
Abstract
Background: CXCR4, the chemokine receptor for CXCL12, has recently been involved in the metastatic process of several neoplasms. Materials and methods: The expression of CXCR4 was evaluated by immunohistochemistry of colorectal tissue samples and by flow cytometry on Caco2, GEO, SW480, SW48, Lovo and SW620 human colon carcinoma cell lines. Correlations with pathological characteristics of the specimens were analysed with chi-square test. To verify the functional status of CXCR4, cell lines were tested in adhesion, migration, and proliferation assays. Results: We studied the expression of CXCR4 in 88 human colorectal tissues and we found that CXCR4 was expressed in >10% of epithelial cells in 50% of normal mucosae (7/14), in 55% of polyps (29/53), in all of carcinomas (16/16) and hepatic metastasis (5/5). Notably, CXCR4 was significantly over-expressed in cancerous lesions (carcinomas and metastasis) compared to non-cancerous lesions (normal mucosa and polyps) (P=0.003) and in adenomatous polyps versus hyperplastic polyps (P=0.009). The diameter of a polyp was also significantly associated with CXCR4 expression (P=0.031). SW480, SW48 and SW620 cell lines showed the highest levels of CXCR4 (60–80% of positive cells). Adhesion, migration, and proliferation increased in response to the CXCL12 chemokine. These effects were abrogated by the addition of anti-CXCR4 antibodies. Further, CXCL12 activated ERK1/2 in SW480 cells. Conclusions: These data suggest that CXCR4 might play a role in colon cancer cell properties and that anti-CXCR4 antibodies could have therapeutic effects against colorectal cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Malignant neoplasms have a tendency to spread from primary tissues to different secondary locations. The migration of tumour cells from their primary location to a secondary one is a key event in cancer metastasis. Much debate exists about the mechanisms that arrest and/or attract malignant cells to specific metastatic sites. Many studies suggest that the mechanisms used for the homing of leukocytes and haematopoietic progenitors may be applied for the dissemination of tumours via the bloodstream and lymphatics. Recent evidence suggests an important role for CXCL12 (a CXC chemokine) and its receptor (CXCR4) in the “metastatic homing” and establishment of tumour cells [1]. It is interesting that CXCL12 is normally produced by the stromal cells of lymph nodes, lung, liver, and bone marrow, the organs that are the most frequent sites for metastasis [2].
Originally characterized as a pre-B-cell stimulatory factor, CXCL12 is a chemotactic factor for T cells, monocytes, pre-B cells, dendritic cells, and haematopoietic progenitor cells. It also increases proliferation of CD34+ cells [3]. More recently, numerous authors have reported CXCR4 expression in tumours and demonstrated that the CXCR4/CXCL12 axis is able to trigger cell adhesion, directional migration, and proliferation in tumour cells. These phenomena have been demonstrated with different methodological approaches in breast cancer [4, 5], small cell lung cancer [6], ovarian cancer [7, 8], pancreatic cancer [9], melanoma [10, 11], lymphoma [12–14], neuroblastoma [15], glioblastoma [16, 17], renal cell carcinoma [18], thyroid cancer [19, 20], rabdomyosarcoma [21], and prostate cancer [22]. Furthermore, a mechanism for CXCR4 activation during tumour-cell evolution has been described in patients with the von Hippel-Lindau (VHL) syndrome. In these patients, inactivation of the VHL tumour suppressor gene in incipient tumour cells results in over-expression of CXCR4 conferring a selective survival advantage and the tendency to home to selected organs [23].
Colorectal cancer is a common neoplasm and represents the third leading cause of cancer deaths worldwide [24]. This tumour frequently metastasizes to the liver, so that hepatic metastases are initially present in 30–40% of patients. In an advanced state of the disease, the liver is involved in up to 95% of patients, and the mortality of colorectal cancer is principally attributable to the spread of tumour cells to the liver. Nevertheless, 50% of patients develop extrahepatic metastases, most often in the lungs, peritoneum, and lymph nodes. However, our knowledge of the factors involved in colorectal cancer spreading to secondary sites is still limited.
In this report, we show that CXCR4 is over-expressed in human colon cancer tissue compared to normal mucosa and benign lesions. CXCR4 expression was also detected in human colon cancer cell lines. In SW480 cells with elevated CXCR4 expression, we studied adhesion, migration, and proliferation. CXCL12 was able to activate these processes. Moreover, as expected of a functional pathway, CXCR4 stimulation also induced ERK activation. We conclude that CXCR4 is over-expressed in colon carcinomas and is also functional as is evident from the activation of downstream pathways making it a potential target in colorectal cancer therapy.
Materials and methods
Reagents and materials
Anti-phospho-ERK1/2, anti-ERK, anti-phospho Akt (Ser473), and anti-Akt antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-CXCR4 clone 12G5 and anti-CXCL12 clone 79014.111 were purchased from R&D Systems (Minneapolis, MN, USA). Types I and III human collagen, and human fibronectin were purchased from BD Biosciences (Bedford, MA, USA). PD98059 was purchased from Calbiochem (San Diego, CA, USA). Human CXCL12 was purchased from Upstate (Lake Placid, NY, USA).
Cell lines and cell culture
SW620, SW480, SW48, GEO, Caco2, Lovo, human colorectal cancer cell lines, MDA-MB-231, a human breast cancer cell line, and HT1080, a human fibrosarcoma cell line, were purchased from the American Type Culture Collection (Rockville, MD, USA) and maintained in DMEM supplemented with 10% (v/v) heat-inactivated FCS.
Tissue samples and immunohistochemistry
Formalin-fixed, paraffin-embedded 4-μm tissue sections of normal colorectal mucosa, hyperplastic polyps, dysplastic polyps (well-, moderately, and poorly differentiated), primary carcinomas (well-, moderately, and poorly differentiated) and hepatic metastases were immunostained using a biotin-streptavidin-peroxidase method (YLEM kit, Rome, Italy). Sections were subjected to routine deparaffinization and rehydration. Slides were then immersed in 10 mM sodium citrate buffer (pH 6.0), boiled for 10 min on a hot plate, and then allowed to cool for 20 min. Sections were then incubated for 10 min in 3% hydrogen peroxide in distilled water, washed in PBS three times for 5 min and incubated with 10% normal horse serum in PBS for 30 min. After three washes in PBS buffer, the sections were incubated overnight at 4°C with 2 μg/ml of primary anti-CXCR4 antibody, clone 12G5 (R&D system). The sections were then incubated with a biotin-labelled secondary antibody (1:30) and streptavidin-peroxidase (1:30) for 20 min each. Slides were stained for 5 min with 0.05% 3,3′-diaminobenzidine tetrahydrochloride freshly prepared in 0.05 M Tris–HCl buffer (pH 7.6) containing 0.024% hydrogen peroxidase and then counterstained with haematoxylin, dehydrated and mounted in Diatex. All series included positive controls (melanoma tissues). Negative controls were obtained by substituting the primary antibody with a mouse myeloma protein of the same subclass, at the same concentration as the monoclonal antibody. All controls gave satisfactory results. Staining was categorized into five semiquantitative classes (−, −/+, +, ++, +++) based on the percentage of stained tumour cells (0%, 0 to <10%, 10 to <50%, 50 to <70% and 70–100%, respectively). Negative or equivocal results were repeated two times. We decided that the specimens should be regarded as positive when the percentage of stained tumour cells was ≤10%. In normal mucosa and polyps, CXCR4 expression was evaluated in epithelial cells and in carcinomas it was evaluated in tumour cells.
Flow cytometry
To evaluate the expression of CXCR4, adherent cancer cells at subconfluency (50–70% confluent) were detached with 2 mM EDTA in PBS, washed, resuspended in ice-cold PBS, and incubated for 30 min at 4°C with anti-CXCR4 antibody (mAb 12G5, 10 μg/ml). After three washes in PBS, FITC-labelled goat anti-mouse (GAM) was added, followed by a further 30-min incubation. For intracellular flow cytometry, cells were resuspended in 100 μl of Cytofix/Cytoperm solution (Pharmingen) for 20 min at 4°C. Cells were then washed two times in Perm/Wash (Pharmingen) solution (1 ml/wash for staining in tubes). Cells were incubated with the primary antibody (mAb 79014.111, 50 μg/ml) in Perm/Wash solution for 30 min at 4°C. After three washes with Perm/Wash solution (1 ml/wash for staining in tubes), cells were incubated with FITC-conjugated GAM antibodies for 30 min. Finally, the cells were washed three times in PBS and analysed by FACS (Becton Dickinson). Negative controls were obtained by omitting the primary antibody.
Migration assays
Migration was studied in collagen-coated (human collagen type I/III) 24-well cell-culture chambers using inserts with 8-μm pore membrane. Test cells were placed in the upper chamber (5×105 cells/well) in DMEM containing 1% BSA (migration media), and 50 or 100 ng/ml CXCL12 or liver-derived proteins were added in triplicate to the lower chamber. After 8 h of incubation, cells on the upper surface of the filter were removed using a cotton wool swab. MDA-MB-231 cells, in the same conditions, were used as positive control; migration of cells in migration media alone was compared with migration in media containing 100 ng/ml CXCL12. When indicated, anti-CXCR4 12G5 (10 μg/ml) was added to the SW480 cells for 5 min before CXCL12 stimulation and during the experiment. Migrated cells on the lower surface were fixed in 10% acetic acid/90% methanol and stained with H&E. Migrating cells in five high-power fields per filter were counted microscopically (×400 magnification). Data were normalized as the migration index, defined as the number of migrating cells in an experimental group divided by the number of migrating cells in control groups without chemokines.
Adhesion assays
For adhesion assays, the wells of a 24-well tissue culture plate (Corning-Costar, Cambridge, MA, USA), precoated with 10 μg/ml human plasma fibronectin or human collagen type I/III overnight at 4°C, were washed with PBS twice and blocked for 1 h at 37°C with DMEM containing 1% BSA (adhesion media) before plating cells. SW480 cells (5×105) were resuspended in adhesion media with or without CXCL12 (50 and 100 ng/ml) and plated in quadruplicate. When indicated, anti-CXCR4 12G5 (10 μg/ml) was added to the SW480 cells for 5 min before CXCL12 stimulation and during stimulation. Unattached cells were removed after incubation for 2 h at 37°C by gentle washing with adhesion media. Adherent cells were fixed in 10% acetic acid/90% methanol, stained with H&E and counted using a microscope by adding the number of cells in five high-power fields (×200 magnification).
Western blotting
Cells were lysed in RIPA buffer (phosphate-buffered saline solution, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium docecyl sulphate, 1 mmol/l phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 1 mmol/l Na3VO4), and the supernates were obtained. Cell lysates (10 μg total protein) were resolved by sodium docecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10% or 4–20% gradient gels (Invitrogen, San Diego, CA, USA) and transferred to Immobilon P membranes. The blots were probed with primary antibodies specific to the following proteins: phospho-ERK1/2, total ERK, phosphorylated Akt, and Akt. The detection of immunocomplex was performed using the ECL detection (Amersham Pharmacia Biotech, London, UK).
Cell proliferation assay
Cells (1×104) were cultured in 96-well flat-bottom plates in quadruplicate and allowed to adhere overnight. The media was then replaced with DMEM supplemented with 1% BSA ± 50 or 100 ng/ml CXCL12. The proliferative activity was determined by the WST-1 colorimetric assay (Boehringer Mannheim) using a microtiter plate reader (Bio-Tek Instruments) at 450 nm (a reference wavelength was subtracted at 600 nm). The background control (blank) was obtained by adding the same volume of culture medium (100 μl) and WST-1 (10 μl) as used in the experimental wells. When indicated, PD98059 (40 μM) or anti-CXCR4 12G5 (10 μg/ml) was added to the SW480 cells for 5 min before CXCL12 stimulation and during the stimulation. Growth stimulation/inhibition was calculated as the percentage of growth stimulation/inhibition: [1− (A/B)]×100, where A is the absorbance of treated cells and B is the absorbance in untreated control cells.
Immunofluorescence microscopy
To detect F-actin, cells were grown sequentially in DMEM 10% FCS and overnight with DMEM 1% BSA on coverslips in 24-well plates. After removing the medium, the samples were treated at 37°C with or without CXCL12 (100 ng/ml) in DMEM containing 1% BSA. After 1, 2, 5, 30, 60 and 240 min cells were washed with PBS and fixed with 3.7% formaldehyde in PBS, rinsed, and permeabilized with 0.5% Triton X-100 in PBS. Samples were blocked with 1% BSA and cells were labelled with FITC-phalloidin (Molecular Probes, Eugene, OR, USA) for 30 min at 37°C. Coverslips were washed and mounted on slide glasses with Mowiol, and observations were performed by an investigator (MVB) who was blind to the treatment conditions of the samples she examined with a fluorescence microscope connected to a laser scanning cytometer (CompuCyte Corp., Cambridge, MA, USA).
ELISA for CXCL12
Proteins from homogenized normal human liver were extracted in Tris–HCL and protease inhibitor as described [25]. SW480 cells were plated to an initial density of 1×104 cells/ml in DMEM supplemented with 10% (v/v) heat-inactivated FCS in 24-well plates; and conditioned medium was collected after 24, 48 and 96 h of culture. CXCL12 protein levels in liver extracts and in conditioned medium of SW480 cells were measured using the antibody sandwich system (R&D system) with a detection range of 62.5–5,000 pg/ml CXCL12.
Statistical analyses and data presentation
All the analyses performed were descriptive. Associations between CXCR4 immunohistochemical scores and clinicopathological variables of tissue specimens were evaluated by the chi-square test. P values <0.05 were considered statistically significant. For functional assays (migration, adhesion, proliferation), data are representative of at least three experiments with comparable results. Bar graphs show the mean ± SD of data from a representative experiment. Student’s t test was used for comparing the means and P values <0.05 were considered statistically significant.
Results
Expression of the CXCR4 receptor in human colorectal cancer tissues
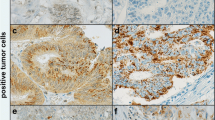
In order to identify a possible prognostic marker in colorectal cancer, we studied the expression of CXCR4 in colorectal tumours by performing immunohistochemical analysis of 88 different tissues including 14 normal mucosae, 53 polyps, and 21 carcinomas (16 primary carcinomas and 5 hepatic metastasis) (Table 1). Staining of CXCR4 was identified in the cytoplasm and/or the cell membrane of the cells. Figure 1 shows representative immunostainings of normal mucosa (a), dysplastic polyps (b–d) and carcinomas (e, f). Carcinomas were intensively stained (11 of 16, or 68.7% were categorized as ++ or +++). There was a significant correlation between the expression of the CXCR4 protein and several clinicopathological variables examined (Table 1). In fact, CXCR4 was significantly over-expressed in cancerous lesions (carcinomas and metastasis, 21/21) compared to non-cancerous samples (normal mucosa and polyps, 36/67) (P=0.003). Dysplastic polyps showed a higher expression of CXCR4 (66.7%) than hyperplastic polyps (21.4%) (P=0.009). A polyp diameter of >4 mm was significantly correlated with a higher expression of CXCR4 (P=0.031). Thus, CXCR4 expression significantly correlates with malignant transformation.

Immunohistochemical study of CXCR4 expression in human colorectal tissues. Representative results of CXCR4 staining in normal mucosa (a, ×40), adenomatous polyps with G2/G3 dysplasia (b–d, ×20), and two carcinomas (e, f, ×20). Staining of CXCR4 was identified in the cytoplasm and/or cell membrane of the cells. The normal mucosa represented in (a) is essentially negative while the stromal cells express CXCR4. In G2/G3 polypi (b–d), CXCR4 staining was higher in high-grade dysplastic cells (indicated by the arrows) compared to adjacent low-grade dysplastic cells (b, c) or to the normal mucosa (d). CXCR4 staining was highly positive in carcinomas (e, f). Negative or positive staining was also observed in a majority of the infiltrating cells in the specimens. Secretions and extracellular matrix are negative
Expression of the CXCR4 receptor in human colorectal cancer cell lines
In order to understand the role of CXCR4 in the biology of colon cancer we analysed six human colon cancer cell lines for the expression of CXCR4. Three different patterns of receptor expression were observed. GEO cells express very low amounts of the receptor while SW620 (~40–60% of cells positive), SW48 (~50–60% of cells positive) and SW480 (~50–80% of cells positive) cell lines were strongly positive. CXCR4 was moderately expressed in Caco-2 (~20% of cells positive) and Lovo cells (~20% of cells positive) (Fig. 2a). Expression of both CXCR4 and its ligand CXCL12 was detected by RT-PCR in SW480 colorectal cancer cells but only CXCR4 was expressed in SW620 cells (data not shown). Moreover, the chemokine CXCL12 was detected in the conditioned medium of SW480 cells by ELISA (data not shown) and by intracellular flow cytometry (Fig. 2b).

Expression of CXCR4 receptor in human colon cancer cell lines. As indicated in a, the expression of the CXCR4 receptor was evaluated with anti-CXCR4 antibody by flow cytometry (empty histograms) in two non-colon cancer cell lines (HT1080, a human fibrosarcoma cell line and MDA-231, a human breast cancer cell line) and in six human colon cancer cell lines (Lovo, GEO, SW48, Caco2, SW480, and SW620). Negative controls were obtained by omitting the primary antibody (filled histograms). In b, CXCL12 expression in SW480 cells has been evaluated with anti-CXCL12 by intracellular flow cytometry. As indicated by different colours, cells were fixed, permeabilized and labelled after 24, 48, 72 and 96 h. Shown is one out of three representative experiments
CXCL12 regulates adhesion, chemotaxis and cell shape in SW480 cells
In SW480 and SW620 (~50–80% of cells positive for CXCR4), we performed functional assays to test the state of the receptor. In the adhesion assay to fibronectin and collagen type I/III, the cells (5×105 cells/well) were resuspended in adhesion media with or without CXCL12 (50 and 100 ng/ml) and seeded in 24-well plates. After 2 h of incubation, unattached cells were removed by gentle washing with adhesion media. Adherent cells were fixed, stained with H&E and counted microscopically. As shown in Fig. 3a, CXCL12 enhanced the adhesion of SW480 cells ~2- and ~3-fold at 50 and 100 ng/ml, respectively; pre-treatment with 10 μg/ml anti-CXCR4 mAb 12G5 strongly reduced CXCL12-dependent adhesion of SW480 cells (Fig. 3a, b). Next, we tested the ability of 100 ng/ml CXCL12 and liver-derived proteins to induce the migration of SW480 cells in collagen-coated cell-culture chambers. SW480 (5×105 cells/well) were plated in the upper chamber. CXCL12 or liver-derived proteins were added to the lower chamber. After 8 h of incubation, cells on the upper surface of the filter were removed using a cotton wool swab. Migrated cells on the lower surface were fixed, stained and counted microscopically. Four different liver-derived extracts were produced and assayed with ELISA to show the presence of the CXCL12 chemokine (mean ± SD pg/ml, A: 2100±73, B: 842±26, 384±44, 271±37). CXCL12 and the liver-derived extracts were able to induce an ~11-fold and a ~6-fold increase in the migration index of SW480 cells, respectively (Fig. 3c). As with the adhesion assay, the chemotaxis induced by CXCL12 could be blocked by pre-treating the cells with anti-CXCR4 antibodies with a marked but not complete inhibition of the chemotactic effect of liver-derived proteins (Fig. 3c). Similar results were obtained with SW620 cells in terms of migration and adhesion while no chemotaxis towards CXCL12 was detected with GEO cells (data not shown). Finally, the ability of CXCL12 to alter the cytoskeleton of SW480 cells was assessed by FITC-phalloidin using a fluorescence microscope. Treatment with 100 ng/ml CXCL12 was associated with the acquisition of a spindle shape, and the formation of pseudopodia and neurite-like projections, as well as increased membrane ruffling (Fig. 3d). The cell shape was changing dramatically from time to time (d, right panels), but no increase in the overall fluorescence was observed. Two representative fields of unstimulated cells were also shown (d, left panels).

CXCL12 regulates adhesion, chemotaxis and cell shape. SW480 cells were plated on human plasma fibronectin (a) or human collagen type I/III (b). When indicated, SW480 cells were resuspended in adhesion media with or without CXCL12. Anti-CXCR4 12G5 was added to the SW480 cells for 5 min before CXCL12 stimulation and during the stimulation. Adherent cells were fixed, stained with H&E and counted microscopically. Migration of SW480 cells was studied using collagen-coated chambers. The cells were placed in the upper chamber and, when indicated below each column, 100 ng/ml CXCL12, or liver-derived proteins were added to the lower chamber in triplicate (c). When indicated, anti-CXCR4 12G5 (10 μg/ml) were added to the SW480 cells for 5 min before and during the experiment (c). Migrated cells on the lower surface were fixed, stained with H&E and counted microscopically. In order to study the cell shape, cells were treated at 37°C with or without CXCL12 (100 ng/ml). As indicated in d, after 10, 30, 60 min and 4 h cells were fixed, rinsed, permeabilized and labelled with FITC-phalloidin. Observations were performed with a fluorescence microscope connected to a laser scanning cytometer. The arrows indicate some morphological changes (neurite-like projections, membrane ruffling, filopodia and uropods formations) in treated cells. Data (a–c) (mean ± SD values of triplicate samples) represent one representative result of four independent experiments. Asterisk significant increase versus control (P<0.05, t test), dagger significant inhibition by anti-CXCR4 (P<0.05, t test)
CXCL12 causes proliferation of SW480 and SW620 cells
CXCL12 was added to SW480 and SW620 cells maintained in serum-free medium and proliferation was measured after 24 h. There was a significant increase in SW480 cell growth with 100 ng/ml CXCL12 (P<0.05) (Fig. 4). Addition of anti-CXCR4 12G5 (10 μg/ml) or PD98059 (40 μM), a MEK inhibitor, strongly blocked the CXCL12-dependent stimulation of cell proliferation (Fig. 4). Interestingly, anti-CXCR4 12G5 (10 μg/ml) inhibited proliferation of unstimulated cells below control (Fig. 4). This evidence suggests an autocrine loop is present in the SW480 cells that might confer a selective advantage to the CXCR4-expressing cells. Similar results were obtained with SW620 cells (data not shown). Furthermore, to study whether CXCL12 could activate a downstream pathway, we tested the activation of ERK1/2 in SW480 cells. SW480 cells were serum-starved for 48 h and then incubated with CXCL12 (100 ng/ml). Changes in the phosphorylation of p44/42 (Erk1/2) kinases were analysed by Western blotting. CXCL12-induced rapid (5 min) phosphorylation of the ERK kinases, increasing to 6–7 the ratio of phospho-ERK1/2 to unphosphosphorylated forms. This ratio then decreased after 15–30 min before increasing again to 3–4 after 24 h (Fig. 5). These results suggest that there is a biphasic pattern of ERKs stimulation in SW480 cells. We were not able to detect induction of phospho-Akt which remains high after serum starvation (data not shown).

CXCL12 induces proliferation of colorectal cancer cells. To measure CXCL12-induced proliferation, SW480 cells were cultured in 96-well flat-bottomed plates and allowed to adhere overnight. As indicated beside the bars, the media was then replaced with DMEM serum-free ±100 ng/ml CXCL12. When indicated, PD98059 (40 μM) or anti-CXCR4 12G5 (10 μg/ml) was added to the SW480 cells for 5 min before CXCL12 stimulation and during the stimulation (filled bars). Anti-CXCR4 12G5 (10 ug/ml) was added to SW480 cells cultured in DMEM 10% FCS (empty bar). The proliferative activity was determined after 24 h by the WST-1 colorimetric assay. Data are presented as mean ± SD values of quadruplicate samples. This is a representative experiment of three done in quadruplicate. Asterisk significant increase versus control (P<0.05, t test) dagger significant inhibition (P<0.05, t test)

CXCL12 induces phosphorylation of extracellular signal-regulated kinases (ERK) 1 and 2 in SW480 cells. To study whether CXCL12 could induce phosphorylation of ERK1/2 in colon cancer cells, SW480 cells were serum-starved for 48 h and then incubated with CXCL12 (100 ng/ml). Changes in the phosphorylation of p44/42 (Erk1/2) kinases were analysed by Western blotting using antibodies specific for the following proteins: phospho-ERK1 and 2, total ERK (a). In b, the ratio of phospho-ERK1/2 to unphosphosphorylated forms is shown based on scanning densitometry values. CXCL12 rapidly (5 min) up-regulated the phosphorylation of ERK kinases increasing to 6–7 the ratio of phospho-ERK1/2 to unphosphosphorylated forms. This ratio then decreased after 15–30 min before increasing to 3–4 after 24 h. Results are representative of three independent experiments
Discussion
In the present study, we focused on the role of CXCR4, the receptor for the CXCL12 chemokine, in colorectal cancer. We have addressed the hypothesis that the CXCR4/CXCL12 axis could be involved in promoting colorectal cancer progression. First, expression of CXCR4 in colorectal tissues was examined by immunohistochemistry. CXCR4 was significantly over-expressed in cancerous lesions (carcinomas and metastasis, 21/21) compared to non-cancerous lesions (normal mucosa and polyps, 36/67). We also observed that dysplastic and larger polyps showed a higher expression of CXCR4 compared to hyperplastic and smaller polyps. The expression of the CXCR4 receptor in six colorectal cancer cell lines (SW480, SW48, GEO, Lovo, SW620, and Caco2) was compared to two non-colorectal human cell lines (HT1080 and MDA231). We found high expression of CXCR4 in three colorectal cancer cell lines (SW480, SW48, and SW620). Then, we determined the biological effects (adhesion, chemotaxis, cell shape, and proliferation) of stimulating the CXCR4 receptor in vitro with its natural ligand, CXCL12.
Previously, Jordan et al. [26] reported CXCR4 expression in both normal and inflamed colonic mucosa and in human colon cancer cells, HT29 by RT-PCR, and Mitra et al. [27] showed CXCR4 expression in colon cancer. We observed immunoreactivity for CXCR4 in the cytoplasm and cell membrane of human intestinal epithelial cells (mAb 12G5 also reacts with intracellular CXCR4) [28], and a heterogeneous staining (from negative to intense staining) in a majority of the infiltrating stromal lymphocytes in the specimens. In contrast to breast cancer tissue [4], these data suggest that the expression of CXCR4 is not a specific characteristic of the malignant colorectal tissue. Interestingly, we observed that in grade 2/3 polyps, CXCR4 staining was higher in high-grade dysplastic cells compared to adjacent low-grade dysplastic cells. Notably, CXCR4 staining in colorectal tumours was significantly higher than in normal mucosa in terms of intensity and the percentage of positive cells.
There is evidence that colon cancer can progress from normal tissue to adenoma and carcinoma through an accumulation of multiple genetic alterations. Thus, the colorectal tumour is an interesting model to study the progression of the neoplastic transformation [29, 30]. With an explorative and predominantly descriptive aim, we studied the expression of CXCR4 in normal mucosa, polyps with different clinicopathological characteristics and carcinomas. Significant correlations were found between the expression of CXCR4 and a variety of clinicopathological features. Surprisingly, adenomatous polyps had significantly higher immunostaining for CXCR4 than hyperplastic polyps. This finding suggests that the alteration of the proliferation status (hyperproliferation is the hallmark of hyperplastic polyps) is not associated with over-expression of CXCR4. The grading of dysplasia of adenomatous polyps was not significantly associated with CXCR4 expression although a slight trend was observed (positive immunostaining: grade 1, 60% vs. grades 2/3, 76.5%). Surprisingly, the diameter of the polyps was correlated with CXCR4 expression, with staining observed to be more intense in larger (>4 mm) polyps. While this association between CXCR4 expression and lesion diameter could reflect an activation of proliferation induced by CXCR4, these data should be interpreted with caution due to the small number of specimens. In fact, we cannot rule out the hypothesis that the higher expression of CXCR4 in adenomatous polyps was reflective of a larger proportion of adenomas in larger polyps. More detailed studies are needed to address correlations between clinicopathological characteristics (various types of polyps, disease stage of carcinomas, p53 expression, APC mutations, etc.), colon cancer progression and CXCR4 expression.
Colorectal cancer is remarkable for its tendency to metastasize to the liver. In the present study, we observed that both CXCL12 and liver extracts are able to induce chemotaxis. However, an anti-CXCR4 antibody did not completely block the chemotactic effect of the liver extracts suggesting that, in addition to the CXCL12 chemokine, other soluble factors may have important roles.
In addition to attraction and adhesion, CXCL12 may also help to establish metastases by acting as a growth paracrine/autocrine factor. This is particularly interesting because CXCL12 is expressed by most cell types [2, 3] and is likely produced by tumour stromal cells; thus, it may be present in the microenvironment of both primary and metastatic colorectal tumours. To evaluate this possibility, we evaluated the importance of CXCL12 on SW480 and SW620 colorectal cancer cells cultured in serum-free conditions. We observed that the addition of CXCL12 stimulated proliferation and resulted in phosphorylation of ERK1/2. As SW480 cells produce CXCL12, they could function as autocrine growth factors. This thesis is supported by the observations that the anti-CXCR4 mAb 12G5 was able to affect CXCL12-stimulated proliferation and that treatment of SW480 cells with mAb 12G5 inhibited the growth of unstimulated cells cultured in the presence of serum (below control levels). In this context, we would note the observations of Zeelenberg et al. [31] on the role of CXCR4 in promoting the outgrowth of colon carcinoma cell metastases. These authors showed inhibition of CXCR4 function in CT-26 colon carcinoma cells using a CXCL12 containing a KDEL sequence that binds to and retains CXCR4 protein in the endoplasmic reticulum. CXCL12-KDEL expressing cells did not form metastases in the liver (relative to the control cells), and also had a reduction in lung micro- and macrometastases, demonstrating a role for the CXCR4/CXCL12 axis in these colon cancer cells. Recently, it has been shown that the PI3K/Akt signalling pathway is frequently involved in survival and migration of tumour cells. However, no induction of phospho-Akt was observed in SW480 cells [32, 33].
Previously, Aihua et al. [34] reported that colon cancer cells migrate and proliferate in response to IL-8, a factor that has been shown to bind to CXCR1 and CXCR2. These receptors are also higher in metastatic/invasive colon carcinoma cells than in non-metastatic colon carcinoma cells. Altogether, these data suggest that chemokines might play an important role in promoting the aggressiveness of colon carcinoma cells. Interestingly, it has recently been shown that CXCR4 expression in colorectal cancer could be associated with high risk of liver metastasis. Comparison of CXCR4 expression levels at all stages by quantitative real-time PCR revealed significant upregulation of CXCR4 expression in patients who developed liver metastases. Analysis of patients in the early stage (I/II) revealed that 30% (n=3/10) of patients with high CXCR4 expression developed liver localizations, whereas no patient with low expression (n=0/23) developed liver metatsases during a mean follow-up of 39 months (P=0.022) [35]. A lot of data have been collected addressing the role of antibodies directed against CXCR4 in blocking its biological activity. We showed that neutralizing anti-CXCR4 antibodies inhibited chemotaxis, adhesion and proliferation of colorectal tumour SW480 cells at a concentration of 10 μg/ml. We think that further studies are warranted in order to produce humanized/chimeric antibodies to test against human colorectal neoplasms. AMD3100, a small CXCR4 antagonist, has been evaluated as an HIV inhibitor because the virus uses CXCR4 as a co-receptor [36] and is entering clinical trials for AIDS [37]. Further pre-clinical studies are required to evaluate its efficacy against cancer in combination with biological and chemotherapeutic agents. On the other hand, our data suggest that CXCL12 may promote colorectal cancer progression at different levels so that agents directed at CXCR4 (antagonists, blocking antibodies, etc.) could have some therapeutic effects against colorectal cancer.
References
Murphy PM (2001) Chemokines and the molecular basis of cancer metastasis. N Engl J Med 345:833–835
Rollins BJ (1997) Chemokines. Blood 3:909–928
Christopherson K, Hromas R (2001) Chemokine regulation of normal and pathological immune responses. Stem Cells 19:388–396
Muller A, Homey B, Soto H et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56
Bachelder RE, Wendt MA, Mercurio AM (2002) Vascular endothelial growth factor promotes breast carcinoma invasion in an autocrine manner by regulating the chemokine receptor CXCR4. Cancer Res 62:7203–7206
Kijima T, Maulik G, Ma PC et al (2002) Regulation of cellular proliferation, cytoskeletal function, and signal transduction through CXCR4 and c-Kit in small cell lung ancer cells. Cancer Res 62:6304–6311
Scotton C, Milliken D, Wilson J, Raju S, Balkwill F (2001) Analysis of CC chemokine and chemokine receptor expression in solid ovarian tumors. Br J Cancer 85:891–897
Scotton CJ, Wilson JL, Scott K et al (2002) Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res 62:5930–5938
Koshiba T, Hosotani R, Miyamoto Y et al (2000) Expression of stromal cell-derived factor 1 and CXCR4 ligand receptor system in pancreatic cancer: a possible role for tumor progression. Clin Cancer Res 6:3530–3535
Robledo MM, Bartolome RA, Longo N et al (2001) Expression of functional chemokine receptors CXCR3 and CXCR4 on human melanoma cells. J Biol Chem 276:45098–45105
Murakami T, Maki W, Cardones AR et al (2002) Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res 62:7328–7334
Bertolini F, Dell’Agnola C, Mancuso P et al (2002) CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin’s lymphoma. Cancer Res 62:3106–3112
Corcione A, Ottonello L, Tortolina G et al (2000) Stromal cell-derived factor 1 as a chemoattractant for follicular center lymphoma B cells. J Natl Cancer Inst 92:625–628
Arai J, Yasukawa M, Yakushijin Y, Miyazaki T, Fujita S (2000) Stromal cells in lymph nodes attract B lymphoma cells via production of stromal cell-derived factor-1. Eur J Haematol 64:323–332
Geminder H, Sagi-Assif O, Goldberg L et al (2001) A possible role for CXCR4 and its ligand, the CXC chemokine stromal cell-derived factor-1, in the development of bone marrow metastases in neuroblastoma. J Immunol 167:4747–4757
Rempel SA, Dudas S, Ge S, Gutierrez JA (2000) Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res 6:102–111
Barbero S, Bonavia R, Bajetto A et al (2003) Stromal cell-derived factor 1a stimulates human glioblastoma cell growth through the activation of both extracellular signal-regulated kinases 1 and 2 and Akt. Cancer Res 63:1969–1974
Schrader AJ, Lechner O, Templin M et al (2002) CXCR4/CXCL12 expression and signalling in kidney cancer. Br J Cancer 86:1250–1256
Aust G, Steinert M, Kiessling S, Kamprad M, Simchen C (2001) Reduced expression of stromal-derived factor 1 in autonomous thyroid adenomas and its regulation in thyroid derived cells. J Clin Endocrinol Metab 86:3368–3374
Hwang JH, Hwang JH, Chung HK et al (2002) CXC chemokine receptor 4 expression and function in human anaplastic thyroid cancer cells. J Clin Endocrinol Metab 88:408–416
Libura J, Drukala J, Majka M et al (2002) CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 100:2597–2606
Taichman RS, Cooper C, Keller ET et al (2002) Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res 62:1832–1837
Staller P, Sulitkova J, Lisztwan J et al (2003) Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 425:307–313
Ries LAG, Eisner MP, Kosary CL (eds) (2002) SEER cancer statistics review, 1973–1999. National Cancer Institute, Bethesda
Hujanen ES, Terranova VP (1985) Migration of tumor cells to organ-derived chemoattractants. Cancer Res 45:3517–3521
Jordan NJ, Kolios G, Abbot SE et al (1999) Expression of functional CXCR4 chemokine receptors on human colonic epithelial cells. J Clin Invest 104:1061–1069
Mitra P, Shibuta K, Mathai J et al (1999) CXCR4 mRNA expression in colon, esophageal and gastric cancers and hepatitis C infected liver. Int J Oncol 14:917–925
Dwinell MB, Eckmann L, Leopard JD, Varki NM, Kagnoff MF (1999) Chemokine receptor expression by human intestinal epithelial cells. Gastroenterology 117:359–367
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87:159–170
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61:759–767
Zeelenberg IS, Ruuls-Van Stalle L, Roos E (2003) The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res 63:3833–3839
Brader S, Eccles SA (2004) Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori 90:2–8
Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M (2004) PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 30:193–204
Aihua Li, Michelle LV, Rakesh KS (2001) Expression of interleukin 8 and its receptors in human colon carcinoma cells with different metastatic potentials. Clin Cancer Res 7:3298–3304
Kim J, Takeuchi H, Foshag L, Bilchik A, Hoon DSB. Colorectal cancer expression of chemokine receptor CXCR4 promotes liver metastasis. ASCO 2004 annual meeting, Abstract number 3582
De Clercq E (2003) The bicyclam AMD3100 story. Nat Rev Drug Discov 2:581–587
Hendrix CW, Flexner C, MacFarland RT et al (2000) Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother 44:1667–1673
Acknowledgements
We thank Dr Maria Vittoria Barone (NCI—Naples) for her help in chemotaxis and adhesion experiments. We also thank Antonio Barbato and Fernando Caccavello for their technical assistance in the laboratories of the Department of Surgical Pathology. This work was supported by grants from Novartis (Dr Nora Pigna and Dr Saverio Valerio) and AstraZeneca (Dr Nino Sala).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ottaiano, A., di Palma, A., Napolitano, M. et al. Inhibitory effects of anti-CXCR4 antibodies on human colon cancer cells. Cancer Immunol Immunother 54, 781–791 (2005). https://doi.org/10.1007/s00262-004-0636-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-004-0636-3