaw VE, Gabitass RF, Billingham L, Corrie P, Eatock M, Valle J, Smith D, Wadsley J, Cunningham D, Pandha H, Neoptolemos JP, Middleton G (2014) The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother 63:175–183
Takahashi S (2011) Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol Pharm Bull 34:1785–1788
Chung AS, Ferrara N (2011) Developmental and pathological angiogenesis. Annu Rev Cell Dev Biol 27:563–584
Dirkx AE, Oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC, Wagstaff J, Griffioen AW (2003) Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res 63:2322–2329
Griffioen AW, Damen CA, Mayo KH, Barendsz-Janson AF, Martinotti S, Blijham GH, Groenewegen G (1999) Angiogenesis inhibitors overcome tumor induced endothelial cell anergy. Int J Cancer 80:315–319
Johnson BF, Clay TM, Hobeika AC, Lyerly HK, Morse MA (2007) Vascular endothelial growth factor and immunosuppression in cancer: current knowledge and potential for new therapy. Expert Opin Biol Ther 7:449–460
Murdoch C, Muthana M, Coffelt SB, Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8:618–631
Castelli C, Tazzari M, Negri T, Vergani B, Rivoltini L, Stacchiotti S, Pilotti S (2013) Structured myeloid cells and anti-angiogenic therapy in alveolar soft part sarcoma. J Transl Med 11:237
Lazar AJ, Das P, Tuvin D, Korchin B, Zhu Q, Jin Z, Warneke CL, Zhang PS, Hernandez V, Lopez-Terrada D, Pisters PW, Pollock RE, Lev D (2007) Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin Cancer Res 13:7314–7321
Stacchiotti S, Negri T, Zaffaroni N, Palassini E, Morosi C, Brich S, Conca E, Bozzi F, Cassinelli G, Gronchi A, Casali PG, Pilotti S (2011) Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol 22:1682–1690
Mir O, Boudou-Rouquette P, Larousserie F, Blanchet B, Babinet A, Anract P, Goldwasser F (2012) Durable clinical activity of single-agent bevacizumab in a nonagenarian patient with metastatic alveolar soft part sarcoma. Anticancer Drugs 23:745–748
Azizi AA, Haberler C, Czech T, Gupper A, Prayer D, Breitschopf H, Acker T, Slavc I (2006) Vascular-endothelial-growth-factor (VEGF) expression and possible response to angiogenesis inhibitor bevacizumab in metastatic alveolar soft part sarcoma. Lancet Oncol 7:521–523
Kummar S, Allen D, Monks A, Polley EC, Hose CD, Ivy SP, Turkbey IB, Lawrence S, Kinders RJ, Choyke P, Simon R, Steinberg SM, Doroshow JH, Helman L (2013) Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol 31:2296–2302
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Ellis LM, Reardon DA (2010) Is there really a yin and yang to VEGF-targeted therapies? Lancet Oncol 11:809–811
Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J (2013) VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res 73:539–549
Huang Y, Goel S, Duda DG, Fukumura D, Jain RK (2013) Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res 73:2943–2948
Linehan WM, Srinivasan R, Garcia JA (2013) Non-clear cell renal cancer: disease-based management and opportunities for targeted therapeutic approaches. Semin Oncol 40:511–520
Adotevi O, Pere H, Ravel P, Haicheur N, Badoual C, Merillon N, Medioni J, Peyrard S, Roncelin S, Verkarre V, Mejean A, Fridman WH, Oudard S, Tartour E (2010) A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. J Immunother 33:991–998
Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R (2008) Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res 14:6674–6682
Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH (2009) Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 15:2148–2157
Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H (2009) Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 69:2506–2513
van Cruijsen H, van der Veldt AA, Vroling L, Oosterhoff D, Broxterman HJ, Scheper RJ, Giaccone G, Haanen JB, van den Eertwegh AJ, Boven E, Hoekman K, de Gruijl TD (2008) Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res 14:5884–5892
Clemente CG, Mihm MC Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310
Schatton T, Scolyer RA, Thompson JF, Mihm MC Jr (2014) Tumor-infiltrating lymphocytes and their significance in melanoma prognosis. Methods Mol Biol 1102:287–324
Thomas NE, Busam KJ, From L, Kricker A, Armstrong BK, Anton-Culver H, Gruber SB, Gallagher RP, Zanetti R, Rosso S, Dwyer T, Venn A, Kanetsky PA, Groben PA, Hao H, Orlow I, Reiner AS, Luo L, Paine S, Ollila DW, Wilcox H, Begg CB, Berwick M (2013) Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J Clin Oncol 31:4252–4259
Parmiani G, Castelli C, Santinami M, Rivoltini L (2007) Melanoma immunology: past, present and future. Curr Opin Oncol 19:121–127
Kawakami Y, Yaguchi T, Sumimoto H, Kudo-Saito C, Tsukamoto N, Iwata-Kajihara T, Nakamura S, Nishio H, Satomi R, Kobayashi A, Tanaka M, Park JH, Kamijuku H, Tsujikawa T, Kawamura N (2013) Cancer-induced immunosuppressive cascades and their reversal by molecular-targeted therapy. Ann N Y Acad Sci 1284:80–86
Sumimoto H, Imabayashi F, Iwata T, Kawakami Y (2006) The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med 203:1651–1656
Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, Mitra D, Boni A, Newton LP, Liu C, Peng W, Sullivan RJ, Lawrence DP, Hodi FS, Overwijk WW, Lizee G, Murphy GF, Hwu P, Flaherty KT, Fisher DE, Wargo JA (2013) BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 19:1225–1231
Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, Zhang M, Cooper ZA, Frederick DT, Li Y, Zhang M, Joseph RW, Bernatchez C, Ekmekcioglu S, Grimm E, Radvanyi LG, Davis RE, Davies MA, Wargo JA, Hwu P, Lizee G (2012) Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res 18:5329–5340
Sparmann A, Bar-Sagi D (2004) Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6:447–458
Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA (2012) Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 18:1386–1394
Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, Sharpe AH, Fisher DE, Flaherty KT, Wargo JA (2013) BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology 2:e26615
Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L (2007) Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol 25:2546–2553
Schilling B, Sucker A, Griewank K, Zhao F, Weide B, Gorgens A, Giebel B, Schadendorf D, Paschen A (2013) Vemurafenib reverses immunosuppression by myeloid derived suppressor cells. Int J Cancer 133:1653–1663
Wilmott JS, Haydu LE, Menzies AM, Lum T, Hyman J, Thompson JF, Hersey P, Kefford RF, Scolyer RA, Long GV (2014) Dynamics of chemokine, cytokine, and growth factor serum levels in BRAF-mutant melanoma patients during BRAF inhibitor treatment. J Immunol 192:2505–2513
Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, Graeber TG, Ribas A, McArthur GA, Smyth MJ (2013) Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest 123:1371–1381
Allavena P, Mantovani A (2012) Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol 167:195–205
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, Rugo HS, Hwang ES, Jirstrom K, West BL, Coussens LM (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1:54–67
De Palma M, Lewis CE (2013) Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 23:277–286
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, Hewitt S, Udupi GM, Gallagher WM, Wegner C, West BL, Wang-Gillam A, Goedegebuure P, Linehan DC, DeNardo DG (2013) Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73:1128–1141
Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JA (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19:1264–1272
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, Erba E, Uboldi S, Zucchetti M, Pasqualini F, Nebuloni M, van Rooijen N, Mortarini R, Beltrame L, Marchini S, Fuso Nerini I, Sanfilippo R, Casali PG, Pilotti S, Galmarini CM, Anichini A, Mantovani A, D’Incalci M, Allavena P (2013) Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 23:249–262
Grosso F, Jones RL, Demetri GD, Judson IR, Blay JY, Le Cesne A, Sanfilippo R, Casieri P, Collini P, Dileo P, Spreafico C, Stacchiotti S, Tamborini E, Tercero JC, Jimeno J, D’Incalci M, Gronchi A, Fletcher JA, Pilotti S, Casali PG (2007) Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoid liposarcomas: a retrospective study. Lancet Oncol 8:595–602
Le Cesne A, Cresta S, Maki RG, Blay JY, Verweij J, Poveda A, Casali PG, Balana C, Schoffski P, Grosso F, Lardelli P, Nieto A, Alfaro V, Demetri GD (2012) A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur J Cancer 48:3036–3044
Acknowledgments
This study was supported by the Associazione Italiana Ricerca sul Cancro (CC IG-10615; LR IG-10727; MR IG-9030 and PA IG-12051). M. Tazzari and C. Belgiovine are supported by a fellowship from FIRC (Fondazione Italiana Ricerca sul Cancro).
Conflict of interest
No conflict of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
This paper is a Focussed Research Review based on a presentation given at the Eleventh Meeting of the Network Italiano per la Bioterapia dei Tumori (NIBIT) on Cancer Bio-Immunotherapy, held in Siena, Italy, 17th–19th October 2013. It is part of a CII series of Focussed Research Reviews and meeting report.
Rights and permissions
About this article

Cite this article
Castelli, C., Rivoltini, L., Rodolfo, M. et al. Modulation of the myeloid compartment of the immune system by angiogenic- and kinase inhibitor-targeted anti-cancer therapies. Cancer Immunol Immunother 64, 83–89 (2015). https://doi.org/10.1007/s00262-014-1576-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-014-1576-1