
Abstract
Introduction
We hypothesize that adenosine and PGE2 could have a complementary immunosuppressive effect that is mediated via common cAMP-PKA signaling.
Materials and methods
To test this hypothesis, the effect of adenosine and PGE2 on the cytotoxic activity and cytokine production of lymphokine activated killer (LAK) cells was investigated.
Results
PGE2 and adenosine inhibited LAK cells cytotoxic activity and production of INF-γ, GM-CSF and TNF-α. In combination they showed substantially higher inhibition than each modality used alone. Using agonists and antagonists specific for PGE2 and adenosine receptors we found that cooperation of PGE2 and adenosine in their inhibitory effects are mediated via EP2 and A2A receptors, respectively. LAK cells have 35-fold higher expression of EP2 than A2A. Combined PGE2 and adenosine treatment resulted in augmentation of cAMP production, PKA activity, CREB phosphorylation and inhibition of Akt phosphorylation. Wortmannin and LY294002 enhanced the suppressive effects of adenosine and PGE2. In contrast, Rp-8-Br-cAMPS, an inhibitor of PKA type I blocked their immunosuppressive effects, suggesting that the inhibitory effects of PGE2 and adenosine are mediated via common pathway with activation of cAMP-PKA and inhibition of Akt.
Conclusion
In comparison to other immunosuppressive molecules (TGF-β and IL-10), adenosine and PGE2 are unique in their ability to inhibit the executive function of highly cytotoxic cells. High intratumor levels of adenosine and PGE2 could protect tumor from immune-mediated destruction by inactivation of the tumor infiltrating functionally active immune cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostaglandin E2 (PGE2 ) is a major product of cyclooxygenases (COX-1 and COX-2) and plays an important role in regulating inflammation, as well as various other biological processes. PGE2 exerts biological effects by binding to four receptors, termed EP1, EP2, EP3 and EP4 that belong to the family of G protein-coupled receptors and are differentially expressed on divergent types of cells [1]. EP2 and EP4 receptors are coupled to Gs, and PGE2 binding to these receptors therefore results in stimulation of cAMP production. In addition, some evidence suggests that EP4 signaling also activates the PI3K pathway [2]. Signaling via the EP1 receptor leads to increases in intracellular calcium. EP3 receptor-effector coupling is more complex and involves multiple splice variants, some of which could inhibit or stimulate cAMP production and increase IP3 generation [1].
Emerging evidence indicates that PGE2 plays an important role in malignant processes. Over-expression of COX-2 and elevated levels of PGE2 are found in various types of human malignancies and are often associated with a poor prognosis [3–5]. Moreover, selective and nonselective COX-2 inhibitors appear to be beneficial in cancer patients and inhibit tumor development in experimental animals [3, 6–10]. Numerous studies indicate that COX-2 and its products are directly involved in primary carcinogenesis. For example, pharmacological or genetic ablation of COX-2 inhibits skin carcinogenesis, development of intestinal polyps in APCMin mice and colonic polyps in APC716 mice [11, 12]. Conversely, over-expression of COX-2 in mammary glands results in increased breast cancer formation [13]. PGE2 affects carcinogenesis via different cognate EP receptors [14–16]. Several mechanisms participate in PGE2-mediated stimulation of tumor growth and metastasis. PGE2 directly stimulates tumor cell proliferation, migration and invasiveness [17], stimulates production of VEGF and angiogenesis [18–20] and inhibits functional activity of immune cells, thereby aiding the escape of tumor cells from immune-mediated destruction [1, 21].
Adenosine is another endogenous molecule that is over-produced during inflammation in normal and malignant tissues. Adenosine is a nucleoside generated from metabolism of purine precursors and mediates a broad range of biological functions via four specific G protein-coupled receptors (termed A1, A2A, A2B and A3) [22]. Numerous studies demonstrate that adenosine has potent anti-inflammatory effects and provides a “stop” signal to inflammatory cells and thus prevents normal tissue from excessive damage during inflammation [22–26]. Therefore, it is conceivable that, like PGE2, adenosine protects malignant tissue from immune-mediated destruction [25, 26]. In support of this concept, pharmacological or genetic blocking of A2A receptors results in a more efficient immune response with inhibition of tumor growth and metastasis formation, indicating that adenosine produced within the tumor suppresses the host-mediated immune response via A2A-receptor signaling [27]. Consistent with this conclusion, our previous studies demonstrate that adenosine substantially inhibits the cytotoxic activity and cytokine production of murine and human LAK cells and show that these inhibitory effects are mediated via adenosine A2A receptors, leading to activation of cAMP and PKA type I signaling [28–30] We also found that adenosine is able to inhibit the cytotoxic activity and cytokine production of human anti-melanoma specific CD8+ and CD4+ T cells [31].
Malignant cells, as well as stromal cells (macrophages, fibroblasts, endothelial cells), are able to produce adenosine and PGE2, resulting in high levels of these substances in the tumor microenvironment. PGE2 can activate cAMP signaling via either EP2 or EP4 receptors [1, 2], and adenosine also stimulates cAMP production via A2A and A2B receptors [22–26]. cAMP provides inhibitory signaling that could be proportional to the amount of produced cAMP. Because both adenosine and PGE2 can activate cAMP signaling, we postulate that adenosine and PGE2 cooperate and cause additive inhibitory effects on the cytotoxic activity of immune cells. We further hypothesize that this cooperation is mediated via cAMP-elevating receptors triggered by adenosine and PGE2, resulting in more profound increases in cAMP production and enhanced activation of PKA. In the present study we tested our hypothesis by analyzing the combined effects of adenosine and PGE2 on the cytotoxic activity of LAK cells. The ability of other immunosuppressive molecules, such as TGF-β and IL-10 to potentiate the inhibitory effects of adenosine and PGE2 was also evaluated.
Materials and methods
Mice
C57BL/6 and BALB/c (6–8 weeks old) females were purchased from the Jackson Laboratory (Bar Harbor, Maine). Experiments were performed in accordance with the approved institutional protocol and guidelines of the Institutional Animal Care and use Committee.
Reagents
Adenosine, 2-chloroadenosine (CADO), erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA), Rolaprim, CGS21680, sulprostone, butaprost, AH23848, SC19220, H89 and myristoylated PKI14–22 peptide were purchased from Sigma-Aldrich (St Louis, MO). AH6809 was purchased from Cayman Chemicals (Ann Arbor, MI) and Rp-8-Br-cAMPS was from Alexis Biochemicals (San Diego, CA). TGF-β1 and IL-10 were from PeproTech (Rocky Hill, NJ). When DMSO was used as a solvent for various chemicals it was added to LAK cells as an additional control. In all experiments DMSO at used concentrations showed no effects on LAK cells functional activity.
LAK cell generation
LAK cells were generated from spleens of C57BL/6 or BALB/c mice by incubation with IL-2 (6,000 IU/ml) and purified for their increased adhesion to plastic as previously described [28, 31]. Briefly, a single cell suspension of spleen cells (50 × 106) was cultured in T-75 flasks for 3 days in the presence of IL-2 (6,000 IU/ml), and non-adherent spleen cells were removed. The flasks were washed with pre-warmed (37°C) complete medium to remove those cells that were not firmly attached to the plastic. Plastic-adherent cells were cultured for an additional 3–8 days and were transferred to another flask for further expansion. This approach generated large numbers of purified highly cytotoxic LAK cells [28, 32].
Cytotoxic activity of LAK cells
The cytotoxic activity of LAK cells was tested against 51Cr-labeled 3LL Lewis lung carcinoma cells. LAK cells were distributed into V-bottom 96-well plates with and without test agents, and 30 min. later 51Cr-labeled 3LL tumor cells (5 × 103/well) were added. LAK cell cytotoxicity was determined in triplicate at the various effector:target (E:T) ratios. After 4 h of incubation at 37°C, supernatants (25 μl) were transferred into yttrium silicate scintillator-coated white microplates (LumaPlateTM-96, PerkinElmer, Boston, MA) and the level of β-emission released by 51Cr was measured in a β-counter. The percentage of cytotoxicity was calculated [29]. Data presented as mean ± SD. All experiments were repeated 2–3 times.
Cytokine production
LAK cells from C57BL/6 mice were stimulated with anti-Ly49D mAb as described [28, 33]. Anti-Ly49D mAb was a gift from Dr. John Ortaldo (NCI, NIH, Frederic, MD). Costar 96-well plates were precoated with rabbit anti-rat IgG (2 μg/well) and blocked with RPMI 1640 medium containing 10% FCS. LAK cells were washed and rested for 2 h in the absence of IL-2. LAK cells were incubated with anti-Ly49D rat mAb and seeded into 96-well plates precoated with rabbit anti-rat IgG (0.5 × 106 LAK cells/well) in the presence or absence of tested chemicals. In some experiments LAK cells were stimulated with immobilized NK1.1. mAb. After 16 h of incubation at 37°C, supernatants (0.1 ml) were collected and concentrations of various cytokines were analyzed using Luminex LabMAP technology.
Multiplex bead-based cytokine analysis
We used murine multiplex antibody bead kit (Biosource International, Camarillo, CA) for Luminex xMAP analysis that allows simultaneous testing of IFN-γ, GM-CSF and TNF-α. The multiplexed assay was performed at the University of Pittsburgh Cancer Institute Luminex Core Facility as described [28, 34]. Data are expressed as mean ± SD pg/ml.
Analysis of cAMP production
LAK cells (0.5 × 106 cells/0.5 ml/tube) were incubated with CADO or PGE2 for 10 min. Culture medium was removed and 1 ml of ice-cold 1-propanol was added to cells. cAMP levels in the cellular extracts were analyzed using a ThermoFinnigan high-pressure liquid chromatographic system coupled to a ThermoFinnigan LCQ Duo ion trap mass spectrometer equipped with an electrospray ionization source (Thermo Electron Corporation, Walthan, MA) by adapting a method recently described for purines [35]. All experiments were repeated 2–3 times.
Western blot analysis
LAK cells were incubated with CADO(5 μM) and PGE2 (100 nM) for 30 min. Cells were lysed with RIPA buffer supplemented with 1 mM PMSF and 1% CLAP cocktail (anti-pain, leupeptin, pepstatin and chymostatin). Protein extracts (50 μg) were resolved using 10% SDS-PAGE and transferred to PVDF membranes. The membranes were blocked and incubated with antibodies against phosphorylated CREB (anti-phospho S129 + S133; 1:1,000, Abcam, England) or anti-total CREB (1:2,000, Abcam) and then with horseradish peroxidase-labeled secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
To assess Akt phosphorylation, LAK cells were rested overnight in the absence of IL-2 and then were stimulated with IL-2 in the presence of CADO (25 μM) and/or PGE2 (500 nM) for 30 min. Prepared protein extracts were resolved using 10% SDS-PAGE and transferred to PVDF membranes. The membranes were also incubated with phospho-AKT(Ser473), dilution 1:100 or antibody for total AKT, dilution 1:500 (Cell Signaling, Billerica, MA) then with horseradish peroxidase-labeled secondary antibody (Santa Cruz). Films were scanned and analyzed by ImageQuanT data analysis software (Molecular Dynamics).
Analysis of EP receptors by LAK cells
LAK cells lysates were prepared as described above and Western blot analysis of protein was performed using 10% SDS-PAGE. After transfer, membranes were incubated with anti-EP1, anti-EP2, anti-EP3 or anti-EP4 rabbit polyclonal antibodies (Cayman Chemicals, Ann Arbor, MI), followed by horseradish peroxidase-labeled secondary antibody. Western blot analysis was repeated using LAK cells from C57Bl/6 and BALB/c mice with similar results.
Quantative real-time RT-PCR
Mouse LAK cell RNA was prepared using the Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA was produced from 2 μg of RNA using the SuperArray ReactionReady First Strand cDNA Synthesis kit (SuperArray Bioscience, Frederick, MD). Real-time PCR was carried out using the SuperArray RT2 Real-Time TM SYBR Green PCR Master Mix (SuperArray Bioscience) and was performed on the ABI PrismTM 7700 sequence detector real-time PCR system (AB Applied Biosystems). PCR conditions were as follows: denaturation at 95°C for 10 min, 40 cycles at 95°C for 15 s, and 60°C for 1 min. Primers for A2A, forward: 5′-gtgactctcccctccacaccc-3′; reverse: 5′-catagtttctgtcttccagccc-3′; for EP2, forward: 5′-agcaggacctctatctccttg-3′; reverse: 5′-ccaccagggtctggtttcttg-3′. Standard curves were generated from five 10-fold serial dilutions of LAK cDNA, and no product could be seen in the no-template control. The cycle threshold (C t) value for A2A and EP2 expression was defined as the number of PCR cycles required for the fluorescence signal to exceed the detection threshold value (background noise). Differences in gene expression were calculated using the ΔΔC t method according to the manual from SuperArray Bioscience.
Statistics
Statistical analysis of the data was performed using the Student’s t test. The significance level was set up at P < 0.05.
Results
Inhibitory effects of PGE2 and adenosine on the cytotoxic activity of LAK cells
In the first set of experiments, we compared the ability of PGE2, adenosine and its stable analog 2-chloroadenosine (CADO) to inhibit the cytotoxic activity of LAK cells. LAK cells were incubated with 51Cr labeled 3LL tumor cells for 4 h in the absence or presence of CADO (0.125–2 μM). Significant (P < 0.05) inhibition of LAK cell cytotoxicity was observed at 0.5 μM CADO, and the level of inhibition was further increased at 2 μM (Fig. 1a).

a The inhibitory effects of 2-chloroadenosine (CADO) and PGE2 on LAK cell cytotoxicity. The cytotoxic activity of LAK cells was tested against 51Cr-labeled 3LL tumor cells at the effector-to-target ratio of 30:1 in the presence of CADO (0.125–2 μM) or PGE2 (0.006–2 μM). b Combined inhibitory effect of adenosine (ADO) and PGE2 on LAK cell cytotoxicity. LAK cell cytotoxicity was tested in the presence of adenosine (ADO) (2 μM) plus an inhibitor of adenosine deaminasae (EHNA; 30 μM) and/or PGE2 (6–240 nM). PGE2 as well as ADO plus EHNA significantly (P < 0.05) inhibited LAK cell cytotoxicity. EHNA used alone did not show significant effects on LAK cell cytotoxicity (data not shown). The combined inhibitory effect of ADO and PGE2 was significantly (P < 0.05) higher than when each modality was used alone
Unlike CADO, adenosine is rapidly metabolized by adenosine deaminase (ADA), which is produced by tumor cells and LAK cells. Therefore, the effect of adenosine on LAK cell cytotoxicity was analyzed in the presence of EHNA, an inhibitor of ADA. LAK cells killed 61.8% of tumor cells at the effector-to-target ratio of 30:1. In the presence of 2, 10 and 50 μM of adenosine, the cytotoxic activity of LAK cells reduced to 34.8, 19.1 and 15.6%, respectively (data not shown).
Whereas adenosine and CADO exerted inhibitory effects at micromolar concentrations, PGE2 profoundly inhibited LAK cell cytotoxicity at nanomolar concentrations (Fig. 1a). Even at 6 nM, PGE2 significantly (P < 0.05) inhibited LAK cell cytotoxicity, and the level of inhibition further increased with higher PGE2 concentrations. PGE2 at 1–2 μM almost completely inhibited the cytotoxic activity of LAK cells (Fig. 1a).
Next we analyzed the combined effects of adenosine (ADO) and PGE2. In the presence of adenosine (2 μM) or PGE2 (6 nM), LAK cell cytotoxicity was reduced from 63.9 to 34.8 and 38.1%, respectively (Fig. 1b). In combination, ADO (2 μM) plus PGE2 (6 nM) reduced cytotoxicity to 18.4%, a response that was similar to that observed with PGE2 alone at a concentration of 240 nM (15.2%). A combination of 2 μM of ADO plus 240 nM of PGE2 caused even more severe inhibition of the cytotoxic activity of LAK cells (4.1%) (Fig. 1b). These results indicate that adenosine and PGE2 could be potent factors limiting the efficacy of LAK cells.
Analysis of PGE2 and adenosine receptors involved in the inhibition of LAK cells functional activity
PGE2 mediates its biological effects by binding to four receptors (EP1, EP2, EP3 and EP4). To address which receptor subtypes of PGE2 were involved in the imunosuppressive effects of PGE2, we tested the ability of various agonists and antagonists of EP receptors to affect the cytotoxicity of LAK cells. Suprostone, an agonist of EP1 and EP3 receptors, did not affect the cytotoxic activity of LAK cells (Table 1). Butaprost is a specific agonist of EP2 receptors, although it has an affinity ten times lower than that for PGE2 [36]. Butaprost at 5 μM significantly inhibited LAK cell activity, suggesting involvement of EP2 receptors in the inhibitory signaling (Table 1). PGE2 at 50 nM showed a substantial inhibitory effect that was not blocked by SC19220, an antagonist of EP1 receptors. Similarly, an antagonist of EP4 receptors AH2384 failed to block the inhibitory effect of PGE2. In contrast, A6809, an antagonist of EP2 receptors, significantly (P < 0.05) blocked the inhibitory effect of PGE2 on LAK cells (Table 1). Thus, these results indicate that PGE2 inhibits LAK cell cytotoxic activity via EP2-receptor signaling.
We next analyzed what receptors are involved in enhancement of the immunosuppressive signals mediated by adenosine and PGE2. Previously we showed that adenosine inhibits LAK cell activity via A2A receptors [29]. Therefore, we tested whether CGS21680 (CGS) (a selective agonist of A2A receptors) could work in concert and potentiate the inhibitory effects of butaprost (a selective agonist of EP2 receptors). Both CGS and butaprost significantly (P < 0.05) inhibited the cytotoxic activity of LAK cells (Fig. 2a). A combination of butaprost with CGS manifested a more efficacious inhibitory effect than each modality used alone. A combination of butaprost, with CADO or CGS with PGE2 also had a complementary inhibitory effect (Fig. 2a). In contrast, sulprostone, an agonist of EP1 and EP3 receptors, at all tested concentrations (0.05–5 μM) did not affect the cytotoxic activity of LAK cells and did not potentiate the inhibitory effects of CADO (data not shown). These results indicate that simultaneous signaling via adenosine A2A and EP2 receptors resulted in augmented suppression of LAK cell cytotoxic activity.

a Combined inhibitory effect of agonists of A2A and EP2 receptors. LAK cell cytotoxicity was tested in the presence of CGS21680 (CGS, a selective agonist of adenosine A2A receptors; 50 μM), butaprost (a selective agonist of EP2 receptors; 0.5 μM), CADO (2 μM) or PGE2 (6 nM). In some groups, LAK cell cytotoxicity was tested in the presence of a combination of agonists. In all groups, the combined inhibitory effects were significantly (P < 0.05) higher than when each agent was tested separately. b Western blot analysis of EP receptor expression by LAK cells. LAK-cell lysates were prepared and western blot analysis of protein was performed using 10% SDS-PAGE. After transfer, membranes were incubated with anti-EP1, anti-EP2, anti-EP3 or anti-EP4 rabbit polyclonal antibodies (Cayman Chemicals), followed by horseradish peroxidase-labeled secondary antibody. c Real-time PCR of A2A and EP2 gene expression in murine LAK cells. Real-time PCR was carried out using the SuperArray RT2 Real-Time TM SYBR Green PCR Master Mix (SuperArray Bioscience) and was performed on the ABI Prism TM 7700 sequence detector real-time PCR system. Standard curves were generated from five [5] 10-fold serial dilutions of LAK cDNA. The cycle threshold (C t) value for A2A and EP2 expression was defined as the number of PCR cycles required for the fluorescence signal to exceed the detection threshold value (background noise). Differences in gene expression were calculated using the ΔΔC t method
In all our experiments, PGE2 inhibited LAK cell cytotoxic activity at nanomolar concentrations, whereas adenosine and CADO inhibited at micromolar concentrations. These differences may result from differences in expression of EP2 and A2A receptors. We analyzed expression of PGE2 receptors by LAK cells. Our western blot analysis revealed that LAK cells express all four EP receptors with high expression of EP2 and EP4 receptors and low expression of EP1. EP3 receptor, as previously reported [1] had several splice variants (Fig. 2b).
Our western blot analysis failed to detect adenosine A2A receptors using antibodies from different commercial sources, suggesting lower levels of A2A expression.
To evaluate EP2 and A2A expression by LAK cells, we compared the levels of their mRNA using real-time quantitative RT-PCR. The difference between the cycle threshold (C t) values for A2A and EP2 was 5.14 cycles. Relative quantitative expression of adenosine A2A and EP2 receptors was calculated by the ΔΔC t method. The results of this calculation indicate that in murine LAK cells, EP2 mRNA expression was higher than A2A mRNA by 35.3 ± 2.76 fold (Fig. 2c). Thus, higher levels of EP2 receptors expression are most likely responsible for the observed greater ability of PGE2 versus adenosine to inhibit the cytotoxic activity of LAK cells.
Effect of CADO and PGE2 on cytokine production by LAK cells
Previously we demonstrated that adenosine and CADO are able dramatically inhibit production of various cytokines and chemokines by LAK cells [28]. Now we investigated whether this inhibitory effect can be augmented by PGE2. Rested LAK cells were stimulated with immobilized Ly49D mAb for 16 h in the presence of CADO and/or PGE2. Analysis of cytokine concentrations in culture media revealed that CADO at 5 μM substantially inhibited production of IFN-γ, GM-CSF and TNF-α (Table 2). PGE2 at nanomolar concentrations showed a very profound dose dependent inhibition of cytokine production (Table 2). When LAK cells were treated by combination of CADO (5 μM) and PGE2 (6 nM) a higher inhibitory effect was observed. CADO (5 μM) further potentiated already strong inhibitory effects of PGE2 at higher concentrations (Table 2). Thus, CADO and PGE2 are able to enhance their inhibition of two major functions of activated NK cells: cytotoxic activity and cytokine production.
Amplification of cAMP–PKA signaling by PGE2 and CADO
Because both A2A and EP2 receptors are positively coupled to adenylyl cyclase, we hypothesized that the complementary effect of adenosine and PGE2 on cytotoxicity is mediated via increased production of cAMP. The results of our analyses revealed that PGE2 at concentrations ranging from 6 to 100 nM inhibited cytotoxicity and increased cAMP production by LAK cells in a concentration-dependent manner (Fig. 3a, b). PGE2 at concentrations 25 and 100 nM was more efficient in the production of cAMP and inhibition of LAK cytotoxicity than CADO at 2 μM. When LAK cells were treated with PGE2 in combination with CADO, a more profound increase in cAMP production and inhibition of cytotoxicity was observed (Fig. 3a, b).

Combined effects of CADO and PGE2 on the cytotoxic activity (a) and cAMP production (b) by LAK cells. a The cytotoxic activity of LAK cells against 3LL tumor cells was tested in the presence of CADO (2 μM) and/or PGE2 (6, 25 or 100 nM). b Production of cAMP was evaluated by incubation of LAK cells (0.5 × 106 cells/0.5 ml) with CADO (2 μM) and/or PGE2 (6, 25 or 100 nM) for 10 min following treatment with ice-cold 1-propanol. The levels of cAMP in cell extracts were analyzed using mass spectrometry. The experiments were repeated two times with similar results. c Rolaprim, an inhibitor of phosphodiestyerase 4 (PDE4) potentiates the inhibitory effects of CADO and PGE2. The cytotoxic activity of LAK cells against 3LL tumor cells (E:T ratio 30:1) was tested in the presence of CADO (5 μM), PGE2 (6 nM) and/or rolaprim (25 μM). All groups significantly (P < 0.01) differ from control. The combined inhibitory effects in all groups were significantly (P < 0.05) higher than each agent tested separately
To bring additional confirmation that increased production of cAMP is responsible for the increased inhibitory effects, we evaluated the effect of phosphodiesterases (PDE) inhibitor on the inhibitory effects of CADO or PGE2. Phosphodiesterases control the levels of cellular cAMP and quickly metabolize cAMP terminating the cyclic nucleotide secondary message signaling [37]. PDE4 is a major form operating in lymphocytes and can be inhibited by the specific inhibitor rolaprim [37]. PDE4 by metabolization of cAMP induced by CADO and PGE2 could restrict their inhibitory effects. If so, inhibition of PDE4 by rolaprim could increase the inhibitory effects of CADO and PGE2. To test this prediction, the cytotoxic activity of LAK cells was tested in presence of CADO, PGE2 and rolaprim (Fig. 3c). Our titration experiments revealed that rolaprim at 25 μM significantly (P < 0.05) inhibited the cytotoxic activity of LAK cells as a result of accumulation of the spontaneously generated cAMP. When rolaprim was used in combination with CADO or PGE2 their inhibitory effects on LAK cytotoxicity was substantially augmented (Fig. 3c). These data indicate that PGE4 is a potent regulator of LAK cytotoxicity as well as the activity of adenosine and PGE2.
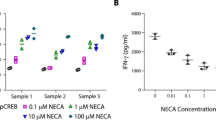
cAMP induced by adenosine or PGE2 could activate PKA that could phosphorylate serine and threonine residues on specific substrate proteins, including cAMP responding element-binding protein (CREB) [38]. Western blot analysis (Fig. 4a) showed that treatment of LAK cells with CADO or PGE2 increased CREB phosphorylation. When LAK cells were treated with a combination of CADO and PGE2, the level of CREB phosphorylation was further increased (Fig. 4a).

Western blot analysis of the effects of CADO and/or PGE2 on CREB (a) or Akt (b) phosphorylation. a LAK cells were incubated for 30 min with CADO (5 μM) and/or PGE2 (50 nM). Protein extracts (50 μg) were resolved by 10% SDS-PAGE and transferred to PVDF membranes, and the levels of total and phosphorylated CREB were assessed using specific antibodies. Corresponding densitometric analysis of optic density ratios between pCREB and total CREB is presented. b Effect of CADO and/or PGE2 on AKT phosphorylation. LAK cells were rested overnight without IL-2 and then were incubated with IL-2 (5,000 IU/ml) in the presence of CADO (25 μM) and/or PGE2 (500 nM) for 30 min. Protein extracts (50 μg) were prepared and resolved by 10% SDS-PAGE and transferred to PVDF membranes. The levels of total and phosphorylated AKT were assessed using specific antibodies. Corresponding densitometric analysis of optic density ratios between p-AKT and total AKT is presented. In rested LAK cells AKT was not phosphorylated (data not shown). c Wortmanin, an inhibitor of Akt phosphorylation poteniates the inhibitory effects of CADO and PGE2. The cytotoxic activity of LAK cells against 3LL tumor cells (E:T ratio 30:1) was tested in the presence of wortmanin (25 μM), CADO (5 μM) or PGE2 (6 nM). All groups significantly (P < 0.01) differ from control. The combined inhibitory effects in all groups were significantly (P < 0.05) higher than when each agent tested separately
It was shown that activation of Akt is triggered by NK cells interaction with the target cells and it is essential for NK lysis of tumor cells. Inhibition of AKT phosphorylation by wortmannin or LY294002 results in inhibition of NK cell cytotoxicity [39]. We next tested whether CADO and PGE2 are able to affect Akt phosphorylation in NK cells. LAK cells were rested overnight and incubated with IL-2 in the presence of CADO and PGE2. After 30 min the levels of Akt phosphorylation was evaluated using Western blot analysis (Fig. 4b). High level of Akt phosphorylation was induced by restimulation of resting LAK cells with IL-2. CADO and PGE2 substantially inhibited Akt phosphorylation. This inhibition further increased when LAK cells were treated with combination of CADO and PGE2 (Fig. 4b). Thus, these data indicate that CADO and PGE2 inhibit NK cells activity via c-AMP mediated activation of PKA and inhibition of Akt activation.
Next we tested whether wortmannin that inhibit Akt activation are able to potentiate the inhibitory effects of CADO and PGE2 on IFN-γ production. LAK cells of C57BL/6 mice were rested for 2 h and were restimulated with immobilized NK1.1 mAb overnight in the presence of CADO, PGE2 and wortmannin (Fig. 4c). CADO and PGE2 substantially inhibited IFN-γ production. Our titration experiments determined that 25 nM is the lowest concentrations of wortmannin causing a significant inhibition of INF-γ production by LAK cells. With increase concentrations of these inhibitors the levels of inhibition increased (data not shown). When LAK cells were treated with wortmanin in combination with CADO or PGE2 the levels of inhibition of INF-γ production was further significantly (P < 0.05) increased. The level of inhibition was similar to those induced by the combination of CADO and PGE2 (Fig. 4c). Experiments with LY294002 showed similar results (data not shown). Wortmannin and LY294002 also potentiated the inhibitory effects of CADO and PGE2 on LAK cell cytotoxicity (data not shown).
Blocking the inhibitory effects of CADO and PGE2 by PKA inhibitors
Activation of PKA is probably essential for the inhibitory effects of adenosine and PGE2. Therefore, it was of interest to investigate the ability of PKA inhibitors to block the suppressive effects of PGE2 and CADO. There are two types of PKA. We previously showed that PKA type I, but not type II, is involved in the inhibitory effects of adenosine on LAK cells [28]. PKA consist of a tetrameric structure including two regulatory (R) and two catalytic (C) subunits [40, 41]. PKA type I and II differ in their regulatory subunits but not catalytic subunits. Activity of PKA type I and II can be inhibited by H89 or myristoylated peptide PKI14–22, that inactivate the released catalytic subunits [40, 41]. Alternatively, PKA activity can be inhibited by blocking the binding of cAMP to R subunits by the specific antagonists. Rp-8-Br-cAMPs specifically blocks binding of cAMP to regulatory subunits of PKA type I and prevents the dissociation of the PKAI holozyme and the release of catalytic subunits [42]. To assess the ability of PKA inhibitors to block the suppressive effects of PGE2, LAK cells were pre-incubated with H89, myristoylated peptide PKI14–22 or Rp-8-Br-cAMPS and their cytotoxic activity was tested against 3LL tumor cells. The PKA inhibitor H89 alone at 10–2.5 μM severely inhibited LAK cell cytotoxicity. When it was applied in combination with CADO or PGE2, H89 amplified their inhibitory effects (data not shown). PKI14–22 protein alone did not affect LAK cell cytotoxicity and failed to block the inhibitory effects of CADO or PGE2 (Fig. 5a). In contrast, Rp-8-Br-cAMPS completely prevented the inhibitory effects of CADO and PGE2. CADO in combination with PGE2 had more profound inhibitory effects on LAK cell cytotoxicity than each agent used separately (Fig. 5a). Pre-incubation of LAK cells with Rp-8-Br-cAMPS, but not with PKI14–22 peptide, significantly (P < 0.05) attenuated the combined inhibitory effect of CADO and PGE2 (Fig. 5a). This blocking effect was partial, most likely because of the very high production of cAMP that was incompletely antagonized by Rp-8-Br-cAMPS.

Blocking the inhibitor effects of CADO and PGE2 with Rp-8-Br-cAMP, an antagonist of regulatory (RI) subunits of PKA type I. a LAK cells were pre-incubated with Rp-8-Br-cAMPS (1 mM; antagonist of RI subunits of PKA type 1) or myristoylated PKI14–22 peptide (20 μM; inhibitor of catalytic subunits of PKA type 1) for 30 min before CADO (2 μM ) and/or PGE2 (25 nM) were added. The cytotoxic activity of LAK cells against 3LL tumor cells (E:T ratio 30:1) was tested in the 4 h 51Cr release assay. Rp-8-Br-cAMP significantly (P < 0.05) blocked the inhibitory effects of CADO, PGE2 and CADO plus PGE2. PKA inhibitors PKI14–22 or Rp-8-Br-cAMPS used alone did not affect LAK cell cytotoxiocity (data not shown). b LAK cells of C57BL/6 mice were rested for 2 h without IL-2 and then they were seeded into 96-well plate (2 × 105 cells/well) with immobilized Ly49D mAb. Cells were pre-incubated with Rp-8-Br-cAMPS (1 mM; antagonist of RI subunits of PKA type 1) or myristoylated PKI14–22 peptide (20 μM; inhibitor of catalytic subunits of PKA type 1) for 30 min before CADO (2 μM) and/or PGE2 (25 nM) were added. Supernatants were collected after 16 h of culture and concentration of IFN-γ, GM-CSF and TNF-α were analyzed using a multiplex kit. Only values of IFN-γ are presented. Rp-8-Br-cAMP significantly (P < 0.05) blocked the inhibitory effects of CADO, PGE2 and CADO plus PGE2. PKA inhibitors PKI14–22 or Rp-8-Br-cAMPS used alone did not affect LAK cell cytokine production (data not shown)
We next tested the ability of PKA inhibitors to protect IFN-γ production by LAK cells incubated with CADO and PGE2. PKI14–22 peptide failed to abrogate the inhibitory effects of CADO and PGE2 (Fig. 5b). Pretreatment of LAK cells with Rp-8-Br-cAMPS significantly (P < 0.05) blocked the inhibitory effects of CADO and PGE2 on the IFN-γ production, although these blocking effects were partial.
Comparison of the inhibitory effects of CADO, PGE2, TGF-β and IL-10
TGF-β and IL-10 are considered as the major immunosuppressive factors in tumor microenvironment capable of inhibiting antitumor immune response [43, 44]. It was of interest to compare their immunosuppressive ability with adenosine or PGE2. We tested whether TGF-β and IL-10 are able to inhibit the cytotoxic activity and cytokine production of LAK cells and whether they could enhance the inhibitory effects of CADO and PGE2.
As it was shown above and confirm in this experiment, CADO or PGE2 alone inhibited the cytotoxic activity of LAK cells and this inhibition was further increased by combination of CADO and PGE2 (Fig. 6a). In contrast, the cytotoxicity of LAK cells was not impaired by TGF-β or IL-10 and they did not potentiate the inhibitory effects of CADO or PGE2 (Fig. 6a).

Failure of TGF-β and IL-10 to inhibit LAK cells cytotoxicity (a) and IFN-γ production (b) and potentiate the inhibitory effects of CADO or PGE2. a The cytotoxic activity of LAK cells was tested against 3LL tumor cells (E:T ratio 30;1) in the presence of CADO (5 μM) and/or PGE2 (5 nM), TGF-β (30 ng/ml) or IL-10 (30 ng/ml). In some groups TGF-β or IL-10 were tested in combination with CADO or PGE2. TGF-β or IL-10 alone or in combination with CADO or PGE2 did not significantly (P > 0.05) affected LAK cell cytotoxicty. b LAK cells were rested for 2 h without IL-2 and stimulated with the immobilized NK1.1 mAb for overnight in the presence of CADO (5 μM) and/or PGE2 (5 nM), TGF-β (30 ng/ml) or IL-10 (30 ng/ml). Concentrations of IFN-γ in the supernatants were analyzed. TGF-β significantly (P < 0.05) increased IFN-γ production. Only combination of CADO and PGE2 significantly (P < 0.05) blocked the stimulatory effect of TGF-β. IL-10 had no significant (P > 0.05) effects of CADO and/or PGE2 on IFN-γ production
Next we tested whether TGF-β and IL-10 are able to inhibit LAK cells cytokine production. Rested LAK cells were stimulated for 16 h with the surface bound NK1.1 mAb in the absence or presence of tested molecules and concentration of IFN-γ in the collected media was analyzed. CADO and PGE2 strongly inhibited IFN-γ production by LAK cells and this inhibition further increased when these molecules were used in combination. In contrast, TGF-β (30 ng/ml) significantly increased production of IFN-γ (Fig. 5b). CADO or PGE2 just blocked TGF-β-induced stimulation and amount of produced IFN-γ was similar to the control level. Only combination of CADO and PGE2 is able to antagonize the effect of TGF-β and reduce IFN-γ below the control levels. Thus, TGF-β counterbalances the inhibitory effects of CADO and PGE2 on IFN-γ production. The same effects were observed with TGF-β at lower concentration (10 ng/ml) (facts not shown).
IL-10 did not affect production of IFN-γ. When IL-10 was used in combination with CADO and/or PGE2 it did not change the inhibitory effects of CADO and PGE2 (Fig. 6b). The same effects were observed with IL-10 used at lower concentration (10 ng/ml). These data indicate that CADO and PGE2, in contrast to TGF-β or IL-10 have unique ability to quickly inhibit LAK cells cytotoxic activity and cytokine production.
Discussion
Our data demonstrate that both adenosine and PGE2 impair the ability of LAK cells to kill tumor cells and produce cytokines. Combined treatment with adenosine and PGE2 results in a substantial augmentation of their ability to inhibit LAK cell functional activity. Previous studies suggest that cAMP is a mediator of PGE2-induced suppression of NK cell activity [42]. Consistent with this concept, the present study demonstrates that activation of A2A and EP2 receptors by their ligands results in cAMP production and PKA mediated phosphorylation of CREB proportionally to the level of inhibition of LAK cell activity.
One goal of the present study was to elucidate the receptor subtypes mediating inhibition by PGE2. In this regard, our western blot analysis show that LAK cells express all four PGE2 receptor subtypes. Moreover, experiments with agonists and antagonists of EP receptors indicate that the PGE2 inhibitory signal is mediated via EP2 receptors that are positively coupled with adenylyl cyclase and are able to stimulate cAMP production.
Because EP4 receptors are also positively coupled with adenylyl cyclase [1] and are expressed by LAK cells, it is surprising that EP4 receptors are not involved in PGE2-mediated inhibition of LAK cell cytotoxicity. There are at least three non-mutually exclusive explanations for this enigma. First, EP2 receptors more efficiently stimulate cAMP production than do EP4 receptors [2]. Second, EP4 receptors internalize very quickly following PGE2 binding, whereas EP2 receptors do not internalize [45]. Third, stimulation of EP4, but not EP2, receptors results in the activation of the PI3K signaling pathway that could inhibit the activity of PKA [2, 46].
The experiments with butaprost (specific agonist of EP2 receptors) and CGS (specific agonist of adenosine A2A receptors) indicate that the suppressive effects of PGE2 and adenosine are mediated via EP2 and A2A receptors signaling, respectively. Importantly, nanomolar concentrations of PGE2 inhibit LAK cell activity, whereas significant inhibition with adenosine requires micromolar concentrations. Greater LAK cell inhibition by PGE2 is consistent with a higher level of cAMP induction by PGE2. Because adenosine A2A receptors and EP2 receptors have only slight differences in affinity for their respective ligands [42, 45], it is conceivable that the differences between adenosine and PGE2 with regard to potency and efficacy for cAMP production and inhibition of LAK cell activity is mostly due to a higher density of EP2, compared with A2A receptor expression on LAK cells. In corroboration of this hypothesis, real-time PCR demonstrates that EP2 gene expression by LAK cells is 35.3 times higher than A2A receptor expression. Lower number of receptors might require higher proportion of them to be activated and for this higher concentration of ligand is considered necessary in order to provide sufficient signal. Indeed, it was shown that decreased number of adenosine receptors in A1+/− mice requires twice more adenosine to signal [47].
Our previous studies showed that specific agonists of PKA type I, but not PKA type II, inhibit activity of human and murine LAK cells [28, 30], suggesting that PKA type I is involved in the inhibitory effects of adenosine and PGE2. In support of this hypothesis, we show in the present study that the inhibitory effects of CADO and PGE2 are blocked by Rp-8-Br-cAMPS that prevents binding of cAMP to A and B sites of regulatory I subunits of PKA type I. In contrast, inhibitors of catalytic subunits of PKA (H89 or myristoylated PKI14–22 peptide) fail to block the inhibitory effects of PGE2 and CADO. The ability of Rp-8-Br-cAMPS to block the inhibitory effects depends on the levels of inhibition induced by CADO and PGE2. More profound inhibition of LAK cytotoxicity induced by combine treatment with CADO and PGE2 that is due to higher level of cAMP production was only partially blocked by Rp-8-Br-cAMPS. In general, cytokine production in comparison to the cytotoxic activity of LAK cells was more sensitive to the inhibitory effects of CADO and PGE2. Overall, CADO at 5 μM and PGE2 at 6 nM inhibited LAK cells cytotoxicity by 30–40%, whereas cytokine production of IFN-γ was inhibited by 50–70%, suggesting that cytokine production in comparison to cytotoxic activity has lower threshold sensitivity to cAMP-mediated inhibitory signaling in LAK cells. Therefore, Rp-8-Br-cAMPS was less efficient in blocking CADO and PGE2-mediated inhibition of cytokine production than cytotoxic activity.
It remains unclear why PKI14–22 peptide that inhibit the activity of catalytic subunits of PKA do not abrogate the inhibitory effects of CADO and PGE2. Rp-8-Br-cAMPS and PKI14–22 peptide at the tested concentrations show similar ability to inhibit PKA activity as measured by the StressXpress non-radiactive PKA activity kit (Stressgen Bioreagents, Victoria, BC, Canada). Although cAMP activates both PKA type I and II, causing the release of catalytic subunits capable of phosphorylating CREB, only blocking cAMP binding to RI subunits of PKA I with Rp-8-Br-cAMPS abrogates the inhibitory effects of adenosine as well as PGE2. It is possible that regulatory subunits type I has independent functional activity and could take part in the inhibitory effects mediated by adenosine and PGE2. Some studies using mice with knock out regulatory subunits of PKA I or type II showed different and multiple phenotypic changes, suggesting that regulatory subunits of PKA could participate in regulation of various biological processes [40, 41].
Numerous experimental data indicate that various immunosuppressive molecules, such as IL-10, TGF-β, VEGF, and indoleamine 2,3-dioxygenase (IDO) could help tumor to escape the immune destruction [43, 48]. Comparative analysis of their ability to inhibit the functional activity of immune cells was not performed. Although it was shown that TGF-β inhibits the induction of the cytolytic machinery by preventing expression of five cytolytic products, such as perforin, granzyme A and B, FasL and IFN-γ in murine T cells stimulated by anti-CD3 and anti-CD28 antibodies [49], TGF-β as well as IL-10 failed to inhibit the cytotolytic activity of CTLs in the cytotoxicity assay [50]. In our experiments TGF-β and IL-10, in contrast to adenosine and PGE2, failed to inhibit the cytotoxic activity and cytokine production by LAK cells. Furthermore, IL-10 and TGF-β failed to potentiate the inhibitory effects of CADO or PGE2.
Analysis of the effects of TGF-β and IL-10 on IFN-γ production revealed that TGF-β increased production of IFN-γ by NK1.1 stimulated LAK cells and antagonized the inhibitory effects of CADO and PGE2. Although TGF-β is a potent inhibitor of IFN-γ production, the ability of TGF-β to enhance IFN-γ production by murine T cells was also observed [51]. IL-10 did not affect IFN-γ production and the inhibitory effects of CADO and PGE2. These studies demonstrate that PGE2 and adenosine are unique in their ability to quickly inhibit the execution phase of already cytotoxic cells.
In summary, the present study reveals several novel findings with important implications. This work demonstrates for the first time that PGE2 and adenosine act in concert such that the immunosuppressive effects of PGE2 plus adenosine are greater than the inhibitory effects of PGE2 alone or adenosine alone. This additivity of effects applies to both inhibition of cytotoxic activity and inhibition of cytokine production by IL-2 activated NK cells.
Over-expression of COX-2 and increased production of PGE2 are essential properties of various malignancies [3]. Tumors also contain high levels of adenosine as a result of hypoxia and inflammation [26, 28, 52]. It is possible that tumor produced PGE2 and adenosine could inhibit the efficacy of adoptive transfer of cytotoxic LAK cells and could be contributing factors responsible for the failure of adoptive immunotherapy using LAK cells as well as active antitumor vaccinations. Indeed, pharmacological blockade or genetic disruption of adenosine A2A receptors prevents the immunosuppressive effects of adenosine and inhibits tumor growth and metastasis formation [27]. Some studies demonstrate that nonselective or selective inhibitors of COX-2 improve the efficacy of NK cell- or T cell-mediated immunity and inhibit local or metastatic growth [8–10]. Our findings indicate that inhibition of PGE2 production (even by 90% or more) following administration of COX inhibitors might not lead to a complete blockade of PGE2-mediated immunosuppression because the reduced amounts of PGE2 could combine with adenosine to maintain a strong immunosuppressive tumor environment.
Blocking regulatory subunits of PKA type I with Rp-8-Br-cAMPS attenuates the inhibitory effects of both PGE2 and adenosine. These results indicate that the immunosuppressive effects of PGE2 and adenosine are mediated via a common signal transduction pathway involving increased production of cAMP and activation of PKA. The implication of this concept is that inhibition of PKA type I in immune cells before their adoptive transfer might be a novel therapeutic approach to enhance the effectiveness of cancer immunotherapy.
This report represents the first comparative analysis of the immunosuppressive effects of four different immunosuppressive molecules (PGE2, adenosine, TGF-β and IL-10). Our data indicate that PGE2 and adenosine are unique in their ability to rapidly inactivate the effector function of highly cytotoxic LAK cells. Similar data were obtained when human anti-melanoma specific CD8+ T cells were used (unpublished observations). Over-expression of COX-2 and increased production of PGE2 are essential properties of various malignancies. Moreover, intratumor hypoxia and inflammation are the most potent stimuli of adenosine production. Therefore, blocking the immunosuppressive properties of both PGE2 and adenosine, either at the receptor level or in the downstream signaling pathway, may be essential for improving cancer immunotherapy.
References
Hata AN, Breyer RM (2004) Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther 103:147–166
Fujino H, Salvi S, Regan JW (2005) Differential regulation of phosphorylation of the cAMP response element-binding protein after activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. Mol Pharmacol 68:251–259
Dannenberg AJ, Subbaramaiah K (2003) Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell 4:431–436
Denkert C, Winzer KJ, Hauptmann S (2004) Prognostic impact of cyclooxygenase-2 in breast cancer. Clin Breast Cancer 4:428–433
Mrena J, Wiksten JP, Thiel A, Kokkola A, Pohjola L, Lundin J, Nordling S, Ristimaki A, Haglund C (2005) Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor HuR. Clin Cancer Res 11:7362–7368
Eisenthal A (1990) Indomethacin up-regulates the generation of lymphokine-activated killer-cell activity and antibody-dependent cellular cytotoxicity mediated by interleukin-2. Cancer Immunol Immunother 31:342–348
Kundu N, Fulton AM (2002) Selective cyclooxygenase (COX)-1 or COX-2 inhibitors control metastatic disease in a murine model of breast cancer. Cancer Res 62:2343–2346
Kundu N, Walser TC, Ma X, Fulton AM (2005) Cyclooxygenase inhibitors modulate NK activities that control metastatic disease. Cancer Immunol Immunother 54:981–987
Lala PK, Parhar RS (1988) Cure of B16F10 melanoma lung metastasis in mice by chronic indomethacin therapy combined with repeated rounds of interleukin 2: characteristics of killer cells generated in situ. Cancer Res 48:1072–1079
Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R (2000) Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res 60:4705–4708
Chun KS, Akunda JK, Langenbach R (2007) Cyclooxygenase-2 inhibits UVB-induced apoptosis in mouse skin by activating the prostaglandin E2 receptors, EP2 and EP4. Cancer Res 67:2015–2021
Langenbach R, Loftin CD, Lee C, Tiano H (1999) Cyclooxygenase-deficient mice. A summary of their characteristics and susceptibilities to inflammation and carcinogenesis. Ann N Y Acad Sci 889:52–61
Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM (1996) Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87:803–809
Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T (2001) Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem 276:18563–18569
Ma X, Kundu N, Rifat S, Walser T, Fulton AM (2006) Prostaglandin E receptor EP4 antagonism inhibits breast cancer metastasis. Cancer Res 66:2923–2927
Majima M, Amano H, Hayashi I (2003) Prostanoid receptor signaling relevant to tumor growth and angiogenesis. Trends Pharmacol Sci 24:524–529
Sung YM, He G, Fischer SM (2005) Lack of expression of the EP2 but not EP3 receptor for prostaglandin E2 results in suppression of skin tumor development. Cancer Res 65:9304–9311
Rozic JG, Chakraborty C, Lala PK (2001) Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor cell migration, invasiveness and angiogenesis. Int J Cancer 93:497–506
Chang SH, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, Lane TF, Hla T (2004) Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA 101:591–596
Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ, Seibert K (2000) Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res 60:1306–1311
Harris SG, Padilla J, Koumas L, Ray D, Phipps RP (2002) Prostaglandins as modulators of immunity. Trends Immunol 23:144–150
Burnstock G (1997) The past, present and future of purine nucleotides as signalling molecules. Neuropharmacology 36:1127–1139
Cronstein BN (1994) Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol 76:5–13
Hasko G, Cronstein BN (2004) Adenosine: an endogenous regulator of innate immunity. Trends Immunol 25:33–39
Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M (2004) Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol 22:657–682
Sitkovsky MV, Ohta A (2005) The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol 26:299–304
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA 103:13132–13137
Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E (2006) Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res 66:7758–7765
Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E (2005) Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol 175:4383–4391
Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E (2006) Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res 36:91–100
Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, Jackson EK, Gorelik E (2007) Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase A type I signaling. Cancer Res 67:5949–5956
Gunji Y, Vujanovic NL, Hiserodt JC, Herberman RB, Gorelik E (1989) Generation and characterization of purified adherent lymphokine-activated killer cells in mice. J Immunol 142:1748–1754
Ortaldo JR, Bere EW, Hodge D, Young HA (2001) Activating Ly-49 NK receptors: central role in cytokine and chemokine production. J Immunol 166:4994–4999
Gorelik E, Landsittel DP, Marrangoni AM, Modugno F, Velikokhatnaya L, Winans MT, Bigbee WL, Herberman RB, Lokshin AE (2005) Multiplexed immunobead-based cytokine profiling for early detection of ovarian cancer. Cancer Epidemiol Biomarkers Prev 14:981–987
Jackson EK, Zacharia LC, Zhang M, Gillespie DG, Zhu C, Dubey RK (2006) cAMP-adenosine pathway in the proximal tubule. J Pharmacol Exp Ther 317:1219–1229
Fedyk ER, Ripper JM, Brown DM, Phipps RP (1996) A molecular analysis of PGE receptor (EP) expression on normal and transformed B lymphocytes: coexpression of EP1, EP2, EP3beta and EP4. Mol Immunol 33:33–45
Bender AT, Beavo JA (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58:488–520
Skalhegg BS, Tasken K (2000) Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front Biosci 5:D678–693
Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, Djeu JY (2000) Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol 1:419–425
Chin KV, Yang WL, Ravatn R, Kita T, Reitman E, Vettori D, Cvijic ME, Shin M, Iacono L (2002) Reinventing the wheel of cyclic AMP: novel mechanisms of cAMP signaling. Ann N Y Acad Sci 968:49–64
Cho-Chung YS, Nesterova M, Becker KG, Srivastava R, Park YG, Lee YN, Cho YS, Kim MK, Neary C, Cheadle C (2002) Dissecting the circuitry of protein kinase A and cAMP signaling in cancer genesis: antisense, microarray, gene overexpression, and transcription factor decoy. Ann N Y Acad Sci 968:22–36
Torgersen KM, Vaage JT, Levy FO, Hansson V, Rolstad B, Tasken K (1997) Selective activation of cAMP-dependent protein kinase type I inhibits rat natural killer cell cytotoxicity. J Biol Chem 272:5495–5500
Gorelik L, Flavell RA (2001) Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med 7:1118–1122
Kim R, Emi M, Tanabe K, Arihiro K (2006) Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res 66:5527–5536
Desai S, April H, Nwaneshiudu C, Ashby B (2000) Comparison of agonist-induced internalization of the human EP2 and EP4 prostaglandin receptors: role of the carboxyl terminus in EP4 receptor sequestration. Mol Pharmacol 58:1279–1286
Fujino H, Regan JW (2006) EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol Pharmacol 69:5–10
Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB (2001) Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci USA 98:9407–9412
Gabrilovich D, Pisarev V (2003) Tumor escape from immune response: mechanisms and targets of activity. Curr Drug Targets 4:525–536
Thomas DA, Massague J (2005) TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8:369–380
Koehler H, Kofler D, Hombach A, Abken H (2007) CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res 67:2265–2273
Smeltz RB, Chen J, Shevach EM (2005) Transforming growth factor-beta1 enhances the interferon-gamma-dependent, interleukin-12-independent pathway of T helper 1 cell differentiation. Immunology 114:484–492
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57:2602–2605
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Su, Y., Huang, X., Raskovalova, T. et al. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP–PKA signaling and immunosuppression. Cancer Immunol Immunother 57, 1611–1623 (2008). https://doi.org/10.1007/s00262-008-0494-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-008-0494-5