
Abstract
Despite the registration of over 1,000 clinical trials assessing the activity of therapeutic cancer vaccines in human patients with multiple cancer types, only a single vaccine has received FDA approval for clinical use. Nonetheless, the therapeutic potential of immune modulation for treating cancer has continued to be validated with both preclinical and clinical studies, most recently in studies investigating so-called checkpoint inhibitory antibodies targeting CTLA-4 and PD-1. One important class of therapeutic cancer vaccines seeks to generate therapeutic immunity based on the combined adjuvant and antigen delivery characteristics of heat-shock proteins. Heat-shock protein-based vaccines are unique among other approaches due to the unique ability of certain heat-shock proteins to dually activate antigen-presenting cells and specifically deliver tumor antigens to cytotoxic CD8+ T cells via the antigen cross-presentation pathway. The enclosed chapter provides a comprehensive overview of heat-shock protein-based cancer vaccines assessed in human clinical trials within the context of parallel progress in understanding the interactions between a developing tumor and the human immune system.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Renal Cell Carcinoma
- Major Histocompatibility Complex
- Antitumor Immunity
- Major Histocompatibility Complex Molecule
- Distinct Tumor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Heat-Shock Proteins, Sterile Inflammation, and Immunosurveillance
Molecular alarm systems are an essential component of vertebrate immunity and function to signify the occurrence of an event which threatens the survival of the host. One such system operates through the family of receptors known as “Toll-like” receptors (TLRs), which evolved to recognize common pathogen-associated molecular patterns (PAMPs) such as bacterial cell wall sugars, single-stranded viral DNA, and flagella [1, 2]. Other pattern recognition molecules include C-type lectin receptors (CLRs), caspase-recruitment domain (CARD), and nucleotide-binding domain (NOD) family members [3]. There are currently 13 known TLRs that recognize PAMPs derived from many of the most common human pathogens [4, 5]. The predetermined specificity of the TLR/PAMP warning system provides a very efficient mechanism for host notification of an invading pathogen but performs this function principally by promoting inflammation and is incapable of directly stimulating antigen-specific immunity. Instead, TLR ligation signals the maturation of antigen-presenting cells (APCs) via upregulation of costimulatory molecules including CD80 and CD86, production of inflammatory cytokines including interleukins-12 and -18, and migration of activated APCs to local lymphoid organs [6–8].
The limitation inherent to screening for PAMPs is that such a system requires a unique receptor for each PAMP. Likely as a mechanism to increase efficiency in this process, the immune system evolved an antigen-specific presentation system to screen not just for pathogen-specific patterns but also for individual peptide sequences that are specific to the pathogen in question. Such a candidate system would ideally have at least several of the following properties: (1) abundant expression in all cell types, (2) ability to bind a diverse array of proteins, (3) ability for specific detection by immune cells, and (4) ability to inform immune cells as to the identity of the pathogen in question. These requirements describe precisely the role of the major histocompatibility complex (MHC) in the evolution of adaptive immunity. Reviewed extensively elsewhere, the MHC consists of two complexes (MHC I and MHC II) which provide a division of labor for defense against intracellular (MHC I) and extracellular (MHC II) pathogens [9–11]. The expression patterns of these receptors follow the behavioral patterns of the pathogens to which they provide defense, with the MHC I molecules being ubiquitously expressed by all cells (save erythrocytes) and the MHC II molecules being restricted to cells capable of engulfing or directly binding extracellular pathogens such as dendritic cells, macrophages, and B cells. Together, the MHC molecules and TLR system provide an integrated, but parallel, system of antigen presentation and expression of costimulatory molecules which lead to the antigen-specific activation of adaptive immunity (exerted by T and B cells) in response to the dual presence of both “danger signals” and specific antigens presented by MHC I and/or MHC II.
Survival to reproductive age is threatened by not only invasion from foreign pathogens but also maladaptive mutations throughout development. Adaptation itself is a process afforded by the acquisition of individual mutations in the human genome which may lead to cellular progeny with differential fitness from the parental cell. For this to occur DNA replication must, by necessity, be an imperfect process. Taking into account all DNA proofreading mechanisms, the fidelity of eukaryotic DNA replication is estimated to be on the order of 10−10, which predicts that a cell will progressively and randomly acquire a single mutation every 1–2 cell divisions (in a human genome containing approximately 6.6 billion nucleotides) even in the absence of genotoxic stress and in proportion to the overall rate of cell division throughout development. Thus, the evolutionary trade-off for adaptation is the acquisition of mutations during development that may lead to the dysregulated growth and potentially transformation of otherwise normal cells into cancer cells. This is a process against which both cell-intrinsic and -extrinsic defense systems have developed; the extrinsic defense system is known today as cancer immunosurveillance [12, 13].
An immunological defense against transformed cells is fundamentally distinct from defense against exogenous pathogens in that the immune response must be initiated under the so-called sterile conditions for non-virus-associated malignancies. Sterile inflammation is detected through a distinct group of molecules known collectively as “damage-associated molecular patterns” (DAMPs) and their receptors [14, 15]. DAMPs include a range of endogenous molecules including heat-shock proteins, HMGB1, S100 proteins, as well as nucleic acids and extracellular matrix components [16]. In general, DAMPs are molecules that are released as a result of cell necrosis as occurs during conditions of extreme cellular stress or trauma. Many DAMPs are also recognized by the TLR system and are important for mediating inflammatory cytokine production in response to tissue damage that may contribute to recruitment of innate immune cells and wound healing [17–19]. Certain DAMPs, including HMGB1, may play critical roles in the efficacy of cancer chemotherapy and radiotherapy by generating inflammation within the tumor microenvironment via TLR and RAGE interactions [20]. Dysregulation of DAMP-mediated immune activation is also associated with a variety of pathological conditions including atherosclerosis, pseudogout, type 1 diabetes, and Alzheimer’s disease, which may represent the evolutionary trade-off for a DAMP-mediated sensor system to detect necrotic cell death.
In addition to TLRs, DAMPs can also interact with several other receptors, of which CD91 and CLEC9 are unique in bridging sterile inflammation to antigen cross-presentation [21–25]. CD91 and CLEC9 are both expressed by CD11c+ dendritic cells and in particular by the CD8α+ subset of dendritic cells that play a critical role in antigen cross-presentation [22, 25–27]. The ligand for CLEC9 was recently identified as F-actin [28, 29], and the ligands for CD91 include well-described members of the heat-shock protein family, which constitutes the oldest and most abundant class of protein in all mammalian cells [25, 30, 31]. Because the adaptive immune response is developmentally programmed to recognize foreign antigens [32], the existence of a linkage between sterile inflammation and adaptive immunity implies that certain antigens may arise in metabolically stressed “self” cells that are sufficiently nonself to engage the adaptive immune response and that such a pathway provides a survival advantage to the host at large. It has been suggested in the “neo-ligand” hypothesis that such a linkage is purely maladaptive and contributes only to autoimmunity [33]; however, the possibility that this pathway provides a survival advantage via tumor immunosurveillance must also be considered. This linkage may also be important for defense against the introduction of exogenous antigens during traumatic tissue damage; however, it is clear that a role of HSP/CD91 in this situation would be redundant with the PAMP/MHC system. Such redundancy may provide benefit in response to infection with pathogens that have developed mechanisms to evade (low-frequency CpG DNA by adenoviruses for example) or thwart (V-proteins by paramyxoviruses for example) innate immune activation by TLRs; however, this may not be the only benefit. The recent identification of antigen cross-presentation as a critical mechanism for tumor immunosurveillance supports a specialized role of the HSP/CD91 system in this process [26, 34].
Heat-shock proteins are an abundant family of intracellular proteins that collectively facilitate protein folding, trafficking, localization, and degradation [35–37]. The classification of this family of proteins as being related to “heat shock” dates to their accidental discovery as molecular mediators of cell stress, and the name has persisted despite the knowledge that their primary role is to chaperone protein folding and trafficking [38]. The ability of a relatively small number of HSP to function as protein chaperones for a large number of unique proteins expressed across all cell types requires that these HSP have unusual promiscuity in peptide binding specificity. This property has been confirmed by several groups, all seeking to identify the source of immunogenicity of different HSP. Most comprehensively shown for HSP gp96, efforts to identify specific HSP peptide-binding motifs have failed to elucidate a defined peptide profile based on amino acid content or peptide length that defines HSP binding capacity. In the specific case of gp96, nearly every peptide analyzed has been found in association with gp96 and the binding of these peptides has surprisingly high affinity, surviving SDS-PAGE and only weakly released by high temperature or high salt conditions in vitro [39]. The peptide binding promiscuity of HSP70 is slightly more limited than for gp96, being specific for aliphatic amino acid motifs and extremely sensitive to peptide release in the presence of ATP [31, 40]. This promiscuity in peptide binding is likely the source of evolutionary efficiency in APC adaptation to screen for extracellular HSP via CD91 as a sensor for necrotic cell death. CD91 is the endocytic receptor for all known heat-shock proteins, including HSP70, HSP90, gp96, and calreticulin [5, 9]. Among DAMP receptors, CD91 is also the primary endocytic receptor, which indicates that among DAMPs, HSPs are highly specialized adjuvants that can provide APCs with both a maturation signal (via TLRs) and a source of antigen via endocytosis of HSP/antigen complexes. The remarkable efficiency of HSP/peptide complex uptake by CD91 facilitates the induction of antigen-specific immunity at femto-molar concentrations of antigen, which represent physiologic concentrations [37, 41]. The evolution of HSP proteins as dual-purpose adjuvants may have taken place as a specific immunosurveillance mechanism in cancer, because linkage of adaptive immunity to sterile inflammation in diseases other than cancer is usually maladaptive.
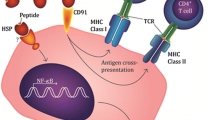
The combined adjuvant properties of APC activation via TLRs and antigen delivery via CD91 are what make HSP ideal candidates for vaccine development. The hypothesis that the dual adjuvant role of HSP evolved specifically as an antigen-cross-presenting mechanism for immunosurveillance against cancer arising under conditions of sterile inflammation remains to be experimentally confirmed; however, this evidence would heighten the validity of utilizing HSP as cancer immunotherapy. Such findings would provide an elegant circularity to the original description of HSP as the critical tumor rejection “antigens” (now understood to be HSP/antigen complexes) for sarcoma tumors in mice [42]. To date, 20 clinical trials have been conducted in the United States with HSP-based oncology vaccines. Of these, 13 utilized gp96-based approaches and 7 HSP70-based approaches. There are not yet any immunotherapy trials testing calreticulin, HSP90, or grp170 listed on clinicaltrials.gov. The major focus of the following sections is geared toward those approaches that have been studied in human patients. A schematic overview of the core attributes of autologous and allogeneic HSP vaccines is illustrated in Fig. 1, using gp96 as the archetypal HSP.

Schematic overview of the key characteristics of autologous and allogeneic HSP based vaccines in clinical development
2 Autologous Purified HSP Vaccines
The initial discovery of HSP gp96 as a “tumor rejection antigen” demonstrated that purified preparations of gp96 provided T cell-mediated protection against parental, but not unrelated, sarcoma tumors [42]. In these experiments, the immunogenic component within individual chemically induced murine sarcoma cell lysates was meticulously chased using several fractionation strategies into a fraction of glycoproteins of approximately 96 kDa molecular weight. Subsequent immunization of mice with these 96 kDa molecular weight proteins was protective against a subsequent challenge with the parental, but not unrelated, sarcoma cell lines. The proposed explanation for the immunogenicity of gp96 and the limited protection it provided only to parental tumor cells was that gp96 itself must be uniquely mutated in various sarcoma tumor cell lines. This hypothesis was quickly proven false, and the specific immunogenicity of gp96 was unequivocally demonstrated to be due to the client tumor peptides chaperoned by gp96 [24, 43]. Thus, the apparent restriction of therapeutic immunogenicity to parental but not unrelated tumors was assumed to be due to the unique antigenic “fingerprints” of individual sarcomas [44]. Because purified gp96 is assumed to remain bound to the full antigenic fingerprint of an individual sarcoma cell, the failure of one sarcoma preparation to protect against a challenge with a distinct sarcoma cell line predicted that the antigenic fingerprints of one were sufficiently distinct from another as to provide no benefit.
These observations provided the scientific basis for the first HSP-based vaccine trials in humans, which were performed using autologous preparations of gp96 isolated from surgical specimens from a small safety trial and subsequently a larger study in patients with advanced melanoma. The strategy used for these trials was similar to the initial murine studies, wherein individual patient tumor specimens were surgically collected, shipped, and processed at a centralized facility and then returned to the physician for re-administration of the purified tumor-derived gp96 preparation to the original patient. In the first human trial, performed in Germany, 16 patients with various tumor types were enrolled and treated postsurgically in the setting of residual disease [45]. This study demonstrated that the autologous gp96 vaccine was safe, induced an immune response in 50 % of patients tested (as measured by tumor antigen-specific CD8+ T cells), and produced interesting, albeit anecdotal, tumor responses in at least one patient with coincident hepatocellular and breast carcinoma.
This safety study set the stage for the next human trial, performed in Italy: 39 patients with stage IV melanoma were treated with at least one cycle (four vaccinations) of autologous gp96 starting 5–8 weeks after surgical resection of at least one lesion by intradermal or subcutaneous injection [46]. Patients who did not progress were eligible to continue on a second cycle of vaccinations and continue with monthly injections thereafter until progression or exhaustion of the autologous gp96 preparation. The vaccine was observed to be safe in all patients tested, and 10/21 evaluable patients demonstrated a positive and specific immune response to melanoma antigens by the ELISPOT assay [47]. Of the 28 patients with residual disease post surgery, there were 2 complete responses (CR) and another 3 patients with stable disease (SD) for varying intervals. Of the two patients with CRs, one responded extremely quickly to the vaccine, with resolution of lung metastasis after only the first cycle, and remained disease free for 24 months after vaccination. In the second patient the immune response took over a year to resolve the metastatic lesions, which extended well beyond the period of vaccination and led to a CR in excess of 38 months in duration. These two patients in the very early studies serve to highlight the variability in the time required for an immune response to manifest in patients, which is a phenomenon that is only now becoming accepted by clinicians and well highlighted by recent data with anti-CTLA-4 antibodies [48, 49]. In total, this initial trial demonstrated that autologous gp96 was safe, feasible in at least 60 % of patients enrolled, and warranted further testing in humans [46]. A separate phase I/II study performed in the United States on a similar population of patients with advanced melanoma obtained similar findings, including the intriguing, but unconfirmed, suggestion that patients fared better following treatment in the adjuvant setting [50].
In a subsequent phase II clinical trial, also conducted in Italy, autologous gp96 preparations were prepared from a similar population of patients with stage IV melanoma; however, the trial design was significantly modified. A total of 20 patients completed the first cycle of vaccinations and were evaluable in the study for immune response and survival. Once again, no safety issues were observed in any patients and only minor injection-site reactions including erythema and induration were common. In this study each weekly vaccination was performed together with GM-CSF injection and patients received two injections of interferon-alpha (IFNα) between vaccinations [51]. A greater number of patients achieved SD (11/20) than in the phase I study, and a single patient had a CR after the first cycle of vaccination. It remains unclear whether these responses were related to an increased immune response or to the combination therapy with GM-CSF and IFNα because as in the phase I study, approximately half of the patients (7/13) had a positive ELISPOT result. Interestingly, the patient achieving a CR had the lowest expression of the melanoma antigens MART1 and gp100, perhaps indicating that other antigens not highly expressed by the ELISPOT target cells contributed to the clinical response [51]. These data served to extend the safety database, immunological activity, and potential clinical benefit of autologous gp96 for the treatment of melanoma and facilitated testing in a controlled phase III clinical trial.
An international phase III trial of 322 patients with stage IV melanoma was subsequently conducted with autologous gp96 to determine overall survival compared to physicians’ choice [52]. Once again, the study design was revised significantly from the phase II study. In both prior studies, patients were pretreated with a combination of surgery + chemotherapy or radiotherapy and in some patients with IFNα or IL-2. In contrast to the phase II study, patients did not receive peri-vaccination treatment with GM-CSF or intermittent IFNα; however, the patient population was otherwise similar to the two prior studies. As was observed in the phase I and II studies, vaccination was feasible in just over 60 % of patients enrolled, with a significant number of patients not receiving treatment due to quality control failures in manufacturing. Unfortunately, this trial failed to demonstrate a benefit in overall survival for patients treated with autologous gp96 as compared to physicians’ choice. In a subset analysis, a trend toward increased overall survival was observed in all patient subsets depending on the number of vaccine doses administered to each patient. In this analysis, it was reported that patients with stage M1a and M1b disease who received at least ten doses of the vaccine demonstrated a survival benefit as compared to physicians’ choice [52]. Whether or not the failure of this trial was due to feasibility questions related to vaccine production and adequate supply of product to reach a therapeutic dose in a majority of patients remains unclear; however, such a conclusion is supported both by preclinical studies and the overall trends observed in this randomized phase III trial [44, 53]. In addition, it is worth noting that the trend toward increased survival also correlated with earlier stage disease, suggesting that vaccine therapy may be more effective early in the course of disease.
At the same time the first trials in melanoma were running, phase II and III trials were also conducted in patients with metastatic renal cell carcinoma. Renal cell carcinoma was chosen because, similar to melanoma, it was believed to be a relatively immunogenic tumor type that demonstrated intermittent responses to cytokine-based therapy and immunotherapy [54]. In the phase II study, 60 out of 84 enrolled patients were treated and evaluable, demonstrating an improvement in feasibility in this tumor type as compared to melanoma patients, potentially due to increased access to tumor tissue following nephrectomy. Out of these 60 patients, 2 CRs, 2 PRs, and 7 SDs were observed. This trial also included a single patient who developed severe complications that were potentially related to the vaccination. The remaining 59 patients experienced similar injection-site reactions to what was observed in the melanoma trials.
Despite the fact that this study concluded that autologous gp96 was “… relatively ineffective …” a large phase III study was subsequently performed in patients with metastatic renal cell carcinoma. As in melanoma, the design of this phase III trial was a significant departure from the phase II trial and was tested as adjuvant therapy to prevent disease recurrence in non-metastatic patients following nephrectomy [55]. A total of 318 patients were treated with autologous gp96, and both PFS and OS were compared to 367 patients in the observation-only control group. This trial was therefore the first of its kind to examine the efficacy of HSP vaccine therapy in a minimal-residual disease setting but unfortunately also missed its primary endpoint of reducing recurrence-free survival. A post hoc analysis suggested that patients with the earliest stage disease (AJCC stage I and II) may have enjoyed a delayed rate of recurrence; however, this conclusion requires further validation. As in the phase II in renal cell carcinoma, nearly 90 % of the patients randomized to autologous gp96 were able to receive the vaccine, demonstrating that feasibility was significantly improved as compared to melanoma.
In addition to melanoma and renal cell carcinoma, autologous preparations of gp96 have been tested in patients with colorectal and pancreatic cancer as well as non-Hodgkin’s lymphoma. A study including 29 patients with metastatic colorectal cancer treated with autologous gp96 in the adjuvant setting reported impressive increases in MHC I-restricted immune responses in the majority of patients treated [56]. The presence of a positive immune response detected by interferon-γ enzyme-linked immunospot (ELISPOT) assay was significantly correlated with both increased overall survival and increased progression-free survival. As in several previous studies, three different doses of gp96 were tested, with potentially the lowest dose (2.5 μg/injection) providing the most consistent immune response in patients. Another series of phase II trials in patients with non-Hodgkin’s lymphoma also demonstrated safety in all patients and vaccine production feasibility in the majority of patients but was not designed to determine survival benefit or immune response [57, 58]. A small, ten-patient, phase I study in patients with completely resected pancreatic adenocarcinoma treated in the adjuvant setting also demonstrated safety of the approach, with immune responses only in a minority of patients which did not correlate with disease-free survival [59].
To date, over 1,000 patients with multiple tumor types have been safely treated with autologous gp96 but without apparent clinical efficacy. These results in controlled clinical trials are certainly disappointing, but sprinkled throughout these failed trials are individual patients who were observed to have highly unusual “spontaneous” disease remission or subgroups of patients who in post hoc analysis appeared to enjoy a survival benefit. Definitive reasons for these failures are unknown; however, selection of two highly “immunogenic” tumors (melanoma and renal cell carcinoma) for testing in pivotal trials may have played a role [60]. The recent approval studies with anti-CTLA-4 antibodies (ipilimumab) in melanoma support the hypothesis that the most highly immunogenic tumors provide vaccination in situ, which predicts the immunoselection of tumor subclones that either display reduced amounts of critical antigens or contribute to local or systemic immunosuppression [12, 48, 49, 60]. The continued growth of tumors that provide vaccination in situ indicates that a tumor is progressing in spite of an ongoing immune response and that blocking immune regulatory mechanisms is a more critical first strike than attempting to broaden the scope of the immune response with a vaccine. Combinatorial strategies are in development for these tumor types wherein vaccination may play a secondary role to primary therapy with immune regulatory checkpoint inhibitors such as ipilimumab [61]. The overarching themes from these clinical trials also indicate that autologous gp96 is most effective in patients with earlier stage disease, who generate a positive immune response to the vaccine and for whom sufficient vaccine is produced to extend the treatment period well beyond the first four weekly injections. These predictions are generated from only two large, controlled, phase III clinical trials, and it is unfortunate that controlled studies were never run in phase II clinical trials because some of these concepts may have contributed to improved design of phase III clinical trials and been included in predefined endpoint criteria. An ongoing postsurgical adjuvant therapy trial in patients with >90 % resection of brain and central nervous system tumors (NCT00905060) appears poised to enter a pivotal phase III clinical trial and will hopefully incorporate some of these parameters in future trial design.
3 Allogeneic Cell-Based HSP Vaccines
The initial studies by Srivastava and colleagues clearly indicated that the repertoire of antigens bound to gp96 in purified preparations was sufficiently unique to the parental tumor that immunogenicity did not extend to genetically distinct tumor cell lines [42]. In the years since these initial discoveries, a great deal of progress has been made in understanding the specific nature of tumor antigens and in defining those which may or may not be “shared” by genetically distinct tumors. Two classes of tumor antigens have emerged from this work and are now defined as either “tumor-specific antigens” (TSA) or “abnormal self-antigens” (ASA, also referred to as tumor-associated antigens). TSA are those that arise as a direct result of randomly acquired genetic mutations in somatic genes that contribute as “drivers” or stowaway as “passengers” in the oncogenic process. The Cancer Genome Atlas (TCGA) has in recent years provided definitive evidence that dozens of TSA arise in every tumor type investigated and that at least a handful of those TSA appear to have the appropriate characteristics for binding to and presentation by MHC molecules [62–68]. These studies provide unequivocal evidence that except in very rare cases (such as kras in pancreatic adenocarcinoma), somatic mutations do not represent a source of shared antigens between patients with individual tumors. Instead, these studies provide clear evidence that ASA are the much more likely source of shared antigens between patients with related tumors due to common disruptions in core signaling pathways as a result of unique mutations in particular oncogenic “driver” genes [69, 70]. These somatic mutations lead to increases in gene copy number for a range of different proteins that lead to expression patterns not seen in non-transformed cells [71]. It is also clear that acquisition of mutations during oncogenesis leads to re-expression of primitive antigens typically only expressed in germline tissues and which have been broadly named “cancer testis antigens.” This group of antigens is widely understood to represent a source of commonly shared antigens between genetically distinct tumors [72–74]. In fact, the world’s first FDA-approved cancer vaccine is based upon the principle of antigen sharing between genetically distinct tumors and demonstrates that even a single shared ASA (prostatic acid phosphatase) can provide meaningful clinical efficacy [75]. At the same time, preclinical studies demonstrated that shared antigens between several established multiple myeloma cell lines could provide a basis for HSP gp96-mediated immunoprotection against genetically distinct tumors [76]. The antigenic underpinnings of these observations remain to be mechanistically elucidated; however, it is proposed that the spectrum of antigens from individual cell lines that are potentially shared with the antigens expressed by a patient tumor is increased by combining multiple cell lines into the vaccine preparation. Whether these observations reflect a unique antigenic property of myeloma or whether this phenomenon is generalizable to other tumor types also remains to be experimentally proven.
To date, clinical experience with allogeneic heat-shock protein vaccines is limited to a single approach based on a cell-secreted genetically engineered construct of gp96 [77]. This approach seeks to mimic the natural release of gp96 during necrotic cell death by replacing the KDEL endoplasmic reticulum retention sequence on the C-terminus of gp96 with a secretory molecule, in this case the hinge-CH2-CH3 domain from an IgG1 molecule to create a gp96-Ig fusion protein [78]. When transfected cell lines express and secrete gp96-Ig, it was found to chaperone peptides to the cross-presentation pathway similar to autologous gp96 and lead to CD8+ T cell-, NK cell-, and perforin-dependent antitumor immunity [41, 79–81]. Because this construct of gp96 was transfected into mammalian cells in sterile cell culture, required no purification steps, and provided CD8+ T cell-mediated antigen-specific immunity in vivo, this work finally laid to rest the long-standing criticism that HSP-mediated immune activation was simply a consequence of lipopolysaccharide contamination of autologous preparations. Further, preclinical studies demonstrated that immunization with cell-secreted gp96 led to an approximately tenfold increase in the magnitude of antigen-specific CD8+ T cell activation as compared to immunization with an equivalent quantity of cell-purified gp96 [80]. The reasons for this increase likely relate to increased half-life in vivo of a continuously secreted protein. Similar to autologous gp96, cell-secreted gp96-Ig has been shown to stimulate polyclonal and polyfunctional CD8+ T cell responses against all relevant antigens contained within the transfected cells [61, 82, 83].
A phase I clinical trial in patients with advanced non-small-cell lung cancer has examined the safety and immunogenicity of secreted gp96-Ig. NSCLC was selected as a tumor target for this approach because it represents a comparatively non-immunogenic tumor type as compared to melanoma and renal cell carcinoma and because 5-year survival for patients with NSCLC only increased from 14.2 to 18.0 % from 1975 to 2006, indicating that new treatment modalities are necessary [84]. The phase I study was conducted in a total of 18 patients with stage IIIB/IV NSCLC who had failed at least two prior therapies. The drug consisted of an adenocarcinoma cell line that secreted gp96-Ig and which was irradiated and frozen prior to administration to patients by intradermal injection. The cell line provided the source of shared NSCLC antigens for delivery by gp96-Ig and was selected on the basis of cancer/testis antigens that are shared between patients with NSCLC [85–87]. All patients had progressive disease at the time of study enrollment and were divided into three different dosing arms which varied on the basis of frequency of injection but not total dose of vaccine administered. This design was based on preclinical studies indicating that increased frequency of vaccination provided increased antitumor immunity and tumor regression [80, 81]. This study demonstrated that administration of cell-secreted gp96-Ig to patients was safe and stimulated a vaccine-specific immune response in 73 % of patients treated. An analysis of correlation between immune response and overall survival demonstrated a significant association between the two, with nonresponders surviving 4.5 months and responders an average of 16.5 months. These findings remain anecdotal but supported progression to phase II clinical trials which are currently ongoing. This phase II study (NCT01504542) includes a randomized placebo control group, which had not been included in any of the previous HSP trials at the phase II stage and may facilitate appropriate prospective endpoint design for a subsequent phase III study.
Additional clinical trials are needed to demonstrate whether allogeneic approaches with gp96 provide clinical benefit. Potential advantages of this approach relate to feasibility of vaccine production for all patients enrolled in the study. Because the product is identical for all patients and easily scalable, concerns over obtaining sufficient material for vaccine production, which limited feasibility in the phase III melanoma trial to just 60 % of enrolled patients, are significantly reduced. Potential disadvantages surround the issue of whether the antigens expressed by the selected cell line are shared between a sufficient proportion of the treated patient population; the success of a single-antigen vaccine somewhat reduces these concerns [75].
In comparison to other allogeneic cell-based vaccines, HSP constructs provide several distinct advantages. First, no other allogeneic cell-based approach in clinical testing facilitates the delivery of antigens specifically to APCs or to the antigen cross-presentation pathway. In all other cases, stimulation of adaptive immunity first requires destruction of the injected cells by an anti-allogeneic immune response. Killed vaccine cell fragments are then able to be phagocytozed by nearby macrophages, whereupon tumor antigens may be re-presented by those macrophages. In general, this is an antigen presentation pathway that is far more efficient for antigen presentation by MHC II than MHC I and therefore leads to the more potent activation of CD4+ T cells than CD8+ T cells. In addition, because antigens are not delivered to APCs specifically by an HSP, this pathway lacks the efficiency to stimulate CD8+ T cell responses at femto-molar concentrations of antigen as is the case with gp96 and other HSPs. Thus, success of a non-HSP-dependent allogeneic vaccine is predicted to increase the chances that an HSP-dependent approach will also succeed in the clinic.
4 Recombinant and Nucleic Acid-Based HSP Vaccines
The natural immunogenicity of HSP enables the design of recombinant proteins and subsequent loading of those recombinant HSP with antigens of interest. This approach alleviates the feasibility challenges associated with purification of autologous HSP preparations but inherits the efficacy challenges associated with selecting appropriate shared ASA to target. One approach to minimize the ASA-associated shortcoming of this approach is to target cancers with a known viral etiology and where viral antigens may form the foundation of the antitumor immune response. This combination has been examined clinically using a recombinant bacterial Hsp65 (from M. bovis) fused to the E7 protein of human papilloma virus 16 [88].
In the phase II clinical trial, a total of 58 women with cervical intraepithelial neoplasia III (CIN III) were treated with a series of three monthly vaccinations of Hsp65–E7 protein and subsequently monitored by colposcopy. A large proportion of patients enrolled in the trial experienced either a complete or a partial pathologic response to treatment (77.5 %); however, this association was not significantly associated with a history of HPV 16 infection. Because the antigenic nature of the vaccine is predicted to stimulate immunity to HPV 16 E7 antigen, it remains unclear how immunity would develop in patients without HPV 16 infection and in the absence of an appropriate control group no definitive determinations could be made. Nonetheless, this approach was extremely well tolerated and warrants additional testing in an appropriately controlled clinical setting to determine efficacy [88].
Yet another approach to utilize HSP to stimulate antitumor immunity involves the in vivo injection of recombinant DNA molecules encoding a particular HSP of interest. This strategy has been tested in a phase I clinical trial for 21 patients with advanced head and neck squamous cell carcinoma (HNSCC) in Brazil. Escalating doses of recombinant DNA hsp65 (M. bovis) were injected intratumorally to an accessible lesion every 3 weeks for a total of three injections [89]. This phase I study demonstrated that the approach was generally safe but associated with significant pain and edema in a number of patients. It was not possible to determine efficacy in this small, uncontrolled, study, and there was no association found between patient immune response to the hsp65 protein and overall survival [90].
A related strategy to direct intratumoral injection of HSP DNA sequences is to encode particular HSP within viral vectors and attempt to infect tumor cells in vivo with these HSP-expressing virus particles. This has been examined in a phase I clinical trial where a modified group C type 2 adenovirus was genetically engineered to express HSP70 and repeatedly injected in a dose escalation study to 27 patients with multiple advanced-stage solid tumors [91]. All evaluable patients developed an antibody response to the virus; however, no clear evidence of a cellular immune response was found. As in previous trials, anecdotal evidence of tumor response was observed in a minority of patients treated at the highest dose, but it remains unclear whether these responses were associated with the vaccine administration. The vaccine was safe in most patients, with a large number of patients developing fever and a single patient experiencing grade IV thrombocytopenia following treatment at the highest dose level.
5 Conclusions Based on Clinical Evidence
The initial rise in optimism surrounding the use of heat-shock proteins in cancer vaccines resulted from elegant preclinical studies demonstrating that heat-shock proteins are dual-purpose adjuvants that both chaperone the full antigenic repertoire of tumor cells to the cross-presentation pathway via scavenger receptors and simultaneously provide a maturation signal to the receiving APCs via TLR-2 and -4. The subsequent identification of antigen cross-presentation as a critical process for tumor immunosurveillance provided further support for the scientific validity of this approach [26]. This information, combined with an increased understanding of the molecular participants in “sterile” inflammation, helped to clarify that the name “heat-shock proteins” did not appropriately convey the true role of HSP as DAMPs, which in addition to functioning as protein chaperones provide a critical and potentially non-redundant linkage between sterile inflammation and adaptive immunity. Knowledge that this association is mostly maladaptive and contributes to diseases including atherosclerosis, type 1 diabetes, and Alzheimer’s raised the tantalizing possibility that either the linkage between HSP and adaptive immunity was accidental or this association evolved specifically as an immune defense against cellular transformation. Alas, the clinical evidence has clearly demonstrated that the initial wave of optimism was premature.
Incredible effort, expense, and faith on the part of scientists, drug developers, investors, oncologists, and patients have been expended on the development of HSP-based cancer vaccination. Large phase III trial failures in melanoma and renal cell carcinoma may have dampened support for what appeared to be promising early studies in colorectal carcinoma and have no doubt raised the level of skepticism that this approach will eventually lead to an FDA-approved cancer vaccine. Nonetheless, the sporadic and dramatic clinical responses observed in a minority of the patients treated on these trials preserve the belief that HSP-based vaccines will eventually stake their claim as important weapons in a growing immunotherapeutic toolbox available to oncologists in the near future.
These clinical trial results also provide important lessons for how future HSP-based vaccine trials should be designed. In the two largest phase III clinical trials to date, post hoc analysis clearly demonstrated that the dose and duration of vaccination had an important bearing on the clinical response observed in patients and that this clinical response was most apparent in patients with earlier stage disease. Second, the overall absence of placebo-controlled patient groups in phase II clinical trials has likely hampered the clinical success of HSP vaccines. Despite a large number of phase II clinical trials in large number of patients, effectively none of this data provided evidence of an efficacy signal because control groups were not included. This, coupled with the repeated shift in the target patient population between each stage in clinical trials, limited the ability of clinical trial personnel to appropriately select prospective clinical trial endpoints or appropriate patient populations. If controlled phase II studies had been performed in melanoma or renal cell carcinoma they may have enabled phase III designs to determine overall survival in stage M1a/b melanoma patients for whom at least ten doses of vaccine were available or to determine recurrence-free survival in AJCC stage I + II renal cell carcinoma patients. Either of these trials may have led to an FDA-approved HSP-based cancer vaccine and indicate potential strategies for success in future studies.
Ongoing trials may lead to the eventual approval of such a vaccine in the future. The recent approvals of Provenge for treatment of patients with advanced prostate cancer and ipilimumab for patients with advanced melanoma (and potentially with PD-1/PD-L1 blockade in the near future) have renewed enthusiasm in the immunotherapy of cancer [32, 37]. These successes buttress the groundswell of support from the basic scientific community that the immune system plays a dominant role as a cell-extrinsic defense system against cancer [67, 92]. The approval of a single-antigen vaccine also significantly increases the possibility that clinical efficacy of HSP vaccines will not be strictly limited to autologous approaches. If this is indeed the case, then allogeneic, recombinant protein or DNA-based approaches may eventually provide significant advantages in terms of manufacturing cost and scalability given the apparent importance of prolonged treatment for the induction of an effective antitumor immune response. The potential advantages of HSP-based vaccines from a mechanistic perspective provide a compelling rationale for further exploration of the approach. The link between heat-shock proteins and the adaptive immune system may have specifically evolved to provide immunosurveillance against cancer, and through that evolutionary process naturally developed all the core attributes we now understand to be critical for antitumor immunity: poly-antigen specificity, adjuvanticity at physiologic antigen concentrations, and specific stimulation of CD8+ T cell immunity by cross-priming.
References
Medzhitov R, Preston-Hurlburt P, Janeway Jr CA. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7.
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–83.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801.
Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–89.
Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80.
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52.
Reis e Sousa C. Dendritic cells as sensors of infection. Immunity. 2001;14:495–8.
Matis LA. The molecular basis of T-cell specificity. Annu Rev Immunol. 1990;8:65–82.
McDevitt HO. Discovering the role of the major histocompatibility complex in the immune response. Annu Rev Immunol. 2000;18:1–17.
Zinkernagel RM, Pircher HP, Ohashi P, Oehen S, Odermatt B, Mak T, Arnheiter H, Burki K, Hengartner H. T and B cell tolerance and responses to viral antigens in transgenic mice: implications for the pathogenesis of autoimmune versus immunopathological disease. Immunol Rev. 1991;122:133–71.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50.
Quintana FJ, Cohen IR. Heat shock proteins as endogenous adjuvants in sterile and septic inflammation. J Immunol. 2005;175:2777–82.
Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–42.
Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37.
Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343–51.
Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. 2012;13:321–4.
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, Andre F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9.
Belz GT, Vremec D, Febbraio M, Corcoran L, Shortman K, Carbone FR, Heath WR. CD36 is differentially expressed by CD8+ splenic dendritic cells but is not required for cross-presentation in vivo. J Immunol. 2002;168:6066–70.
Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, Rosewell I, Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903.
Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol. 2005;6:593–9.
Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–8.
Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. 2000;1:151–5.
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM. Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–100.
Sancho D, Mourao-Sa D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, Carlyle JR, Reis e Sousa C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest. 2008;118:2098–110.
Zhang JG, Czabotar PE, Policheni AN, Caminschi I, San Wan S, Kitsoulis S, Tullett KM, Robin AY, Brammananth R, van Delft MF, Lu J, O’Reilly LA, Josefsson EC, Kile BT, Chin WJ, Mintern JD, Olshina MA, Wong W, Baum J, Wright MD, Huang DC, Mohandas N, Coppel RL, Colman PM, Nicola NA, Shortman K, Lahoud MH. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity. 2012;36:646–57.
Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, Fletcher G, Durkin C, Postigo A, Skehel M, Batista F, Thompson B, Way M, Reis ESC, Schulz O. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. 2012;36:635–45.
Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–13.
Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425.
Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–401.
Beutler B. Neo-ligands for innate immune receptors and the etiology of sterile inflammatory disease. Immunol Rev. 2007;220:113–28.
Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha} + dendritic cells. J Exp Med. 2011;208:2005–16.
Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–77.
Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45.
Parsell DA, Lindquist S. The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet. 1993;27:437–96.
Ritossa F. Discovery of the heat shock response. Cell Stress Chaperones. 1996;1:97–8.
Blachere NE, Li Z, Chandawarkar RY, Suto R, Jaikaria NS, Basu S, Udono H, Srivastava PK. Heat shock protein-peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J Exp Med. 1997;186:1315–22.
Basu S, Srivastava PK. Heat shock proteins: the fountainhead of innate and adaptive immune responses. Cell Stress Chaperones. 2000;5:443–51.
Oizumi S, Strbo N, Pahwa S, Deyev V, Podack ER. Molecular and cellular requirements for enhanced antigen cross-presentation to CD8 cytotoxic T lymphocytes. J Immunol. 2007;179:2310–7.
Srivastava PK, DeLeo AB, Old LJ. Tumor rejection antigens of chemically induced sarcomas of inbred mice. Proc Natl Acad Sci U S A. 1986;83:3407–11.
Ishii T, Udono H, Yamano T, Ohta H, Uenaka A, Ono T, Hizuta A, Tanaka N, Srivastava PK, Nakayama E. Isolation of MHC class I-restricted tumor antigen peptide and its precursors associated with heat shock proteins hsp70, hsp90, and gp96. J Immunol. 1999;162:1303–9.
Tamura Y, Peng P, Liu K, Daou M, Srivastava PK. Immunotherapy of tumors with autologous tumor-derived heat shock protein preparations. Science. 1997;278:117–20.
Janetzki S, Palla D, Rosenhauer V, Lochs H, Lewis JJ, Srivastava PK. Immunization of cancer patients with autologous cancer-derived heat shock protein gp96 preparations: a pilot study. Int J Cancer. 2000;88:232–8.
Belli F, Testori A, Rivoltini L, Maio M, Andreola G, Sertoli MR, Gallino G, Piris A, Cattelan A, Lazzari I, Carrabba M, Scita G, Santantonio C, Pilla L, Tragni G, Lombardo C, Arienti F, Marchiano A, Queirolo P, Bertolini F, Cova A, Lamaj E, Ascani L, Camerini R, Corsi M, Cascinelli N, Lewis JJ, Srivastava P, Parmiani G. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: clinical and immunologic findings. J Clin Oncol. 2002;20:4169–80.
Rivoltini L, Castelli C, Carrabba M, Mazzaferro V, Pilla L, Huber V, Coppa J, Gallino G, Scheibenbogen C, Squarcina P, Cova A, Camerini R, Lewis JJ, Srivastava PK, Parmiani G. Human tumor-derived heat shock protein 96 mediates in vitro activation and in vivo expansion of melanoma- and colon carcinoma-specific T cells. J Immunol. 2003;171:3467–74.
Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, Macrae S, Nelson M, Canning C, Lowy I, Korman A, Lautz D, Russell S, Jaklitsch MT, Ramaiya N, Chen TC, Neuberg D, Allison JP, Mihm MC, Dranoff G. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008;105:3005–10.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl J Med. 2010;363:711–23.
Eton O, Ross MI, East MJ, Mansfield PF, Papadopoulos N, Ellerhorst JA, Bedikian AY, Lee JE. Autologous tumor-derived heat-shock protein peptide complex-96 (HSPPC-96) in patients with metastatic melanoma. J Transl Med. 2010;8:9.
Pilla L, Patuzzo R, Rivoltini L, Maio M, Pennacchioli E, Lamaj E, Maurichi A, Massarut S, Marchiano A, Santantonio C, Tosi D, Arienti F, Cova A, Sovena G, Piris A, Nonaka D, Bersani I, Di Florio A, Luigi M, Srivastava PK, Hoos A, Santinami M, Parmiani G. A phase II trial of vaccination with autologous, tumor-derived heat-shock protein peptide complexes Gp96, in combination with GM-CSF and interferon-alpha in metastatic melanoma patients. Cancer Immunol Immunother. 2006;55:958–68.
Testori A, Richards J, Whitman E, Mann GB, Lutzky J, Camacho L, Parmiani G, Tosti G, Kirkwood JM, Hoos A, Yuh L, Gupta R, Srivastava PK, C. S. Group. Phase III comparison of vitespen, an autologous tumor-derived heat shock protein gp96 peptide complex vaccine, with physician’s choice of treatment for stage IV melanoma: the C-100-21 Study Group. J Clin Oncol. 2008;26:955–62.
Yedavelli SP, Guo L, Daou ME, Srivastava PK, Mittelman A, Tiwari RK. Preventive and therapeutic effect of tumor derived heat shock protein, gp96, in an experimental prostate cancer model. Int J Mol Med. 1999;4:243–8.
Jonasch E, Wood C, Tamboli P, Pagliaro LC, Tu SM, Kim J, Srivastava P, Perez C, Isakov L, Tannir N. Vaccination of metastatic renal cell carcinoma patients with autologous tumour-derived vitespen vaccine: clinical findings. Br J Cancer. 2008;98:1336–41.
Wood C, Srivastava P, Bukowski R, Lacombe L, Gorelov AI, Gorelov S, Mulders P, Zielinski H, Hoos A, Teofilovici F, Isakov L, Flanigan R, Figlin R, Gupta R, Escudier B, C. R. S. Group. An adjuvant autologous therapeutic vaccine (HSPPC-96; vitespen) versus observation alone for patients at high risk of recurrence after nephrectomy for renal cell carcinoma: a multicentre, open-label, randomised phase III trial. Lancet. 2008;372:145–54.
Mazzaferro V, Coppa J, Carrabba MG, Rivoltini L, Schiavo M, Regalia E, Mariani L, Camerini T, Marchiano A, Andreola S, Camerini R, Corsi M, Lewis JJ, Srivastava PK, Parmiani G. Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin Cancer Res. 2003;9:3235–45.
Younes A. A phase II study of heat shock protein-peptide complex-96 vaccine therapy in patients with indolent non-Hodgkin’s lymphoma. Clin Lymphoma. 2003;4:183–5.
Oki Y, McLaughlin P, Fayad LE, Pro B, Mansfield PF, Clayman GL, Medeiros LJ, Kwak LW, Srivastava PK, Younes A. Experience with heat shock protein-peptide complex 96 vaccine therapy in patients with indolent non-Hodgkin lymphoma. Cancer. 2007;109:77–83.
Maki RG, Livingston PO, Lewis JJ, Janetzki S, Klimstra D, Desantis D, Srivastava PK, Brennan MF. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Dig Dis Sci. 2007;52:1964–72.
Schreiber TH, Raez L, Rosenblatt JD, Podack ER. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. Semin Immunol. 2010;22:105–12.
Ascierto PA, Grimaldi AM, Curti B, Faries MB, Ferrone S, Flaherty K, Fox BA, Gajewski TF, Gershenwald JE, Gogas H, Grossmann K, Hauschild A, Hodi FS, Kefford R, Kirkwood JM, Leachmann S, Maio M, Marais R, Palmieri G, Morton DL, Ribas A, Stroncek DF, Stewart R, Wang E, Mozzillo N, Marincola FM. Future perspectives in melanoma research. Meeting report from the“Melanoma research: a bridge from Naples to the World. Napoli, December 5th–6th2011”. J Transl Med. 2012;10:83.
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, Nikolsky Y, Hartigan J, Smith DR, Gerhard DS, Fults DW, VandenBerg S, Berger MS, Marie SK, Shinjo SM, Clara C, Phillips PC, Minturn JE, Biegel JA, Judkins AR, Resnick AC, Storm PB, Curran T, He Y, Rasheed BA, Friedman HS, Keir ST, McLendon R, Northcott PA, Taylor MD, Burger PC, Riggins GJ, Karchin R, Parmigiani G, Bigner DD, Yan H, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–9.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, Calhoun ES, Kamiyama M, Walter K, Nikolskaya T, Nikolsky Y, Hartigan J, Smith DR, Hidalgo M, Leach SD, Klein AP, Jaffee EM, Goggins M, Maitra A, Iacobuzio-Donahue C, Eshleman JR, Kern SE, Hruban RH, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz Jr LA, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12.
Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, Allison JP. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008;68:889–92.
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C, Onofrio RC, Parkin M, Romkes M, Weissfeld JL, Seethala RR, Wang L, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Winckler W, Ardlie K, Gabriel SB, Meyerson M, Lander ES, Getz G, Golub TR, Garraway LA, Grandis JR. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60.
Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, Baker A, Bergsagel PL, Bernstein BE, Drier Y, Fonseca R, Gabriel SB, Hofmeister CC, Jagannath S, Jakubowiak AJ, Krishnan A, Levy J, Liefeld T, Lonial S, Mahan S, Mfuko B, Monti S, Perkins LM, Onofrio R, Pugh TJ, Rajkumar SV, Ramos AH, Siegel DS, Sivachenko A, Stewart AK, Trudel S, Vij R, Voet D, Winckler W, Zimmerman T, Carpten J, Trent J, Hahn WC, Garraway LA, Meyerson M, Lander ES, Getz G, Golub TR. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–72.
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II B, Ozenberger PJ, Good AC, Chang DG, Beer MA, Watson M, Ladanyi S, Broderick A, Yoshizawa WD, Travis W, Pao MA, Province GM, Weinstock HE, Varmus SB, Gabriel ES, Lander RA, Gibbs MM, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75.
Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905.
Van Der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van Den Eynde BJ, Brasseur F, Boon T. Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev. 2002;188:51–64.
Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–93.
Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–25.
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Engl J Med. 2010;363:411–22.
Qian J, Hong S, Wang S, Zhang L, Sun L, Wang M, Yang J, Kwak LW, Hou J, Yi Q. Myeloma cell line-derived, pooled heat shock proteins as a universal vaccine for immunotherapy of multiple myeloma. Blood. 2009;114:3880–9.
Podack ER, Raez LE. Allogeneic tumor-cell-based vaccines secreting endoplasmic reticulum chaperone gp96. Expet Opin Biol Ther. 2007;7:1679–88.
Yamazaki K, Nguyen T, Podack ER. Cutting edge: tumor secreted heat shock-fusion protein elicits CD8 cells for rejection. J Immunol. 1999;163:5178–82.
Strbo N, Oizumi S, Sotosek-Tokmadzic V, Podack ER. Perforin is required for innate and adaptive immunity induced by heat shock protein gp96. Immunity. 2003;18:381–90.
Oizumi S, Deyev V, Yamazaki K, Schreiber T, Strbo N, Rosenblatt J, Podack ER. Surmounting tumor-induced immune suppression by frequent vaccination or immunization in the absence of B cells. J Immunother. 2008;31:394–401.
Schreiber TH, Deyev VV, Rosenblatt JD, Podack ER. Tumor-induced suppression of CTL expansion and subjugation by gp96-Ig vaccination. Cancer Res. 2009;69:2026–33.
Strbo N, Pahwa S, Kolber MA, Gonzalez L, Fisher E, Podack ER. Cell-secreted Gp96-Ig-peptide complexes induce lamina propria and intraepithelial CD8+ cytotoxic T lymphocytes in the intestinal mucosa. Mucosal Immunol. 2010;3:182–92.
Strbo N, Vaccari M, Pahwa S, Kolber MA, Fisher E, Gonzalez L, Doster MN, Hryniewicz A, Felber BK, Pavlakis GN, Franchini G, Podack ER. Gp96 SIV Ig immunization induces potent polyepitope specific, multifunctional memory responses in rectal and vaginal mucosa. Vaccine. 2011;29:2619–25.
Raez LE, Rosenblatt JD, Podack ER. Present and future of lung cancer vaccines. Expet Opin Emerg Drug. 2006;11:445–59.
Peled N, Oton AB, Hirsch FR, Bunn P. MAGE A3 antigen-specific cancer immunotherapeutic. Immunotherapy. 2009;1:19–25.
Lethe B, Lucas S, Michaux L, De Smet C, Godelaine D, Serrano A, De Plaen E, Boon T. LAGE-1, a new gene with tumor specificity. Int J Cancer. 1998;76:903–8.
Nakagawa K, Noguchi Y, Uenaka A, Sato S, Okumura H, Tanaka M, Shimono M, Ali Eldib AM, Ono T, Ohara N, Yoshino T, Yamashita K, Tsunoda T, Aoe M, Shimizu N, Nakayama E. XAGE-1 expression in non-small cell lung cancer and antibody response in patients. Clin Cancer Res. 2005;11:5496–503.
Einstein MH, Kadish AS, Burk RD, Kim MY, Wadler S, Streicher H, Goldberg GL, Runowicz CD. Heat shock fusion protein-based immunotherapy for treatment of cervical intraepithelial neoplasia III. Gynecol Oncol. 2007;106:453–60.
Michaluart P, Abdallah KA, Lima FD, Smith R, Moyses RA, Coelho V, Victora GD, Socorro-Silva A, Volsi EC, Zarate-Blades CR, Ferraz AR, Barreto AK, Chammas MC, Gomes R, Gebrim E, Arakawa-Sugueno L, Fernandes KP, Lotufo PA, Cardoso MR, Kalil J, Silva CL. Phase I trial of DNA-hsp65 immunotherapy for advanced squamous cell carcinoma of the head and neck. Cancer Gene Ther. 2008;15:676–84.
Victora GD, Socorro-Silva A, Volsi EC, Abdallah K, Lima FD, Smith RB, Moyses RA, Zarate-Blades CR, Michaluart P, Silva CL, Kalil J, Coelho V. Immune response to vaccination with DNA-Hsp65 in a phase I clinical trial with head and neck cancer patients. Cancer Gene Ther. 2009;16:598–608.
Li JL, Liu HL, Zhang XR, Xu JP, Hu WK, Liang M, Chen SY, Hu F, Chu DT. A phase I trial of intratumoral administration of recombinant oncolytic adenovirus overexpressing HSP70 in advanced solid tumor patients. Gene Ther. 2009;16:376–82.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Schreiber, T.H. (2014). Heat-Shock Protein-Based Cancer Immunotherapy. In: Rosenblatt, J., Podack, E., Barber, G., Ochoa, A. (eds) Advances in Tumor Immunology and Immunotherapy. Current Cancer Research. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8809-5_3
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8809-5_3
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8808-8
Online ISBN: 978-1-4614-8809-5
eBook Packages: MedicineMedicine (R0)