
Abstract
Molecules belonging to the Tumor Necrosis Factor (TNF) and TNF receptor superfamilies have explosively expanded through the era of genomics and bioinformatics. Biological investigations of these molecules have explored their potency as attractive targets for cancer therapy. Anti-tumor mechanisms mediated by TNF superfamily molecules (TNFSF) could be classified into direct actions onto tumor cells and indirect effects through immune or non-immune components of tumor-bearing host. In this review, we focus on TRAIL, CD40, 4-1BB (CD137), and LIGHT as promising molecules to mediate powerful and selective anti-tumor responses, and summarize their unique effector mechanisms. In addition, optimal approaches to manipulate these molecules for cancer therapy are also discussed. We try to provide an insight into a role of TNFSF in cancer therapeutics and highlight each of their potency to be an important player in anti-cancer strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tumor necrosis factor (TNF) was originally characterized as a substance mediating necrotic death in various types of tumors in 1975 [4]. As a consequence of the findings, numerous challenges to utilize TNF for the treatment of cancer had been implemented in both experimental animals and clinical trials. Although certain levels of immunological and tumoricidal responses have been observed in experimental tumor models, clinical trials with systemic administration of recombinant human TNF were unsuccessful due to considerable adverse effects without apparent therapeutic benefits [30, 33]. Beginning in 1990s, novel molecules belonging to TNF and TNF receptor superfamily (referred to as TNFSF and TNFRSF hereafter) have been identified and characterized. Developments of worldwide organizations constructing genomic and expressed sequence tags (EST) databases of mammals as well as the emergence of bioinformatics have boosted the expansion of these families. Among novel members of TNFSF and TNFRSF, molecules that mediate powerful anti-tumor effects without inducing severe adverse effects have been detected. Thus, growing TNFSF and TNFRSF molecules have brought us the revival of TNF-related molecules in cancer immunotherapy. In this review, we will discuss the molecules of this superfamily as potential targets for the development of future cancer immunotherapy.
Mechanistic insights of cancer immunotherapy with TNFSF/TNFRSF molecules
Cancer immunotherapies employing TNFSF/TNFRSF molecules exhibit anti-tumor effects through two predominant mechanisms: direct killing of tumor cells and indirect effects by activated anti-tumor immunity. The former mechanism is limited to tumors which express appropriate TNFRSF molecules, while the latter works irrelevant to tumor types so that it may have broad applicability as cancer therapy. These mechanisms are not mutually exclusive, and some TNFSF molecules employ both mechanisms to express anti-tumor effects.
Direct anti-tumor effects through TNFRSF molecules on tumor cells
Signaling from several TNFRSF molecules directly induces cellular phenotypic changes that result in death of tumor cells. One representative mechanism is to deliver apoptotic signal from death domain-containing TNFRSF molecules on tumor cells. Practical application of this strategy to treat cancer, however, was severely hampered by profound adverse effects such as systemic inflammation and liver toxicity triggered by TNFα and Fas signaling, respectively [8, 9, 30, 31, 33, 56]. To overcome this, two alternative approaches have been developed. One is to discover TNFSF molecules capable of delivering death signals on tumor cells, without inducing significant damage in non-malignant cells. In this regard, TRAIL and CD40 are particularly attractive targets. The other strategy is a selective administration technique into tumor sites such as isolated limb perfusion of TNFα.
TRAIL, a new path to selective tumoricidal effects
Among TNFRSF molecules with death domain, a rare example to show selective killing of malignant cells is TNF-related apoptosis-inducing ligand (TRAIL)/TRAIL receptors system. TRAIL is a type-II transmembrane protein expressed as a homotrimer [26, 43], shows high similarity with TNFα and Fas ligand, and is capable of binding to five different receptors [11, 15, 47, 48, 64]. Two of the receptors, death receptor 4 (DR4) and DR5, have cytoplasmic death domains so as to deliver apoptotic signals [47, 48, 64]. The other three receptors are decoy receptor 1 (DcR1), DcR2, and osteoprotegerin (OPG), which are either devoid of functional death domains or produced as secreted protein, and therefore may act as a negative regulator of cell death [11, 15, 47, 64]. TRAIL expression can be induced on activated T, NK, dendritic cells (DC), and monocytes, while a subset of liver NK cells constitutively express [68]. Importantly, many tumor cells are susceptible to TRAIL-induced apoptosis, whereas non-transformed cells are in general resistant to TRAIL. Although the precise mechanisms underlying the differential sensitivity remain unclear, several possibilities have been proposed. First, an obvious hypothesis would suggest a role of functional balance between death domain-containing receptors and decoy receptors. Large-scale screening of different cell lines, however, does not always indicate the correlation between TRAIL sensitivity and receptor expression pattern [34, 85]. Alternative possibility is a distinct transduction of death signal between tumor and normal cells. The primary candidate responsible for the distinct signaling is cellular FLICE-like inhibitory protein (c-FLIP), which prevents apoptosis by blocking caspase-8 activation [28]. Increased expression of c-FLIP in TRAIL-resistant cell lines and a gain of susceptibility by decreasing c-FLIP expression in those lines have been reported [21, 34].
There are numerous studies indicating potential application of TRAIL for cancer immunotherapy. Recombinant TRAIL or anti-DR5 mAb has demonstrated remarkable anti-tumor effects that eradicate established tumors in experimental animals with no or very little adverse effects [27, 75]. The mice genetically deficient of the TRAIL gene exhibit increased susceptibility to experimental and spontaneous tumor [10], suggesting an important role of endogenous TRAIL in tumor surveillance. This notion was further supported by the findings that fibrosarcoma cells grown in the mice treated with anti-TRAIL neutralizing mAb, have increased susceptibility to TRAIL-induced death [69]. Interestingly, TRAIL-deficient T cells exhibit significantly lower activity of graft-versus-leukemia (GVL) effects compared to wild-type T cells, whereas these T cells generate comparable graft-versus-host disease (GVHD) [59]. This study may suggest a potential use of TRAIL to strengthen GVL effects without exacerbating GVHD. In addition, combined usage of chemotherapeutic drugs with TRAIL sensitizes tumor cells otherwise resistant to TRAIL-induced death [24]. Accumulated pre-clinical studies thus clearly indicate a potential of TRAIL for cancer therapy.
Tumoricidal effects of CD40 signaling on tumor cells
Another example for the direct tumoricidal effect through TNFRSF molecules on tumor cells is CD40 and CD40L (CD154) system. CD40 is widely expressed on various types of cancer including hematological malignancies and epithelial cell-derived carcinomas. In hematopoietic tumors, signaling from CD40 leads to diverse outcomes according to cell types. In Hodgkin’s disease and low-grade B cell malignancies such as chronic lymphocytic leukemia, hairy cell leukemia, and follicular lymphoma, CD40 activation contributes to the survival of tumor cells through increased proliferation and inhibited apoptosis [16, 32, 74], as similar to its effects on non-malignant B cells. In contrast, CD40 signaling in high-grade B cell lymphoma, Burkitt lymphoma, and multiple myeloma cells induces growth arrest and apoptosis [17, 50, 57]. In epithelial carcinoma cells, the effects of CD40 signaling appear consistently suppressive, as it mediates growth retardation and apoptosis in breast, ovarian, squamous cell, and lung cancer [19, 25, 52, 86].
Although in vitro studies indicate the direct tumoricidal activity of CD40, in vivo anti-tumor effects could be interpreted as indirect actions to tumor cells because of its broad expression and functions on immune cells [53]. In this regard, recombinant CD154 protein is capable of inhibiting the growth of CD40+ human breast or ovarian tumor xenografted in severe combined immunodeficiency (SCID) mice [19, 25]. These results suggest that in vivo anti-tumor effects by CD40 signaling are, at least in part, mediated independently of adaptive immune systems. Increased expression of apoptotic molecules such as Fas ligand, TNFα, and TRAIL would contribute to the direct tumoricidal effects of CD40 [1, 14, 20]. Alternatively, CD40 signal in B cell lymphoma converts them to suitable antigen-presenting cells (APC) by increasing costimulatory molecule and/or cytokine expressions [61]. This mechanism has been translated into clinic, in which autologous plasma cell leukemia are stimulated with CD154 ex vivo and used as cancer vaccine [62].
Isolated limb perfusion of TNFα
Due to the dose-limiting toxicity, a tolerable dose of systemic TNFα is 10–50 times lower than that required for anti-tumor effects [2]. Isolated limb perfusion, a technique to achieve an elevated concentration of drugs at the isolated extremity without flowing them into systemic circulation, has been successfully applied to local administration of TNFα. Clinical trials by multiple groups have demonstrated that isolated limb perfusion of TNFα with melphalan achieves >70% response rate (complete and partial responses) in patients suffering from unresectable bulk melanoma or soft-tissue sarcomas [13, 35]. This approach thus can avoid the necessity of amputation of limbs. Therapeutic mechanism of isolated limb perfusion of TNFα appears to be destruction of endothelial cells and vasculature of tumors rather than direct killing of tumor cells [46, 54]. In addition, given the profound synergistic effects of TNFα and melphalan, augmented tissue penetration of chemotherapeutic drugs by TNFα would play an important role. Host immune cells may also contribute to the effects since pre-irradiated lymphopenic animals are not susceptible to TNFα perfusion [37]. Taken together, TNFα can be a potent therapeutic reagent for tumors localized in extremity by utilizing the isolated limb perfusion technique.
Indirect anti-tumor effects of TNFRSF expressed on immune cells
Many members of TNFRSF have been shown to function as costimulatory molecules on T lymphocytes [77]. In addition, function and survival of DC can be regulated by signals from TNFRSF molecules [44, 55, 60, 79, 84]. Thus, two major components for T cell immunity, i.e. T cells and APC, are both controlled by TNFSF/TNFRSF interactions. Targeting these pathways, therefore, is a potent strategy for cancer immunotherapy. Here we focus on two representative molecules, 4-1BB (CD137) and LIGHT, based on their capacity to stimulate both T cell and DC, and to generate powerful anti-tumor immunity.
Potent anti-tumor immunity induced by 4-1BB signaling
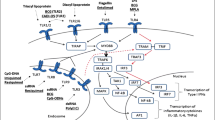
There is ample evidence demonstrating that triggering 4-1BB signaling elicits robust anti-tumor immune responses in vivo [42, 77]. This effect is largely interpreted by 4-1BB signaling on tumor-specific T cells enhancing proliferation and CTL activity, and preventing activation-induced cell death [51, 65, 67]. Recent studies, however, have revealed diverse expression and functions of 4-1BB on immune cells, and have suggested novel mechanisms of its anti-tumor effects (Fig. 1).

Multi-cellular mechanisms of anti-tumor effects through 4-1BB signal. During generation of anti-tumor immunity, 4-1BB signaling is capable of targeting at least three immune cell components, T cells, dendritic cells (DC), and NK cells. 4-1BB signal directly activates tumor-specific T cells, while 4-1BB signal to DC and NK cells indirectly stimulate T cells through cytokines, cognate interaction, or other unknown mechanisms. T cell activation induced by 4-1BB signal confer them the ability to overcome T cell tolerance associated with tumor-bearing conditions, thus leaving secondary lymphoid organs to migrate into the tumor site and attack tumor cells
First, 4-1BB signaling is able to prevent and rescue T cells from immune tolerance [81, 83]. In several animal models that render Ag-specific T cells anergic through tolerogenic Ag immunization, agonistic anti-4-1BB mAb abrogates T cell anergy induction and recovers responsiveness of those T cells [83]. Since there is substantial evidence indicating that tumor-reactive T cells are rendered functionally tolerant in tumor-bearing mice [49], the functional role of 4-1BB signaling in T cell anergy could be crucial to mediate anti-tumor effects. In addition, 4-1BB signal delivery, in conjunction with tumor Ag vaccination, breaks T cell ignorance to poorly immunogenic tumors, leading to an eradication of those tumors [81]. Besides T cell anergy and ignorance, recent studies suggest that 4-1BB signal has a functional role in CD4+CD25+ regulatory T cells, a central player maintaining T cell tolerance and immune homeostasis. 4-1BB is expressed on activated regulatory T cells, and stimulation of regulatory T cells by agonistic 4-1BB mAb abrogates their suppressive function [6]. Taken together, interference with T cell tolerance mechanisms by 4-1BB may play an important role in the anti-tumor effects.
Secondly, 4-1BB expression on non-T cell population including NK cells and DC has been explored, and functional contribution of this pathway to the immune activation is strongly implicated [18, 41, 79, 82]. In vivo depletion of NK cells abrogates anti-tumor effects of anti-4-1BB mAb in some tumor models [41]. 4-1BB signal stimulates in vitro proliferation and cytokine production of NK cells purified from RAG-deficient mice [82]. The NK cells activated with 4-1BB have a positive cross-talk with T cells, in which NK cells accelerate T cell responses to specific Ag and, on the other hand, T cell-derived IL-2 stimulates proliferation of NK cells [82]. 4-1BB expressed on DC may also function as immune modulator since stimulation of DC with 4-1BB ligand triggers IL-12 and IL-6 production and confers them potent Ag-presenting capacity [79]. Collectively, current studies strongly suggest that 4-1BB signaling activate the cross-talk between innate and adaptive immune systems, by which powerful anti-tumor effects are generated.
Finally, 4-1BB stimulation modifies the distribution pattern of tumor-specific T cells in vivo. The number of tumor-specific T cells infiltrating into the tumor sites significantly increases by the administration of agonistic anti-4-1BB mAb [80]. This effect is largely dependent on IFN-γ, since the infiltration of tumor-specific T cells is completely abrogated in mice deficient of IFN-γ, while their number in tumor-draining LN remains unchanged. Consequently, 4-1BB mAb is incapable of inducing anti-tumor effects in the mice deficient of IFN-γ or treated with anti-IFN-γ neutralizing mAb [80]. In addition, blockade of 4-1BB signaling by either anti-4-1BB ligand mAb or gene disruption of 4-1BB, decreases T cell infiltration in cardiac allograft and prolongs the graft survival [5]. Taken together, modification of migratory features in T cells by 4-1BB signaling could be a novel mechanism contributing to the anti-tumor effects.
Dual functions of LIGHT on tumor immunity through two counter-receptors
LIGHT is a potent T cell costimulator in both mouse and human immune systems [70, 71]. It is expressed on activated T cells and immature DC and interacts with two distinct cell-membrane receptors, HVEM and LTβR, and one decoy receptor, TR6/DcR3 [38, 70, 89]. HVEM is expressed on a broad range of hematopoietic cells including T cells, whereas LTβR is mainly detected on non-hematopoietic populations such as stromal cells [3, 23], suggesting a primary role of HVEM in T cell costimulation by LIGHT. In addition, it was also reported that HVEM signaling on DC stimulates their APC function and cytokine production [44]. Genetic disruption of LIGHT results in deficient CD8 T cell functions including impaired graft rejection in allogeneic organ transplantation [58, 72, 87]. Conversely, transgenic expression of LIGHT under T cell-specific promoters results in autoimmune phenotypes associated with the infiltration of activated T cells in multiple organs [63, 76]. These findings indicate that LIGHT–HVEM pathway plays an important role in the competent activation of T cell immunity as well as maintenance of T cell tolerance.
Consistent with its costimulatory functions, expression of LIGHT on tumor cells accelerates anti-tumor T cell immunity, which results in a delayed growth or spontaneous regression of tumors [71, 90]. Increased CTL activity in tumor-draining LN and abrogation of anti-tumor effect by CD8+ T cell depletion, indicates a central role of tumor-reactive CTL in LIGHT-mediated anti-tumor effects. It was shown that inoculation of LIGHT-expressing tumor cells induces profound expression of chemokine CCL21 and adhesion molecule MAdCAM-1 on tumor stromal cells through LIGHT–LTβR interaction [90]. These factors may attract naïve T cells into the site of tumor, where they receive costimulatory signal by LIGHT–HVEM pathway to proliferate and differentiate into effector T cells against tumor (Fig. 2). Although the direct link between this phenomenon and anti-tumor effects by LIGHT has not been established, LIGHT-dependent manipulation of two distinct arms of immunity, i.e. migration and activation of T cells, is an attractive and novel strategy for cancer immunotherapy.

Dual functions of LIGHT for the activation of anti-tumor immunity. LIGHT over-expressed on tumor cells triggers LTβR signal in tumor stromal cells to stimulate chemokine production and adhesion molecule expression such as CCL21 and MAdCAM-1. These factors attract T cells into the tumor site where they receive LIGHT–HVEM costimulatory signal to be activated into anti-tumor effector T cells
Approaches to manipulate TNFSF/TNFRSF molecules for cancer therapy
Gene transfer of costimulatory TNFSF molecules into tumor cells is a powerful cancer vaccine strategy in experimental models [7, 40, 71]. However, this method may encompass obstacles to translate into clinical settings due to requirements of gene transfer into primary tumor cells. Alternatively, recombinant proteins of TNFSF molecules can be prepared in vitro and used as biological adjuvants to enhance T cell responses triggered by cancer vaccines. Based on physiological structure of TNFSF proteins as homo- or heterotrimers [36], protein engineering to trimerize recombinant proteins would be essential for the effects. In fact, conjugation of leucine zipper motif to TNFSF proteins accelerates to form multi-complexes of proteins and results in superior biological effects in vitro and in vivo [22, 45, 75].
Development of agonistic mAb against TNFRSF molecules is one of the most promising strategies for cancer immunotherapy. Administration of agonistic mAb to 4-1BB, CD40, or OX-40 has been shown to generate strong anti-tumor responses in various experimental cancer models [12, 42, 66, 78]. To translate this strategy into clinical settings and to maximize their effects in patients, humanization of mAbs might be necessary. An alternative strategy to employ agonistic mAb for cancer immunotherapy is to construct membrane-bound single-chain Fv fragment (scFv). Recent study indicates that vaccination of anti-4-1BB scFv-expressing tumor cells is capable of treating MHC class I-negative parental tumor in a manner dependent on CD4+ T cells and NK cells, but not CD8+ T cells [88]. These findings suggest a potential use of scFv agonistic to costimulatory receptors to treat low immunogenic or immune-evaded human tumors.
Finally, immunotherapy with TNFSF/TNFRSF molecules can be significantly fortified by a combination with other vaccine strategies. For instance, the effects of agonistic 4-1BB mAb become prominent by concurrent vaccination with tumor-specific Ag or Ag-pulsed DC [29, 73, 81]. In adoptive immunotherapy using tumor-reactive T cells, ex vivo provision of 4-1BB signal induces continuous expansion of T cells and efficient anti-tumor activity after in vivo transfer [39]. In addition, tumoricidal activity of TRAIL has synergistic effects with various chemotherapeutic reagents [24]. Thus, identifying optimal combinations of TNFSF/TNFRSF-based treatments with other anti-cancer therapies would indeed be an important subject in future studies.
Concluding remarks
Wealthy knowledge in molecular nature and immunological functions of the TNF superfamily are now available and ready to be translated into the clinical settings for cancer therapeutics. Manipulation of TNFSF or TNFRSF molecules is an attractive strategy because of their pleiotropic functions on systemic cellular components including T cells, APC, non-hematopoietic cells such as stromal cells, or tumor cells themselves. Their effects include stimulation of anti-tumor immune cells, induction of cytokine and chemokine production, prolonged survival of effector cells, and direct lysis of tumor cells. These functions, if carefully selected and manipulated, represent new and promising strategies for cancer therapy.
References
Afford SC, Randhawa S, Eliopoulos AG, Hubscher SG, Young LS, Adams DH (1999) CD40 activation induces apoptosis in cultured human hepatocytes via induction of cell surface Fas ligand expression and amplifies Fas-mediated hepatocyte death during allograft rejection. J Exp Med 189:441–446
Asher A, Mule JJ, Reichert CM, Shiloni E, Rosenberg SA (1987) Studies on the anti-tumor efficacy of systemically administered recombinant tumor necrosis factor against several murine tumors in vivo. J Immunol 138:963–974
Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, Ambrose CM, Hession C, Miatkowski K, Griffiths DA, Ngam-ek A, Meier W, Benjamin CD, Hochman PS (1997) Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J Immunol 159:3288–3298
Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA 72:3666–3670
Cho HR, Kwon B, Yagita H, La S, Lee EA, Kim JE, Akiba H, Kim J, Suh JH, Vinay DS, Ju SA, Kim BS, Mittler RS, Okumura K, Kwon BS (2004) Blockade of 4-1BB (CD137)/4-1BB ligand interactions increases allograft survival. Transpl Int 17:351–361
Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, Kwon BS (2004) 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J Leukoc Biol 75:785–791
Couderc B, Zitvogel L, Douin-Echinard V, Djennane L, Tahara H, Favre G, Lotze MT, Robbins PD (1998) Enhancement of antitumor immunity by expression of CD70 (CD27 ligand) or CD154 (CD40 ligand) costimulatory molecules in tumor cells. Cancer Gene Ther 5:163–175
Creagan ET, Kovach JS, Moertel CG, Frytak S, Kvols LK (1988) A phase I clinical trial of recombinant human tumor necrosis factor. Cancer 62:2467–2471
Creaven PJ, Plager JE, Dupere S, Huben RP, Takita H, Mittelman A, Proefrock A (1987) Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother Pharmacol 20:137–144
Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168:1356–1361
Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG (1997) The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 7:813–820
Diehl L, den Boer AT, Schoenberger SP, van der Voort EI, Schumacher TN, Melief CJ, Offringa R, Toes RE (1999) CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med 5:774–779
Eggermont AM, Schraffordt Koops H, Klausner JM, Kroon BB, Schlag PM, Lienard D, van Geel AN, Hoekstra HJ, Meller I, Nieweg OE, Kettelhack C, Ben-Ari G, Pector JC, Lejeune FJ (1996) Isolated limb perfusion with tumor necrosis factor and melphalan for limb salvage in 186 patients with locally advanced soft tissue extremity sarcomas. The cumulative multicenter European experience. Ann Surg 224:756–764 (discussion 64–65)
Eliopoulos AG, Davies C, Knox PG, Gallagher NJ, Afford SC, Adams DH, Young LS (2000) CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol Cell Biol 20:5503–5515
Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, Dodds RA, James IE, Rosenberg M, Lee JC, Young PR (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273:14363–14367
Fluckiger AC, Durand I, Banchereau J (1994) Interleukin 10 induces apoptotic cell death of B-chronic lymphocytic leukemia cells. J Exp Med 179:91–99
Funakoshi S, Longo DL, Beckwith M, Conley DK, Tsarfaty G, Tsarfaty I, Armitage RJ, Fanslow WC, Spriggs MK, Murphy WJ (1994) Inhibition of human B-cell lymphoma growth by CD40 stimulation. Blood 83:2787–2794
Futagawa T, Akiba H, Kodama T, Takeda K, Hosoda Y, Yagita H, Okumura K (2002) Expression and function of 4-1BB and 4-1BB ligand on murine dendritic cells. Int Immunol 14:275–286
Ghamande S, Hylander BL, Oflazoglu E, Lele S, Fanslow W, Repasky EA (2001) Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res 61:7556–7562
Grell M, Zimmermann G, Gottfried E, Chen CM, Grunwald U, Huang DC, Wu Lee YH, Durkop H, Engelmann H, Scheurich P, Wajant H, Strasser A (1999) Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. Embo J 18:3034–3043
Griffith TS, Chin WA, Jackson GC, Lynch DH, Kubin MZ (1998) Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J Immunol 161:2833–2840
Gurunathan S, Irvine KR, Wu CY, Cohen JI, Thomas E, Prussin C, Restifo NP, Seder RA (1998) CD40 ligand/trimer DNA enhances both humoral and cellular immune responses and induces protective immunity to infectious and tumor challenge. J Immunol 161:4563–4571
Harrop JA, Reddy M, Dede K, Brigham-Burke M, Lyn S, Tan KB, Silverman C, Eichman C, DiPrinzio R, Spampanato J, Porter T, Holmes S, Young PR, Truneh A (1998) Antibodies to TR2 (herpes virus entry mediator), a new member of the TNF receptor superfamily, block T cell proliferation, expression of activation markers, and production of cytokines. J Immunol 161:1786–1794
Held J, Schulze-Osthoff K (2001) Potential and caveats of TRAIL in cancer therapy. Drug Resist Updat 4:243–252
Hirano A, Longo DL, Taub DD, Ferris DK, Young LS, Eliopoulos AG, Agathanggelou A, Cullen N, Macartney J, Fanslow WC, Murphy WJ (1999) Inhibition of human breast carcinoma growth by a soluble recombinant human CD40 ligand. Blood 93:2999–3007
Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O’Connell M, Kelley RF, Ashkenazi A, de Vos AM (1999) Triggering cell death: the crystal structure of Apo2L/TRAIL in a complex with death receptor 5. Mol Cell 4:563–571
Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, Zhou T (2001) Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med 7:954–960
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388:190–195
Ito F, Li Q, Shreiner AB, Okuyama R, Jure-Kunkel MN, Teitz-Tennenbaum S, Chang AE (2004) Anti-CD137 monoclonal antibody administration augments the antitumor efficacy of dendritic cell-based vaccines. Cancer Res 64:8411–8419
Jones AL, Selby P (1989) Tumour necrosis factor: clinical relevance. Cancer Surv 8:817–836
Kimura K, Taguchi T, Urushizaki I, Ohno R, Abe O, Furue H, Hattori T, Ichihashi H, Inoguchi K, Majima H, et al (1987) Phase I study of recombinant human tumor necrosis factor. Cancer Chemother Pharmacol 20:223–229
Kluin-Nelemans HC, Beverstock GC, Mollevanger P, Wessels HW, Hoogendoorn E, Willemze R, Falkenburg JH (1994) Proliferation and cytogenetic analysis of hairy cell leukemia upon stimulation via the CD40 antigen. Blood 84:3134–3141
Lenk H, Tanneberger S, Muller U, Ebert J, Shiga T (1989) Phase II clinical trial of high-dose recombinant human tumor necrosis factor. Cancer Chemother Pharmacol 24:391–392
Leverkus M, Neumann M, Mengling T, Rauch CT, Brocker EB, Krammer PH, Walczak H (2000) Regulation of tumor necrosis factor-related apoptosis-inducing ligand sensitivity in primary and transformed human keratinocytes. Cancer Res 60:553–559
Lienard D, Eggermont AM, Koops HS, Kroon B, Towse G, Hiemstra S, Schmitz P, Clarke J, Steinmann G, Rosenkaimer F, Lejeune FJ (1999) Isolated limb perfusion with tumour necrosis factor-alpha and melphalan with or without interferon-gamma for the treatment of in-transit melanoma metastases: a multicentre randomized phase II study. Melanoma Res 9:491–502
Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104:487–501
Manusama ER, Nooijen PT, Stavast J, de Wilt JH, Marquet RL, Eggermont AM (1998) Assessment of the role of neutrophils on the antitumor effect of TNFalpha in an in vivo isolated limb perfusion model in sarcoma-bearing brown Norway rats. J Surg Res 78:169–175
Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF (1998) LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8:21–30
Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, Riley JL, June CH (2002) Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol 20:143–148
Melero I, Bach N, Hellstrom KE, Aruffo A, Mittler RS, Chen L (1998) Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BB ligand: synergy with the CD28 co-stimulatory pathway. Eur J Immunol 28:1116–1121
Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L (1998) NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol 190:167–172
Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellstrom KE, Mittler RS, Chen L (1997) Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med 3:682–685
Mongkolsapaya J, Grimes JM, Chen N, Xu XN, Stuart DI, Jones EY, Screaton GR (1999) Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat Struct Biol 6:1048–1053
Morel Y, Truneh A, Sweet RW, Olive D, Costello RT (2001) The TNF superfamily members LIGHT and CD154 (CD40 ligand) costimulate induction of dendritic cell maturation and elicit specific CTL activity. J Immunol 167:2479–2486
Morris AE, Remmele RL Jr, Klinke R, Macduff BM, Fanslow WC, Armitage RJ (1999) Incorporation of an isoleucine zipper motif enhances the biological activity of soluble CD40L (CD154). J Biol Chem 274:418–423
Nooijen PT, Eggermont AM, Schalkwijk L, Henzen-Logmans S, de Waal RM, Ruiter DJ (1998) Complete response of melanoma-in-transit metastasis after isolated limb perfusion with tumor necrosis factor alpha and melphalan without massive tumor necrosis: a clinical and histopathological study of the delayed-type reaction pattern. Cancer Res 58:4880–4887
Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM (1997) An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277:815–818
Pan G, O’Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM (1997) The receptor for the cytotoxic ligand TRAIL. Science 276:111–113
Pardoll D (2003) Does the immune system see tumors as foreign or self? Annu Rev Immunol 21:807–839
Pellat-Deceunynck C, Amiot M, Robillard N, Wijdenes J, Bataille R (1996) CD11a-CD18 and CD102 interactions mediate human myeloma cell growth arrest induced by CD40 stimulation. Cancer Res 56:1909–1916
Pollok KE, Kim YJ, Zhou Z, Hurtado J, Kim KK, Pickard RT, Kwon BS (1993) Inducible T cell antigen 4-1BB. Analysis of expression and function. J Immunol 150:771–781
Posner MR, Cavacini LA, Upton MP, Tillman KC, Gornstein ER, Norris CM Jr (1999) Surface membrane-expressed CD40 is present on tumor cells from squamous cell cancer of the head and neck in vitro and in vivo and regulates cell growth in tumor cell lines. Clin Cancer Res 5:2261–2270
Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ (2004) CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol 22:307–328
Renard N, Lienard D, Lespagnard L, Eggermont A, Heimann R, Lejeune F (1994) Early endothelium activation and polymorphonuclear cell invasion precede specific necrosis of human melanoma and sarcoma treated by intravascular high-dose tumour necrosis factor alpha (rTNF alpha). Int J Cancer 57:656–663
Ridge JP, Di Rosa F, Matzinger P (1998) A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393:474–478
Ryo K, Kamogawa Y, Ikeda I, Yamauchi K, Yonehara S, Nagata S, Hayashi N (2000) Significance of Fas antigen-mediated apoptosis in human fulminant hepatic failure. Am J Gastroenterol 95:2047–2055
Schattner EJ, Mascarenhas J, Bishop J, Yoo DH, Chadburn A, Crow MK, Friedman SM (1996) CD4+ T-cell induction of Fas-mediated apoptosis in Burkitt’s lymphoma B cells. Blood 88:1375–1382
Scheu S, Alferink J, Potzel T, Barchet W, Kalinke U, Pfeffer K (2002) Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med 195:1613–1624
Schmaltz C, Alpdogan O, Kappel BJ, Muriglan SJ, Rotolo JA, Ongchin J, Willis LM, Greenberg AS, Eng JM, Crawford JM, Murphy GF, Yagita H, Walczak H, Peschon JJ, van den Brink MR (2002) T cells require TRAIL for optimal graft-versus-tumor activity. Nat Med 8:1433–1437
Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ (1998) T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393:480–483
Schultze JL, Cardoso AA, Freeman GJ, Seamon MJ, Daley J, Pinkus GS, Gribben JG, Nadler LM (1995) Follicular lymphomas can be induced to present alloantigen efficiently: a conceptual model to improve their tumor immunogenicity. Proc Natl Acad Sci USA 92:8200–8204
Schultze JL, Anderson KC, Gilleece MH, Gribben JG, Nadler LM (2001) A pilot study of combined immunotherapy with autologous adoptive tumour-specific T-cell transfer, vaccination with CD40-activated malignant B cells and interleukin 2. Br J Haematol 113:455–460
Shaikh RB, Santee S, Granger SW, Butrovich K, Cheung T, Kronenberg M, Cheroutre H, Ware CF (2001) Constitutive expression of LIGHT on T cells leads to lymphocyte activation, inflammation, and tissue destruction. J Immunol 167:6330–6337
Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A (1997) Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 277:818–821
Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, Brown TJ, Emswiler J, Raecho H, Larsen CP, Pearson TC, Ledbetter JA, Aruffo A, Mittler RS (1997) 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med 186:47–55
Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI (1999) Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nat Med 5:780–787
Takahashi C, Mittler RS, Vella AT (1999) Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J Immunol 162:5037–5040
Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, Okumura K (2001) Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 7:94–100
Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H, Okumura K (2002) Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med 195:161–169
Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, Chen L (2000) LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol 164:4105–4110
Tamada K, Shimozaki K, Chapoval AI, Zhu G, Sica G, Flies D, Boone T, Hsu H, Fu YX, Nagata S, Ni J, Chen L (2000) Modulation of T-cell-mediated immunity in tumor and graft-versus-host disease models through the LIGHT co-stimulatory pathway. Nat Med 6:283–289
Tamada K, Ni J, Zhu G, Fiscella M, Teng B, van Deursen JM, Chen L (2002) Cutting edge: selective impairment of CD8+ T cell function in mice lacking the TNF superfamily member LIGHT. J Immunol 168:4832–4835
Tirapu I, Arina A, Mazzolini G, Duarte M, Alfaro C, Feijoo E, Qian C, Chen L, Prieto J, Melero I (2004) Improving efficacy of interleukin-12-transfected dendritic cells injected into murine colon cancer with anti-CD137 monoclonal antibodies and alloantigens. Int J Cancer 110:51–60
Umetsu DT, Esserman L, Donlon TA, DeKruyff RH, Levy R (1990) Induction of proliferation of human follicular (B type) lymphoma cells by cognate interaction with CD4+ T cell clones. J Immunol 144:2550–2557
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5:157–163
Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, Tamada K, Chen L, Fu YX (2001) The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest 108:1771–1780
Watts TH (2005) TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol 23:23–68
Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, Urba WJ, Alvord G, Bunce C, Shields J (2000) Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol 164:2160–2169
Wilcox RA, Chapoval AI, Gorski KS, Otsuji M, Shin T, Flies DB, Tamada K, Mittler RS, Tsuchiya H, Pardoll DM, Chen L (2002) Cutting edge: expression of functional CD137 receptor by dendritic cells. J Immunol 168:4262–4267
Wilcox RA, Flies DB, Wang H, Tamada K, Johnson AJ, Pease LR, Rodriguez M, Guo Y, Chen L (2002) Impaired infiltration of tumor-specific cytolytic T cells in the absence of interferon-gamma despite their normal maturation in lymphoid organs during CD137 monoclonal antibody therapy. Cancer Res 62:4413–4418
Wilcox RA, Flies DB, Zhu G, Johnson AJ, Tamada K, Chapoval AI, Strome SE, Pease LR, Chen L (2002) Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest 109:651–659
Wilcox RA, Tamada K, Strome SE, Chen L (2002) Signaling through NK cell-associated CD137 promotes both helper function for CD8+ cytolytic T cells and responsiveness to IL-2 but not cytolytic activity. J Immunol 169:4230–4236
Wilcox RA, Tamada K, Flies DB, Zhu G, Chapoval AI, Blazar BR, Kast WM, Chen L (2004) Ligation of CD137 receptor prevents and reverses established anergy of CD8+ cytolytic T lymphocytes in vivo. Blood 103:177–184
Wong BR, Josien R, Lee SY, Sauter B, Li HL, Steinman RM, Choi Y (1997) TRANCE (tumor necrosis factor [TNF]-related activation-induced cytokine), a new TNF family member predominantly expressed in T cells, is a dendritic cell-specific survival factor. J Exp Med 186:2075–2080
Wuchter C, Krappmann D, Cai Z, Ruppert V, Scheidereit C, Dorken B, Ludwig WD, Karawajew L (2001) In vitro susceptibility to TRAIL-induced apoptosis of acute leukemia cells in the context of TRAIL receptor gene expression and constitutive NF-kappa B activity. Leukemia 15:921–928
Yamada M, Shiroko T, Kawaguchi Y, Sugiyama Y, Egilmez NK, Chen FA, Bankert RB (2001) CD40-CD40 ligand (CD154) engagement is required but not sufficient for modulating MHC class I, ICAM-1 and Fas expression and proliferation of human non-small cell lung tumors. Int J Cancer 92:589–599
Ye Q, Fraser CC, Gao W, Wang L, Busfield SJ, Wang C, Qiu Y, Coyle AJ, Gutierrez-Ramos JC, Hancock WW (2002) Modulation of LIGHT-HVEM costimulation prolongs cardiac allograft survival. J Exp Med 195:795–800
Ye Z, Hellstrom I, Hayden-Ledbetter M, Dahlin A, Ledbetter JA, Hellstrom KE (2002) Gene therapy for cancer using single-chain Fv fragments specific for 4-1BB. Nat Med 8:343–348
Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS (1999) A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem 274:13733–13736
Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, Fu YX (2004) Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol 5:141–149
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tamada, K., Chen, L. Renewed interest in cancer immunotherapy with the tumor necrosis factor superfamily molecules. Cancer Immunol Immunother 55, 355–362 (2006). https://doi.org/10.1007/s00262-005-0081-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-005-0081-y