
Abstract
Approximately 20% of choledochal cysts (CC) present in adult patients and they are commonly associated with a high risk of complications, including malignancy. Additionally, children who underwent internal drainage procedures for CCs can develop complications during adulthood despite treatment. Concepts regarding classification and pathogenesis of the CCs have been evolving. While new subtypes are being added to the widely accepted Todani classification system, simplified classification schemes have also been proposed to guide appropriate management. The exact etiology of CCs is currently unknown. The two leading theories involve either the presence of an anomalous pancreatico-biliary junction with associated reflux of pancreatic juice into the biliary system or, more recently, some form of antenatal biliary obstruction with resulting proximal bile duct dilation. Imaging studies play an important role in the initial diagnosis, surgical planning, and long-term surveillance of CCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Choledochal cysts (CCs) are the congenital anomalies that present as abnormal cystic dilations of the intra and/or extra hepatic bile ducts and comprise of 1% of all benign biliary disorders. In the western world, the incidence of CCs is approximately 1 in 100,000–150,000 live births and is usually diagnosed in childhood; however, about 20% of patients present in adulthood [1]. Given the high risk of complications associated with adult CCs, including the development of cholangiocarcinoma, early diagnosis and treatment is very important. New subtypes of CCs have been added to the widely accepted classification system for CCs, the modified Todani classification [2]. The presence of an anomalous pancreatico-biliary junction with associated reflux of pancreatic juices into the biliary tract is the classically described concept for the etiopathogenesis for CCs though new theories are being proposed to explain pathogenesis of all subtypes [2]. Imaging plays an important role in the diagnosis, characterization, surgical planning, detection of complications, and follow-up.
In this article, we will review the current knowledge regarding the classification and etiopathogenesis of adult CCs; discuss multimodality imaging findings, and the role of imaging studies in patient management.
Choledochal cysts: adult vs. pediatric patients
When compared to pediatric patients, adults with CCs differ in clinical presentation, pathologic findings, and underlying abnormalities of pancreatico-biliary junction (PBJ), associated complications, and prognosis [3]. While children present with jaundice and/or abdominal mass and commonly have higher association with PBJ anomalies, adults commonly have abdominal pain and lower incidence of PBJ anomalies. Adult patients also show increased female sex predilection compared to children [3, 4]. In addition, adults show an increased risk of bile duct stones and a significantly higher risk of developing cholangiocarcinoma (15%–20% in adults vs. 0.7% in pediatric patients) [3, 4]. It is important to know the significant differences between adults and children with CCs and manage accordingly.
Classification of choledochal cysts: current status
The most widely used classification system for CCs is the modified Todani system, which classifies CCs into categories I–V. Type I CCs (solitary extrahepatic cyst) are the most common type comprising approximately 50%–80% and are subdivided into 3 subtypes: Type IA CCs are the most common and are characterized by cystic dilation of the extrahepatic common duct; type IB CCs demonstrate focal, segmental dilation of the extrahepatic common duct and type IC CCs are characterized by smooth, fusiform dilation of the common bile duct (CBD) extending into the common hepatic duct (CHD) (Fig. 1) [2, 5]. Type II CCs are seen in about 2%–3% patients and are defined as discrete diverticula of the CBD and usually projects off right lateral side (Fig. 2) [6]. Type III CCs (choledochoceles) comprise only 4%–6% of all reported cases and represent cystic dilatation of intramural segment of the distal CBD protruding into the duodenal lumen (Fig. 3) [2, 5]. Ziegler et al. have suggested that choledochoceles should not be included in CCs classification due to their unique duodenal histology, location, and associations [7]. Type IV CCs are the second most common type, comprising 15-35% of all cysts and can be further subdivided into two subtypes based on their involvement of the intrahepatic and/or extrahepatic biliary ducts [5]. While type IVA CCs are characterized by multiple cystic dilations of the both intrahepatic and extrahepatic bile ducts, type IVB cysts refer to multiple dilations of the extrahepatic common duct (Figs. 4, 5) [1]. It has been shown that preoperative imaging is unable to accurately predict true intrahepatic involvement in CCs, thus, it may be better to wait to make the distinction between type I and IVA cysts until after the cyst has been excised and postoperative imaging can then be used to determine which patients have true intrahepatic involvement [8]. Also known as Caroli disease or communicating cavernous ectasia, type V CCs are characterized by multifocal saccular dilations of the intrahepatic bile ducts (Fig. 6) [2, 5]. If there is associated congenital hepatic fibrosis, this condition is termed Caroli syndrome, which occurs due to ductal plate malformations [9].

Type I choledochal cyst. Drawing (A) and coronal T2-weighted, unenhanced MR image (B) demonstrate cystic dilatation of the extrahepatic common duct (arrows) consistent with type I choledochal cyst as per the modified Todani classification system.

Type II choledochal cyst. Drawing (A) and coronal T2-weighted, unenhanced MR image (B) demonstrate a discrete diverticulum projecting off right lateral side of the common bile duct (arrow) consistent with type II choledochal cyst.

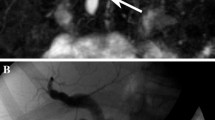
Type III choledochal cyst. Drawing (A) and ERCP image (B) demonstrate cystic dilation of intraduodenal segment of the distal common bile duct (arrow) consistent with type III choledochal cyst.

Type IVA choledochal cyst. Drawing (A) and MRCP image (B) demonstrate multiple cystic dilations of the both intra and extrahepatic bile ducts (arrows) consistent with type IVA choledochal cyst B.

Type IVB choledochal cyst. Drawing (A) and coronal T2-weighted, unenhanced MR image (B) demonstrate multiple cystic dilations of the extrahepatic bile ducts (arrows) consistent with type IVB choledochal cyst.

Type V choledochal cyst (Caroli disease). Drawing (A) and MRCP image (B) demonstrate multifocal, saccular dilations of the intrahepatic bile ducts only (arrows) consistent with Caroli disease.
Recently, there has been significant discussion regarding advantages and disadvantages of classifying CCs in a complex, alphanumerical system, especially since several additional subtypes have been proposed to the Todani system. Type ID and type VI CCs are the newly proposed additions to the Todani system. Type ID is characterized by dilation of the cystic duct in addition to dilated CBD and CHD (type I) resulting in a bicornal configuration of the cyst (Fig. 7) [10, 11]. Type VI CC is manifested as an isolated dilation of the cystic duct without CBD or CHD involvement; this is extremely rare with only few reported cases [12, 13]. “Forme fruste” variant of CCs is another entity, where there is anomalous PBJ with minimal to no dilation of the biliary system; this allows pancreaticobiliary reflux resulting in recurrent pancreatitis and increased risk of gallbladder cancer [14]. Given the increasing complexity of alphanumeric system, surgeons are moving toward a simplified classification scheme of CCs that is more directly relevant to guide management. In this regard, Visser et al. have recently challenged the Todani’s system, arguing that it encompassed several loosely related disease entities with differing etiologies, natural courses, surgical options, and complication profiles [15]. They proposed that type I and type IVA cysts are simply variations of the same disease, citing that in their experience all type I cysts had some element of intrahepatic involvement. Furthermore, they argue that type II cysts are just diverticula of the CBD, more closely resembling gallbladder duplication than true CCs and choledochoceles should be thought of as variants of duodenal duplication cysts. Although Caroli disease resembles CCs morphologically, it has an unrelated etiopathogenesis. Visser et al. advocate that the term “congenital choledochal cyst” be used exclusively for describing congenital dilation of the extrahepatic and intrahepatic bile ducts (apart from Caroli disease) and that CBD diverticula, choledochoceles, and Caroli disease should no longer be thought of as subtypes of CCs [15]. Another simplified classification, which is predominantly based on management approaches, categorizes CCs into intrahepatic cysts, extrahepatic cysts, and intraduodenal cysts [16].

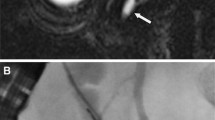
Type ID choledochal cyst. ERCP image demonstrates dilation of the cystic duct (arrow) in addition to the dilated extrahepatic common duct giving to bicornal configuration suggestive of Type ID choledochal cyst.
Pathogenesis of choledochal cysts: evolving concepts
Babbitt’s theory of the ‘common channel’ is the widely accepted theory regarding pathogenesis of CCs; according to this, CCs were thought to develop as a result of an abnormal pancreatico-biliary junction outside the duodenal wall, resulting in an abnormally long common channel between the ampulla of Vater and the insertion of the pancreatic duct on the CBD, predisposing to pancreatico-biliary reflux. In Babbitt’s theory, activation of pancreatic enzymes within bile ducts results in inflammation and weakening of the duct walls leading to subsequent dilation [17]. As there is no sphincter muscle around the origin of the common channel, there is free communication between pancreatic juice and bile. Higher hydrostatic pressure within the pancreatic duct when compared to bile duct will results in the development of pancreatico-biliary reflux [17, 18]. The presence of high amylase levels within the dilated bile ducts has been associated with increased risk of biliary dysplasia and subsequent malignancy, favoring Babbitt’s theory; however, some authors have questioned this concept as anomalous PBJ is only identified in 50%–80% patients with CCs. CCs have also been detected antenatally (about 15% cases) and in infants less than 2 months old, who yet to have pancreatic enzyme activation in their bile aspirate [19, 20]. These findings suggest that the cysts are, at least in part, congenital.
Although the exact etiology and pathogenesis of CCs is still unclear, most theories share the common idea that some form of distal biliary obstruction during fetal life results in high intraductal pressure, surpassing the yield strength of the bile duct, leading to CC formation [2]. Davenport et al. had identified that infantile CCs typically contain fewer neurons and ganglions within the dilated portions of the bile ducts, which results in functional obstruction and proximal dilation similar to Hirschsprung disease [21]. Singham et al. suggested that embryonic over proliferation of epithelial cells within the solid bile ducts during fetal life may result in biliary dilation [1]. Transient dysfunction of sphincter of Oddi intranatally could also result in CCs development [22]. However, it is important to note that the Babbitt and Makin-Davenport theories are not mutually exclusive and some level of both congenital obstruction and pancreatobiliary reflux may be involved.
Another interesting theory regarding pathogenesis of types I and IVA CCs is from Tadokoro et al., who proposed that these entities are congenital anomalies of the pancreas along with anomalous PBJ without biliary dilation (‘Form fruste’ CC) [23]. Types I and IVA CCs may develop due to persistence of left ventral pancreatic anlage and disrupted recanalization of the CBD with subsequent delayed recanalization of intra and extrahepatic bile ducts [23, 24]. Evidence in favor of this concept includes the presence of redundant pancreatic tissue in the head of the pancreas, abnormal shape of the pancreatic head, and abnormal anatomical location of the major duodenal papilla in majority of patients with types I and IVA CCs [24].
Choledochal cysts: role of imaging
Ultrasound (US), multi-detector row computed tomography (MDCT), magnetic resonance imaging (MRI), and magnetic resonance cholangiopancreatography (MRCP), hepatobiliary scintigraphy using technetium-99 iminodiacetic acid (HIDA), and endoscopic retrograde cholangio-pancreaticography (ERCP) are the commonly used imaging techniques in the diagnosis and management of adults with CCs. US is often the initial diagnostic modality that identifies CCs in adults with a sensitivity of 71%–97% [5]. On US, CCs appear as cystic masses in the right upper quadrant separate from the gallbladder with or without intrahepatic biliary dilation depending on the subtype; additionally, US is a preferred modality for long-term surveillance in postsurgical patients (Fig. 8) [5]. MDCT is very helpful in surgical planning, especially in the accurate delineation of extent of the dilated intrahepatic bile ducts prior to segmental lobectomy (Fig. 9). MDCT can also identify cyst wall thickening and intracystic masses that develop secondary to malignancy. In postsurgical patients, MDCT cholangiography (MDCTC) is useful in detecting abnormalities of the bilioenteric anastomosis [25]. HIDA scan helps in the assessment of the spontaneous rupture of the CCs and cyst continuity with adjacent bile ducts; prompt appearance of the radiotracer within the dilated extrahepatic bile ducts is the most common imaging appearance [26].

Color Doppler image of the right upper quadrant in a 37-year-old woman demonstrates a well-defined cystic lesion at the hepatic hilum. This was proven to be type I choledochal cyst on surgical resection.

Axial (A, B) contrast-enhanced CT images of the upper abdomen demonstrate dilated extra and intrahepatic bile ducts (arrows) consistent with type IVA choledochal cyst. CT is very helpful in delineation of extent of dilated intrahepatic ducts before surgery.
MRCP is the current ‘gold standard’ for visualizing CCs and has largely superseded the use CT and ERCP [19, 27]. Unlike MDCT, MRCP can delineate the exact pathologic anatomy, including anomalous PBJ, and detect ductal stones and cholangiocarcinoma (Fig. 10) [28]. ‘Central dot sign’ is the characteristic imaging appearance of Caroli disease on MRI; this feature is secondary to saccular dilation of intrahepatic ducts surrounding portal triad (Fig. 11) [28]. This appearance can also be identified on MDCT. Furthermore, MRI does not carry the risks of ERCP including cholangitis, duodenal perforation, hemorrhage, contrast allergy, biliary sepsis, and pancreatitis [29]. Sacher et al. have identified that MRCP has a 96%–100% detection rate for CCs, a 53%-100% detection rate for diagnosing anomalous PBJ, a 100% detection rate for choledocholithiasis, and a 87% detection rate for cholangiocarcinomas with concurrent CCs, making it the test of choice for pre-operatively evaluation [29]. MRCP is not, however, free of shortcomings. It has limited capacity to detect minor ductal anomalies and small choledochoceles, which may not only be secondary to technical limitations, but also from the lack of ductal distension attained during ERCP [30]. ERCP remains the most widely used diagnostic tool for identification of choledochoceles, with a reported diagnostic sensitivity of 97% (Figs. 3, 10) [31]. This may be in part due to the fact that it simultaneously allows for therapeutic sphincterotomy in these patients [2]. Additionally, ERCP is still considered the reference standard in select cases, where there are questionable findings on MRI. MRCP is also more susceptible to motion artifacts than ERCP; while breath-hold sequences and respiratory triggered scanning can help eliminate motion artifact and increase the fluid signal in the bile duct, sedation may still be required in select patients [19]. In circumstances where sedation is contraindicated or impossible, MDCTC after infusion of meglumine iodoxamate may be used, however, it exposes the patient to radiation [32].

Type I choledochal cyst associated with anomalous pancreatico-biliary junction (PBJ) A MRCP image demonstrates a large cyst arising from the common bile duct (arrows), associated with anomalous PBJ (white arrowhead). B Corresponding ERCP image shows a long common channel at PBJ (black arrowheads).

‘Central dot sign’ in Caroli disease. Axial contrast-enhanced MR image of the liver shows saccular dilations of the intrahepatic ducts surrounding portal triads (arrows) giving to ‘central dot sign’.
Complications
Approximately 60%–80% of adults with CCs can experience one or more of the following complications: biliary stones, biliary strictures, cholangiocarcinoma, ascending cholangitis, pancreatitis, secondary biliary cirrhosis, spontaneous cyst rupture, and rarely gastric outlet obstruction [27, 33]. Among all, formation of stones (cystolithiasis, choledocholithiasis, and hepaticolithiasis) and related inflammation and infections (calculus cholecystitis, cholangitis, intrahepatic abscess) are the common complications of CCs (Fig. 12) [27]. Obstruction and recurrent infection, especially that caused by CCs with intrahepatic involvement, can also lead to secondary biliary cirrhosis in 40%–50% [34]. CCs have also been associated with acute acalculous cholecystitis and spontaneous cyst rupture resulting in sepsis and peritonitis [35, 36]. However, the most serious complication of CCs in adults is the development of biliary tract malignancy.

Choledocholithiasis in type I choledochal cyst. Sagittal T2-weighted MR image shows a dilated common bile duct with multiple filling defects (arrows) consistent with choledocholithiasis
Unresected CCs have been reported to carry up to 30% risk of malignancy [6, 27]. Children with CCs under the age of 10 years carry a 0.7% risk; the risk of malignancy increases to 14.3% in adults over 20 years of age [37]. Patients with CCs develope malignancy earlier than general population [37]. The location of the cancer is most often in the extrahepatic bile duct (50%–62%), followed by gallbladder (38%–46%) and intrahepatic bile ducts (2.5%) [2]. Incidence of cholangiocarcinoma varies depending on the subtype of CC with type I carrying the highest risk (up to 70%), followed by type IV (20%); choledochocele has the least risk of malignancy (<2%) [5]. The risk of malignancy is not reduced in patients with prior cystoenterostomy and incomplete cyst excision [5, 38].
Pathological findings strongly suggest that there is a hyperplasia-dysplasia-carcinoma sequence in carcinogenesis in the biliary tract of the affected patients [27]. Pancreatic enzymes, amylase, bile stasis with bacterial overgrowth, increased levels of phospholipase A2, and increased concentration unconjugated bile acids have been implicated in the dysplastic proliferation of the bile duct epithelium [20, 39–41]. Many molecular factors have been implicated in the development of cholangiocarcinoma. From an everyday clinical standpoint, the most relevant may be the high level of the proliferation activating factor COX-2 found in the bile of patients with anomalous PBJ; this bile has been proven to promote the proliferation of human cholangiocarcinoma QBC939 cells via COX-2 pathway [42–44]. This suggests that COX-2 inhibitors may be effective chemoprevention of biliary carcinoma in patients with anomalous PBJ [42, 43]. K-ras mutations, microsatellite instability, expression of bcl-2, increased telomerase activity, abnormalities of cyclin D1, and p53 are the molecular events responsible for the development of malignancy [27]. MRI with MRCP is the most commonly used non-invasive imaging technique for the diagnosis of cholangiocarcinoma and gallbladder carcinoma; irregular wall thickening of the cyst wall or gallbladder wall, enhancing mass, and papillary nodules are the commonly identified MR findings to suggest biliary malignancy (Figs. 13, 14) [45].

Cholangiocarcinoma in type I choledochal cyst. Axial unenhanced T1- weighted MR image demonstrates irregular wall thickening of the cyst (arrows) concerning for malignancy, which was subsequently proved as cholangiocarcinoma on pathologic examination.

Cholangiocarcinoma in type IVA choledochal cyst. Axial gadolinium-enhanced T1-weighted MR image demonstrates an enhancing mass (arrow) in the common bile duct suggestive of cholangiocarcinoma, which was proven on subsequent surgical resection.
Management
Surgical resection, interventional therapy, and hepatic transplantation are the available treatment options for CCs and choice of management depends on the subtype and extent of biliary tract involvement [46]. Complete excision of the cysts with some form of biliary reconstruction has become the standard of care for most of the extrahepatic CCs. In cysts with intrahepatic involvement, including Caroli disease, segmental hepatectomy or liver transplantation is necessary [16]. For type I and IVB cysts, complete resection of cysts and Roux-en-Y hepaticojejunostomy is the procedure of choice [46]. For type II cysts, some surgeons prefer excision with choledochoduodenostomy, whereas others prefer simple cyst excision with T-tube drainage [47]. However, in patients with a type II cyst and anomalous PBJ, the gallbladder should be removed because of the high risk of gallbladder malignancy, stressing the importance of accurate preoperative imaging [48]. For type III cysts, ERCP helps in diagnosis as well as management; endoscopic sphincterotomy followed by long-term endoscopic surveillance is the management of choice in these patients [46]. For type IVA bile duct cysts a customized approach is needed, with a segmental hepatectomy and wide hilar hepaticojejunostomy for localized intrahepatic involvement and transplantation for symptomatic, diffuse intrahepatic involvement [2, 46, 49]. Similarly, type V cysts are treated with segmental resection for unilobar involvement and liver transplantation for diffuse bilobar involvements complicated with cholangitis and/or portal hypertension [50]. In poor surgical candidates with recurrent hepatolithiasis and cholangitis with type IVA and V cysts, prophylactic antibiotics with endoscopic or percutaneous lithotripsy and ursodeoxycholic acid can also be used palliatively [51]. Cholecystectomy, excision of the malformed ductal tissue with biliary reconstruction is the treatment of choice in patients with ‘form fruste’ CCs [52]. Irrespective of subtype, all postsurgical patients require permanent, meticulous, long-term surveillance given the risk of cholangiocarcinoma and anastomotic strictures involving bilioenteric anastomosis (Fig. 15) [2, 5].

Cholangiocarcinoma developing 9 years after surgical resection of type I choledochal cyst in a 44-year-old man. Axial contrast-enhanced CT image shows a large, heterogeneously enhancing mass involving the entire left hepatic lobe (arrows), which was diagnosed as cholangiocarcinoma on biopsy. This case shows the importance of long-term surveillance in patients with choledochal cysts even after curative surgery.
Conclusion
Adults with CCs differ from pediatric patients in terms of clinical presentation, management, prognosis, and long-term complications. As per the most widely accepted modified Todani classification, five types of CCs have been described; however, adding type I D and type VI has recently been proposed. Simplified classification systems for CCs are being developed to facilitate appropriate patient management. Anomalous PBJ with reflux of pancreatic enzymes into the bile duct resulting in chronic inflammation and subsequent dilatation and, more recently, antenatal biliary obstruction with resulting proximal bile duct dilation are the two most widely accepted concepts regarding the pathogenesis of CCs. Imaging studies are pivotal in the initial diagnosis, early detection of complications, treatment planning, and surveillance of CCs. Thus, the radiologist’s knowledge about evolving concepts in the pathogenesis and classification, multimodality imaging findings, and complications of adult CCs is vital for optimal patient care.
References
Singham J, Yoshida EM, Scudamore CH (2009) Choledochal cysts: part 1 of 3: classification and pathogenesis. Can J Surg 52(5):434–440
Jablonska B (2012) Biliary cysts: etiology, diagnosis and management. World J Gastroenterol 18(35):4801–4810
Huang CS, Huang CC, Chen DF (2010) Choledochal cysts: differences between pediatric and adult patients. J Gastroint Surg 14(7):1105–1110
de Vries JS, de Vries S, Aronson DC, et al. (2002) Choledochal cysts: age of presentation, symptoms, and late complications related to Todani’s classification. J Pediatr Surg 37(11):1568–1573
Bhavsar MS, Vora HB, Giriyappa VH (2012) Choledochal cysts : a review of literature. Saudi J Gastroenterol 18(4):230–236
Tadokoro H (2012) Recent advances in choledochal cysts. Open J Gastroenterol 02(04):145–154
Ziegler KM, Pitt HA, Zyromski NJ, et al. (2010) Choledochoceles: are they choledochal cysts? Ann Surg 252(4):683–690
Acker SN, Bruny JL, Narkewicz MR, et al. (2013) Preoperative imaging does not predict intrahepatic involvement in choledochal cysts. J Pediatr Surg 48(12):2378–2382
Levy AD, Rohrmann CA Jr (2003) Biliary cystic disease. Curr Problems Diagn Radiol 32(6):233–263
Michaelides M, Dimarelos V, Kostantinou D, et al. (2011) A new variant of Todani type I choledochal cyst. Imaging evaluation. Hippokratia 15(2):174–177
Yoon JH (2011) Magnetic resonance cholangiopancreatography diagnosis of choledochal cyst involving the cystic duct: report of three cases. Br J Radiol 84(997):e18–e22
Conway WC, Telian SH, Wasif N, Gagandeep S (2009) Type VI biliary cyst: report of a case. Surg Today 39(1):77–79
De U, Das S, Sarkar S (2011) Type VI choledochal cyst revisited. Singap Med J 52(5):e91–e93
Sarin YK, Sengar M, Puri AS (2005) Forme fruste choledochal cyst. Indian Pediatr 42(11):1153–1155
Visser BC, Suh I, Way LW, Kang SM: Congenital choledochal cysts in adults. Arch Surg 2004, 139(8):855–860; discussion 860–852
Martin RF (2014) Biliary cysts: a review and simplified classification scheme. Surg Clin N Am 94(2):219–232
Babbitt DP (1969) Congenital choledochal cysts: new etiological concept based on anomalous relationships of the common bile duct and pancreatic bulb. Ann Radiol 12(3):231–240
Sugiyama M, Haradome H, Takahara T, et al. (2004) Biliopancreatic reflux via anomalous pancreaticobiliary junction. Surgery 135(4):457–459
Makin E, Davenport M (2012) Understanding choledochal malformation. Arch Dis Child 97(1):69–72
Imazu M, Iwai N, Tokiwa K, Shimotake T, Kimura O, Ono S (2001) Factors of biliary carcinogenesis in choledochal cysts. Eur J Pediatr Surg 11(1):24–27
Davenport M, Basu R (2005) Under pressure: choledochal malformation manometry. J Pediatr Surg 40(2):331–335
Ponce J, Garrigues V, Sala T, Pertejo V, Berenguer J (1989) Endoscopic biliary manometry in patients with suspected sphincter of Oddi dysfunction and in patients with cystic dilatation of the bile ducts. Dig Dis Sci 34(3):367–371
Tadokoro H, Suyama M, Kubokawa Y, Sai JK (2003) Persistence of the left part of the ventral pancreas may cause congenital biliary dilatation. Pancreas 27(1):47–51
Tadokoro H, Takase M (2012) Recent advances in choledochal cysts. Open J Gastroenterol 2:145–154
Lam WW, Lam TP, Saing H, Chan FL, Chan KL (1999) MR cholangiography and CT cholangiography of pediatric patients with choledochal cysts. Am J Roentgenol 173(2):401–405
Camponovo E, Buck JL, Drane WE (1989) Scintigraphic features of choledochal cyst. J Nucl Med 30(5):622–628
Soreide K, Soreide JA (2007) Bile duct cyst as precursor to biliary tract cancer. Ann Surg Oncol 14(3):1200–1211
Lee HK, Park SJ, Yi BH, et al. (2009) Imaging features of adult choledochal cysts: a pictorial review. Korean J Radiol 10(1):71–80
Sacher VY, Davis JS, Sleeman D, Casillas J (2013) Role of magnetic resonance cholangiopancreatography in diagnosing choledochal cysts: case series and review. World J Radiol 5(8):304–312
Park DH, Kim MH, Lee SK, et al. (2005) Can MRCP replace the diagnostic role of ERCP for patients with choledochal cysts? Gastrointest Endosc 62(3):360–366
Law R, Topazian M (2014) Diagnosis and treatment of choledochoceles. Clin Gastroenterol Hepatol 12(2):196–203
Fumino S, Ono S, Kimura O, Deguchi E, Iwai N (2011) Diagnostic impact of computed tomography cholangiography and magnetic resonance cholangiopancreatography on pancreaticobiliary maljunction. J Pediatr Surg 46(7):1373–1378
Atkinson HD, Fischer CP, de Jong CH, et al. (2003) Choledochal cysts in adults and their complications. Int Hepatol Pancreatol Biliary Assoc 5(2):105–110
Singham J, Yoshida EM, Scudamore CH (2009) Choledochal cysts: part 2 of 3: diagnosis. Can J Surg 52(6):506–511
Kiresi DA, Karabacakoglu A, Dilsiz A, Karakose S (2005) Spontaneous rupture of choledochal cyst presenting in childhood. Turk J Pediatr 47(3):283–286
Stipsanelli E, Valsamaki P, Tsiouris S, et al. (2006) Spontaneous rupture of a type IVA choledochal cyst in a young adult during radiological imaging. World J Gastroenterol 12(6):982–986
Voyles CR, Smadja C, Shands WC, Blumgart LH (1983) Carcinoma in choledochal cysts. Age-related incidence. Arch Surg 118(8):986–988
Ohashi T, Wakai T, Kubota M, et al. (2013) Risk of subsequent biliary malignancy in patients undergoing cyst excision for congenital choledochal cysts. J Gastroenterol Hepatol 28(2):243–247
Jeong IH, Jung YS, Kim H, et al. (2005) Amylase level in extrahepatic bile duct in adult patients with choledochal cyst plus anomalous pancreatico-biliary ductal union. World J Gastroenterol 11(13):1965–1970
Shimada K, Yanagisawa J, Nakayama F (1991) Increased lysophosphatidylcholine and pancreatic enzyme content in bile of patients with anomalous pancreaticobiliary ductal junction. Hepatology 13(3):438–444
Reveille RM, Van Stiegmann G, Everson GT (1990) Increased secondary bile acids in a choledochal cyst. Possible role in biliary metaplasia and carcinoma. Gastroenterology 99(2):525–527
Tsuchida A, Itoi T, Aoki T, Koyanagi Y (2003) Carcinogenetic process in gallbladder mucosa with pancreaticobiliary maljunction (review). Oncol Rep 10(6):1693–1699
Tsuchida A, Nagakawa Y, Kasuya K, et al. (2003) Immunohistochemical analysis of cyclooxygenase-2 and vascular endothelial growth factor in pancreaticobiliary maljunction. Oncol Rep 10(2):339–343
Wu GS, Zou SQ, Luo XW, Wu JH, Liu ZR (2003) Proliferative activity of bile from congenital choledochal cyst patients. World J Gastroenterol 9(1):184–187
Liu QY, Lai DM, Gao M, et al. (2013) MRI manifestations of adult choledochal cysts associated with biliary malignancy: a report of ten cases. Abdom Imaging 38(5):1061–1070
Cerwenka H (2013) Bile duct cyst in adults: interventional treatment, resection, or transplantation? World J Gastroenterol 19(32):5207–5211
Ulas M, Polat E, Karaman K, et al. (2012) Management of choledochal cysts in adults: a retrospective analysis of 23 patients. Hepato-Gastroenterology 59(116):1155–1159
Lipsett P, Pitt H (2003) Surgical treatment of choledochal cysts. J Hepato Biliary Pancreat Surg 10:352–359
Ando K, Miyano T, Kohno S, Takamizawa S, Lane G (1998) Spontaneous perforation of choledochal cyst: a study of 13 cases. Eur J Pediatr Surg 8(1):23–25
Mabrut JY, Kianmanesh R, Nuzzo G, et al. (2013) Surgical management of congenital intrahepatic bile duct dilatation, Caroli’s disease and syndrome: long-term results of the French Association of Surgery Multicenter Study. Ann Surg 258(5):713–721 (discussion 721)
Singham J, Yoshida EM, Scudamore CH (2010) Choledochal cysts. Part 3 of 3: management. Can J Surg 53(1):51–56
Miyano G, Yamataka A, Shimotakahara A, et al. (2005) Cholecystectomy alone is inadequate for treating forme fruste choledochal cyst: evidence from a rare but important case report. Pediatr Surg Int 21(1):61–63
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Katabathina, V.S., Kapalczynski, W., Dasyam, A.K. et al. Adult choledochal cysts: current update on classification, pathogenesis, and cross-sectional imaging findings. Abdom Imaging 40, 1971–1981 (2015). https://doi.org/10.1007/s00261-014-0344-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00261-014-0344-1