
Abstract
Heat shock proteins (HSPs) are a large family of proteins with different molecular weights and different intracellular localizations. These proteins undertake crucial functions in maintaining cell homeostasis, and therefore they have been conserved during evolution. Hsp70 and Grp94/gp96, due to their peptide chaperone capacity and their ability to actively interact with professional antigen-presenting cells (APCs), are also endowed with crucial immunological functions. The immunological properties of these proteins and their implications for vaccine in human cancer will be discussed. Immunological and clinical data of phase I/II studies in melanoma and colorectal cancer patients will be reviewed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The last years of research in tumor immunology have witnessed an explosion in the identification of T-cell–defined human tumor antigens that also included tumor-derived peptides recognized by CD4+ T h cells in MHC class II restriction [28]. From a different line of research, initially involving mainly murine studies, heat shock proteins (HSPs)—intracellular molecular chaperones devoted to numerous interrelated functions such as folding and translocation of newly synthesized polypeptides in different subcellular compartments [9, 11]—have been identified as tumor-rejection antigens [30]. In murine tumors, these proteins were responsible for the induction of a protective antitumor immune response whose specificity is determined by the chaperone-assisted peptides [38]. HSPs purified from tumors or virus-infected cells have been shown to induce a CTL response in vivo and in vitro against a variety of antigens (Ags) expressed in the cells from which these immunogenic proteins have been purified. Besides their chaperone activity, three additional features characterize HSPs: first, they are efficiently internalized into antigen-presenting cells by a receptor-mediated endocytosis [36]; second, once internalized, they can traffic into different cellular compartments where chaperoned peptides are released, processed, and made available for assembly to new MHC molecules [7]; third, the receptor-mediated internalization of chaperone proteins induces phenotypic and functional maturation in the APC [31]. By studying the immunological behaviour of chaperone proteins we obtained crucial insights into the requirements associated with the induction of a protective immune response. The features of chaperone molecules could therefore be exploited to engineer new tumor vaccines potentially able to generate a full-fledged immune response overcoming tumor escape and interfering with neoplastic growth.
In this review we will briefly discuss recent reports dealing with the activity of chaperone proteins in human tumors, and we will summarize immunological and clinical data from phase I/II clinical trials conducted in melanoma and colorectal cancer patients.
Heat shock proteins: proteins with chaperone activities induced by cellular stress
The functional, active state of newly synthesized protein is strictly associated with the acquisition of a unique three-dimensional structure. The native fold of a protein compatible with this physiological function is directly encoded inside its amino acid sequence. However, protein folding inside cells in general is not a spontaneous process [17]. Chaperone proteins are necessary to counteract the incorrect aggregation of proteins and actively assist their folding process occurring just immediately after new synthesis or under conditions of stress, such as heat shock, when native proteins have become unfolded [11]. The majority of chaperone proteins, although constitutively expressed, augment their levels of expression upon cellular stress such as heat or nutrient and oxygen deprivations, and are therefore classified as HSPs. HSPs include cytosolic and endoplasmic reticulum (ER) resident proteins, and, for some of them, immunological functions have been clearly demonstrated. When purified from murine tumors and used as vaccine, HSPs were shown to be effective both in prophylactic and therapeutic cancer immunotherapy models [36]. Proteins displaying this activity include the cytosolic heat shock proteins hsp70, hsp90, hsp110, or the ER resident Grp94, also known as gp96, and calreticulin [36]. Not all proteins with chaperone capacity have been found to mediate immunological functions, and the ER residents PDI, BiP, or ERp72, although exerting peptide-binding activity, were unable to elicit a specific CTL response [23].
Among molecular chaperones, hsp70 and Grp94/gp96 are well characterized, and, for these proteins, immunological data are also available in a human setting [10, 25]. Hsp70 has diversified functions, which include being part of the ubiquitin/proteasome system, chaperoning proteins into degradation pathways, binding of new synthesized amino acid chains on ribosomes, and maintaining translocation-competent folding of ER and mitochondrial precursor proteins in the cytosol [27]. The unifying theme of these activities is the ability to bind in an ADP-dependent fashion, short hydrophobic regions of 6–9 amino acids in extended conformation. In hsp70, the peptide-binding domain is positioned at the NH2 termini; the ADP-bound state of hsp70 favors polypeptide binding while the ATP-bound form favors release of peptide. Exploiting these two different functional states, purification procedures have been designed to obtain hsp70 with or without chaperoned peptides [26]. Obviously, the large selection of physiological activity of hsp70 together with its ability to traffic through different cell compartments makes the repertoire of hsp70 chaperone peptides potentially wide and derived from proteins having different subcellular localization.
As for Grp94/gp96, numerous experimental observations have shown that this protein also displays intrinsic polypeptide-binding activity, and it has been found to be associated with different unfolded proteins. Being resident in the ER and able to bind peptides, gp96 could be involved in the antigen-presentation pathways and actively participate in shaping the peptide repertoire then associated with the nascent MHC class I molecules [32, 43]. Indeed, gp96 has been shown to possess trimming activity of amino terminally extended peptides but a direct role of gp96 in antigen presentation is still to be formally proven [21].
Similarly to hsp70, gp96 bind ATP but the role of this nucleotide in peptide loading/unloding is not clear, and the regulation of the peptide-binding activity of gp96 has still to be defined [42]. What is known is that peptide-gp96 complexes are very long-lived and remain stable through all the in vitro procedure for gp96 purification. The structure of the gp96-binding pocket has been recently mapped as positioned at amino acid 624–630 in a highly conserved amino acid region [19]. A model for the peptide-binding groove predicts that peptide binding lies alongside the dimerization domain of gp96. This location may partially explain the extraordinary stability of peptide-gp96 complexes. The general features of the gp96 peptide-binding groove is similar to the structure of the MHC class I–binding pocket possibly suggesting that the repertoire of gp96-bound peptide partially overlaps that of MHC class I [19]. The discovery of a precise region of gp96 involved in peptide binding will allow experiments that, taking advantage of gp96 mutants at selective positions, will potentially define the presence, if any, of structural constraints of gp96-bound peptides. Up to now, not much is known on the structural requirements for peptides bound to gp96 or hsp70. Hsp70 seems to have a broad binding specificity that only requires the presence of hydrophobic regions composed of six or more amino acid residues [14]. The same rules have been found also for peptides bound to gp96, and the presence of charged amino acid inside the primary structure of the peptide abolishs its ability to form a stable complex with gp96 [35]. The primary sequence of very few peptides eluted from hsp70 and gp96 is now available [16, 24], and the overall information is too limited to define more precise and general rules determining the binding capacity of a given peptide.
Immunological evidence that HSPs derived from human tumors do contain immunogenic peptides
In addition to the structural studies reported above, the association of peptides with HSPs has been demonstrated by immunological studies initially conducted in murine systems and more recently also in human settings. Studies in murine systems have clearly shown that peptides associated with HSPs can be derived from self-proteins, tumor, bacterial, viral, or minor histocompatibilty antigens [36]. In humans, more recent data exploiting the recognition capacity of tumor-specific T cells with a defined molecular specificity have provided evidence that hsp70 and gp96 do chaperone tumor-derived peptides in different human tumors [10, 25].
We and others were able to demonstrate that hsp70-peptide complexes purified from human melanoma include peptides derived from normal differentiation proteins and that melanoma-derived hsp70 when loaded on HLA-matched APCs was able to activate melanoma and peptide-specific T cells in an MHC-dependent way. Epitopes found to be associated with the hsp70 chaperone include peptides derived from the differentiation antigens Mart-1, gp100, TRP-2, and tyrosinase [10, 25]. Unfortunately, since T-cell clones specific for unique melanoma antigens were not available, the presence of peptides derived from mutated unique melanoma antigens inside the hsp70-chaperoned peptides could not be assessed and, therefore, the relative abundance of shared versus unique epitopes in the human hsp70-chaperoned peptide repertoire could not be directly compared. Hsp70 chaperoned peptides may include precursors of the final T-cell epitopes, and the sequence of hsp70-complexed peptides are not limited or dictated by the MHC class I ligands of the cells. In fact, in our experiments MHC matching between melanoma cell lines used as a source of hsp70 and responding T cells was not a prerequisite, and the capacity of hsp70 to activate a CTL response was found to work across MHC barriers. Conversely, the peptide-dependent activation of CTLs was affected by the MHC alleles expressed by the APCs loaded with tumor-derived hsp70, and APCs expressing the HLA working as restriction elements for the CTL clones was a strict requirement in order to obtain cytokine release [10]. These observations suggest that the mechanism leading to T-cell activation involved an active trimming of the hsp70-chaperoned precursor peptides by the processing machinery of the APCs, and the final shaped T-cell epitopes are then available for recognition only in the presence of the correct HLA-restriction allele.
Similar data have also been obtained recently in our laboratory for gp96. We showed that peptides derived from Mart-1, gp100, are associated with gp96 purified from melanoma cell lines and that they could be efficiently presented to their cognate CTLs expediting their loading on HLA-matched APCs [29].
Receptor-mediated endocytosis of HSPs by APCs: cross-presentation of HSP-chaperoned peptides and functional activation of APCs
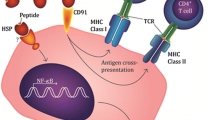
The APC-mediated representation of chaperone-assisted peptides observed for both murine and human systems represents an example of cellular events known as “cross-priming.” The term “cross priming” refers to a complex series of intracellular trafficking mechanisms by which exogenous acquired polypeptides reach specialized intracellular compartments, and, after selective, specific proteolytic processing, their generated peptides are loaded on the MHC class I molecules and presented on the cell surface of APCs where they become available for CD8+ T-cell–mediated recognition [15]. Due to the immunological relevance of CD8+ T-cell activation, cross-presentation should undergo some sort of control in specificity and efficiency. These two main characteristics can be ensured by receptor-mediated processes.
The existence of a receptor specifically mediating the cellular internalization of HSPs was postulated several years ago by Srivastava [37]. However, biological evidence for a receptor-mediated endocytosis of HSPs has been provided only recently, by numerous groups [1, 34, 41]. Applying biochemical techniques, CD91 was the first cell surface molecule identified as a receptor for hsp70, hsp90, and gp96 [4, 8]. This receptor was able to mediate the HSP-peptide complexes’ internalization and cross-presentation. Since then, other cell surface molecules were found to be involved in HSP uptake by APCs [3, 5, 12, 39, 40] (Table 1) and able to mediate other crucial biological functions of HSPs. Indeed, in addition to promoting cross-priming events, hsp70 and gp96 upon specific interaction with their cognate receptors are also endowed with the capacity to promote phenotypic and functional maturation of professional APCs such as dendritic cells or monocytes [2, 18, 33, 39, 40]. Receptor-mediated APC-gp96 or APC-hsp70 interaction leads to up-regulation of MHC class II, CD86, and CD83 expression, and to secretion of activator cytokines such as IL-12 and TNF-α [2, 18, 33, 39, 40]. Therefore, the remarkable immunogenicity of these HSPs is due to two crucial features: HSPs as cross-priming carrier and HSPs as direct activators of professional APCs. Toll-like receptors 2 and 4 are the major receptors involved in transducing hsp70- and gp96-mediated signaling leading to APC activation [3, 39, 40]. The hsp70 or gp96–TLR2/4 interaction results in the activation of the MyD88/NF-ĸB pathway. Notably it has been shown that internalization of gp96 by active endocytosis is required in order to achieve activation of the signaling cascade and consequently the maturation of DCs. However, it is not yet clear what the precise way is by which a gp96/hsp70 internalized via CD91 interacts with TLR2/4. It has been postulated that HSPs, transported in the endocytic vesicles by CD91-mediated internalization, by increasing their local concentration might became able to trigger signaling through the TLR2 and TLR4 present in these vesicles [39]. In addition to TLR2 and TLR4, other cell surface receptors have been found to be potentially involved in transducing activation signals of hsp70 to APCs, among them, CD14 [2] and CD40 [5]; while the scavenger receptor LOX-1 expressed by macrophages and immature DCs has been shown to be involved in hsp70-mediated antigen cross-presentation but not in APC activation [12].
Gp96 as a personalized vaccine for cancer patients: immunological and clinical results
The biological features discussed above make HSPs an appealing candidate for immunotherapeutic approaches to human cancer. The three main points qualifying HSP-based vaccine as having potential to induce a full-fledged immune response able to control tumor growth in vivo could be summarized as follows:
First: being a chaperone of a broad array of peptides, potentially including also unique tumor antigens, HSPs can induce a polyclonal response more likely able to overcome the in vivo selection of antigen-loss variants.
Second: uptake of HSPs by APCs is receptor mediated, thus ensuring specificity and sensitivity of APC antigen loading. HSP-based vaccine therefore preferentially targets the chaperoned antigens to professional APCs, and antigen presentation is likely to occur in an environment endowed with correct costimuli and therefore potentially avoiding anergy induction.
Third: HSPs induce APC activation and provide “danger signals” crucial in the developing of an active immune response.
The in vivo immunogenicity of tumor-derived gp96 and hsp70-peptide complexes has been extensively demonstrated in murine and rat tumors, and HSP-based vaccination has proven efficacious in both prophylactic and therapeutic settings [36].
Based on the general properties of HSPs and on preclinical data available for murine tumors, phase I/II studies have been conducted in melanoma patients with detectable tumor and in colorectal cancer patients rendered disease-free by complete resection of liver metastasis. In these studies, each patient was vaccinated with gp96-peptide complexes isolated from his or her own tumor ensuring therefore an antigenic repertoire as large as possible potentially including also unique tumor-specific antigens [6, 20].
In both studies de novo induction or the augmentation of antitumor-specific T-cell response was achieved in a large proportion of gp96-vaccinated patients. For colorectal cancer, 17 out 29 patients (59%) displayed a statistically significant increase in postvaccination frequency of PBMCs that released INF-γ in response to either autologous or allogeneic HLA-matched colon carcinoma cells. A similar frequency of immunological responder patients was detected in the melanoma vaccination study with 11 out of 23 patients (47.8%) showing an increased number of tumor-specific T cells after gp96 vaccination as evaluated by IFN-γ ELISpot [6]. The antitumor response induced by in vivo gp96 vaccination included T cells specific for shared tumor antigens (gp100 and Melan-A/MART-1 for melanoma, CEA and EpCam for colorectal cancer) [29], while, unfortunately, the presence of T cells directed against individual antigens could not be demonstrated due to the poor viability of fresh tumor cell suspensions and to the difficulty of establishing in vitro cell lines.
Although gp96 vaccinations for both melanoma and colorectal cancer were designed as phase I/II clinical studies with the objectives of evaluating the feasibility, toxicity, and in vivo immunogenicity of tumor-derived gp96, clinical benefits could have been observed in a limited but consistent number of treated patients. For the melanoma study, complete responses involving regression of both cutaneous and visceral metastasis were observed in 2 out of 28 tumor-bearing patients. Taking into account three stable diseases that lasted for more then 5 months, a total response rate of 18% was obtained.
As for the colorectal trial, although the number of patients was too small for any definitive conclusion on possible clinical advantages of adjuvant vaccination with gp96, the results obtained were promising. The disease-free (DSF) and overall survival (OS) rates at 24 months of the 29 vaccinated patients were similar to those reported in the literature for patients who underwent to similar curative surgery [13, 22]. However, a different clinical outcome was observed between the group of patients responding (i.e., showing an increased frequency of tumor-specific T cells in the PBMCs obtained after vaccination) and those not responding to vaccine. Immunologically competent patients had statistically significant survival advantage at 24 months on both OS (100%, versus 50% of nonresponding patients) and DSF (51%, versus 8% of nonresponding patients) (Table 2) [20]. Although, due to the limited number of patients, a possible influence of other prognostic factors could not be excluded, these data suggest that gp96 vaccination not only generated an antitumor response in about half of the treated colorectal cancer patients but that it may also have actively contributed to improvements in the patients’ survival.
A similar association between T-cell–mediated tumor immunity by gp96 vaccination and clinical response was also observed in the melanoma study. The frequency of patients with antitumor immunity was higher in the group who showed a positive clinical outcome than that observed in patients whose disease progressed (Table 3) [6].
Concluding remarks
Due to their immunological functions, HSPs are optimal candidates for vaccines. Gp96 and hsp70 have been proven to work also in human tumors as chaperones for tumor-associated peptides derived from shared antigens such as gp100 and Melan-A/MART-1 for melanoma, or CEA and EpCam for colorectal cancer. When loaded on appropriate APCs by cross-priming mechanisms, HSPs derived from human tumors have been shown to mediate the re-presentation of their chaperoned peptides complexes and lead to the in vitro activation of tumor-specific T cells.
Vaccination studies clearly showed that in two different human cancers—namely, melanoma and colorectal tumors—vaccination with autologus tumor–derived gp96 can successfully induce in vivo antitumor immunity potentially associated with clinical benefit in a percentage of treated patients. The immune responses induced in the vaccinated patients included T cells directed against shared tumor antigens that have been shown to be chaperoned by gp96 and hsp70 by in vitro studies. However, the evidence of immunity directed against unique tumor antigens still remains to be proven. Phase I/II clinical trials lead to the conclusion that vaccination with gp96 derived from autologous tumor represents a feasible and safe approach able to induce an active antitumor response in half of the treated patients. Moreover, the clinical benefit observed in a limited but consistent number of patients who respond to vaccine with an increase of tumor-specific T-cell frequency, warrants further studies evaluating the clinical and immunological outcomes in a larger number of subjects.
References
Arnold-Schild D, Hanau D, Spehner D, Schmid C, Rammensee HG, de la Salle H, Schild H (1999) Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol 162:3757
Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK (2000) HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 6:435
Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK (2002) Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 277:15028
Basu S, Binder RJ, Ramalingam T, Srivastava PK (2001) CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity 14:303
Becker T, Hartl FU, Wieland F (2002) CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. J Cell Biol 158:1277
Belli F, Testori A, Rivoltini L, Maio M, Andreola G, Sertoli MR, Gallino G, Piris A, Cattelan A, Lazzari I, Carrabba M, Scita G, Santantonio C, Pilla L, Tragni G, Lombardo C, Arienti F, Marchianò A, Queirolo P, Bertolini F, Cova A, Lamaj E, Ascani L, Camerini R, Corsi M, Cascinelli N, Lewis JJ, Srivastava P, Parmiani G (2002) Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical and immunologic findings. J Clin Oncol 20:4169
Berwin B, Nicchitta CV (2001) To find the road traveled to tumor immunity: the trafficking itineraries of molecular chaperones in antigen-presenting cells. Traffic 2:690
Binder RJ, Han DK, Srivastava PK (2000) CD91: a receptor for heat shock protein gp96. Nat Immunol 1:151–155
Bukau B, Deuerling E, Pfund C, Craig EA (2000) Getting newly synthesized proteins into shape. Cell 101:119–122
Castelli C, Ciupitu AM, Rini F, Rivoltini L, Mazzocchi A, Kiessling R, Parmiani G (2001) Human heat shock protein 70 peptide complexes specifically activate anti-melanoma T cells. Cancer Res 61:222
Craig EA, Weissman JS, Horwich AL (1994) Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell 78:365
Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, Kawakami-Honda N, Goetsch L, Sawamura T, Bonnefoy J, Jeannin P (2002) Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 17:353
Figueras J, Valls C, Rafecas A, Fabregat J, Ramos E, Jaurrieta E (2001) Resection rate and effect of postoperative chemotherapy on survival after surgery for colorectal liver metastasis. Br J Surg 88:980
Fourie AM, Sambrook JF, Gething MJ (1994) Common and divergent peptide binding specificities of hsp70 molecular chaperones. J Biol Chem 269:30470
Heath WR, Carbone FR (2001) Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 19:47
Ishii T, Udono H, Yamano T, Ohta H, Uenaka A, Ono T, Hizuta A, Tanaka N, Srivastava PK, Nakayama E (1999) Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol 162:1303
Johnson JL, Craig EA (1997) Protein folding in vivo: unraveling complex pathways. Cell 90:201
Kuppner MC, Gastpar R, Gelwer S, Nossner E, Ochmann O, Scharner A, Issels RD (2001) The role of heat shock protein (hsp70) in dendritic cell maturation: hsp70 induces the maturation of immature dendritic cells but reduces DC differentiation from monocyte precursors. Eur J Immunol 3:1602
Linderoth NA, Popowicz A, Sastry S (2000) Identification of the peptide-binding site in the heat shock chaperone/tumor rejection antigen gp96 (Grp94). J Biol Chem 275:5472
Mazzaferro V, Coppa J, Carabba M, Rivoltini L, Schiavo M, Regalia E, Mariani L, Camerini T, Marchiano A, Andreola S. Camerini R, Corsi M, Lewis JJ, Srivastava PK, Parmiani G (2003) Vaccination with autolgous tumor derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin Cancer Res 9:3235
Menoret A, Li Z, Niswonger ML, Altmeyer A, Srivastava PK (2001) An endoplasmic reticulum protein implicated in chaperoning peptides to major histocompatibility of class I is an aminopeptidase. J Biol Chem 276:33313
Minagawa M, Makuuchi M, Torzilli G, Takayama T, Kawasaki S, Kosuge T, Yamamoto J, Imamura H (2000) Extension of the frontiers of surgical indications in the treatment of liver metastases from colorectal cancer: long-term results. Ann Surg 231:487
Nair S, Wearsch PA, Mitchell DA, Wassenberg JJ, Gilboa E, Nicchitta CV (1999) Calreticulin displays in vivo peptide-binding activity and can elicit CTL responses against bound peptides. J Immunol 162:6426
Nieland TJ, Tan MC, Monne-van Muijen M, Koning F, Kruisbeek AM, van Bleek GM (1996) Isolation of an immunodominant viral peptide that is endogenously bound to the stress protein GP96/GRP94. Proc Natl Acad Sci U S A 93:6135
Noessner E, Gastpar R, Milani V, Brandl A, Hutzler PJ, Kuppner MC, Roos M, Kremmer E, Asea A, Calderwood SK, Issels RD (2002) Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J Immunol 169:5424
Peng P, Menoret A, Srivastava PK (1997) Purification of immunogenic heat shock protein 70-peptide complexes by ADP-affinity chromatography. J Immunol Methods 204:13
Pilon M, Schekman R (1999) Protein translocation: how Hsp70 pulls it off. Cell 97:679
Renkvist N, Castelli C, Robbins PF, Parmiani G (2001) A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother 50:3
Rivoltini L, Castelli C, Carabba M, Mazzaferro V, Pilla L, Huber V, Coppa J, Gallino F, Scheibenbogen C, Squarcina P, Cova A, Camerini R, Lewis JJ, Srivastava PK, Parmiani G (2003) Cross-presentation of human tumor-derived heat shock protein gp96 associated peptides leads to in vitro activation and in vivo expansion of melanoma and colon carcinoma-specific T cells. J Immunol 171:3467
Schild H, Arnold-Schild D, Lammert E, Rammensee HG (1999) Stress proteins and immunity mediated by cytotoxic T lymphocytes. Curr Opin Immunol 11:109
Schild H, Rammensee HG (2000) gp96--the immune system’s Swiss army knife. Nat Immunol 1:100
Serwold T, Gaw S, Shastri N (2001) ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol 2:644
Singh-Jasuja H, Scherer HU, Hilf N, Arnold-Schild D, Rammensee HG, Toes RE, Schild H (2000) The heat shock protein gp96 induces maturation of dendritic cells and down-regulation of its receptor. Eur J Immunol 30:2211
Singh-Jasuja H, Toes RE, Spee P, Munz C, Hilf N, Schoenberger SP, Ricciardi-Castagnoli P, Neefjes J, Rammensee HG, Arnold-Schild D, Schild H (2000) Cross-presentation of glycoprotein 96-associated antigens on major histocompatibility complex class I molecules requires receptor-mediated endocytosis. J Exp Med 191:1965
Spee P, Neefjes J (1997) TAP-translocated peptides specifically bind proteins in the endoplasmic reticulum, including gp96, protein disulfide isomerase and calreticulin. Eur J Immunol 27:2441
Srivastava P (2002) Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 20:395
Srivastava PK, Udono H., Blanchere NE, Li Z (1994) Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics 39:93
Suto R, Srivastava PK (1995) A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science 69:1585
Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H (2002) HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem 277:15107
Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, Schild H (2002) The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem 277:20847
Wassenberg JJ, Dezfulian C, Nicchitta CV (1999) Receptor mediated and fluid phase pathways for internalization of the ER Hsp90 chaperone GRP94 in murine macrophages. J Cell Sci 112:2167
Wearsch PA, Nicchitta CV (1997) Interaction of endoplasmic reticulum chaperone GRP94 with peptide substrates is adenine nucleotide-independent. J Biol Chem 272:5152
Wearsch PA, Voglino L, Nicchitta CV (1998) Structural transitions accompanying the activation of peptide binding to the endoplasmic reticulum Hsp90 chaperone GRP94. Biochemistry 37:5709
Acknowledgements
This work was supported in part by Sigma Tau (Rome) and Antigenics (New York). We thank Grazia Barp for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented at the first Cancer Immunology and Immunotherapy Summer School, 8–13 September 2003, Ionian Village, Bartholomeio, Peloponnese, Greece.
Rights and permissions
About this article
Cite this article
Castelli, C., Rivoltini, L., Rini, F. et al. Heat shock proteins: biological functions and clinical application as personalized vaccines for human cancer. Cancer Immunol Immunother 53, 227–233 (2004). https://doi.org/10.1007/s00262-003-0481-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-003-0481-9