
Abstract
The most important intakes of thermal waters within the Sudetic Geothermal Region occur in three separate hydrogeothermal systems: (1) Lądek, (2) Duszniki and (3) Cieplice. All these waters are of meteoric origin and circulate in crystalline rocks to different depths. Their outflow temperatures are between less than 20°C and to about 87°C. To evaluate the geothermal fields in the light of their prospectiveness, to further exploration of thermal energy resources, we took an effort to apply selected isotopic and chemical geothermometers to assess the maximum possible temperatures, which may be found in the reservoirs. The only chemical geothermometers which give a reliable range of reservoir temperatures are SiO2 (chalcedony), Na–Ka–Ca and partly Na–K ones. The oxygen isotopic geothermometer in the SO4–H2O system gives a real range of estimated reservoir temperatures only for deeply circulating waters in the Cieplice area. On the other hand, in the case of CO2 rich waters in the Duszniki area, where outflow temperatures do not exceed 30°C, application of chemical or isotopic temperature indicators always leads to erroneous results due to the lack of equilibrium in the thermodynamic system of water–rock interaction.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
A crucial point in the exploration of thermal water resources is to know the precise temperature at depth, i.e. the temperature of water in a particular geothermal reservoir. The most accurate temperature data are obtained by static bottom-hole temperature survey in exploratory or production well. However, this way of temperature surveying, although often inevitable, is the most expensive one due to the need of costly investment in exploratory drillings. The application of chemical and isotopic groundwater temperature indicators, the so-called geothermometers, allow reducing considerably the costs of geothermal exploration. During the initial phase of thermal water exploration, geothermometers are used to estimate subsurface temperatures expected to be encountered by drillings, using the chemical and isotopic composition of hot spring discharges. However, the majority of known geothermometers were developed and calibrated for high temperature geothermal systems, i.e. those with reservoir temperatures usually exceeding 150°C. Their application to low enthalpy geothermal systems, where reservoir temperatures are even much lower than 100°C, always raises controversies. Many problems connected with the applicability of geothermometers to low temperature systems, concern usually the incorrect understanding of thermodynamic environment for which the particular temperature indicator was developed. This leads to direct and literal application of geothermometric equations to different geothermal systems with no proper corrections.
This paper shows new and reconsidered results of application of selected chemical and isotopic geothermometers to low enthalpy hydrogeothermal systems in the Sudetes Mts, SW Poland. Up to date, a few papers concerning application of geothermometry to Sudetic thermal waters were published by Dowgiałło (1976, 2000, 2002), Leśniak and Nowak (1993), Dowgiałło et al. (2005) and Porowski and Dowgiallo (2008). The paper focuses on important aspects of geochemical data interpretation to show their crucial influence on the results of a geothermometric study.
General characteristics of geothermal systems in the Sudetes Mts
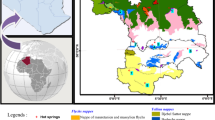
The Sudetic Geothermal Region includes the Sudetes Mts and the Foresudetic block, which is a tectonic step between the uplifted Sudetes Mts. and the lowered Foresudetic Monocline (Fig. 1). The Sudetes Mts consist mainly of Precambrian and early Paleozoic crystalline rocks covered with Paleozoic and Mesozoic sediments.

Geological sketch of the Sudetic Geothermal Region (after Bruszewska 2000)
The most important hydrogeothermal systems within the Sudetes Mts were recognized in the vicinity of thermal spas renown for centuries, namely: (1) Lądek, (2) Duszniki and (3) Cieplice (Fig. 1). Their detailed description can be found in Dowgiałło (1976, 2002).
Thermal water systems under consideration are linked to crystalline formations. At Cieplice, the host rock is fissured Carboniferous monzonitic granite, while at Duszniki and Lądek thermal water circulates in Proterozoic or early Palaeozoic schists and gneisses. The presence of hot springs is always connected with faults and accompanying fissure systems. Areas of faults crossing are particularly favorable to warm springs occurrence (Dowgiałło and Fistek 1995, 2003; Zuber et al. 1995).
In this study, 16 thermal water intakes were sampled for chemical and isotopic analyses during field survey in years 2004–2006. Generalized chemical and isotopic characteristics of thermal waters studied are presented in Tables 1 and 2.
The oxygen and hydrogen isotopic composition of the Sudetic thermal waters indicates their meteoric origin (Fig. 2). Moreover, the tritium content is evidence that modern meteoric water plays an important role in their recharge. Present day tritium content in precipitation in Poland is ca 10 TU. At depths of 700 m and more, thermal waters contain almost no tritium; at moderate depths, up to 200 m, the content of tritium rises considerably, exceeding even values typical for present day precipitation. At springs or shallow wells, the contents of tritium vary significantly from 0.0 to 16.6 TU (Ciężkowski et al. 1996).

Moreover, the content of 14C in the Sudetic thermal waters strongly suggests that some of them may have infiltrated thousands of years ago, probably in colder climatic conditions (Ciężkowski 1990; Ciężkowski et al. 1992). There is agreement among geologists that thermal waters in the Sudetes Mts gain their temperatures due to deep penetration of the meteoric recharge. This deep circulation is a result of the generally deep-reaching fissurity of crystalline rocks and considerable differences in altitude between the recharge area and the warm spring discharge areas.
Application geothermometers
To evaluate the Sudetic geothermal fields in the light of their prospectiveness, to further exploration of thermal energy resources, we took an attempt to apply selected isotopic and chemical geothermometers to assess the maximum possible temperatures, which may be encountered in the reservoirs (Table 3).
Silica geothermometers
Silica (SiO2) geothermometry is widely used in different geothermal systems. It is based on the temperature dependent solubility of various silica minerals, for example: quartz (SiO2−q), chalcedony (SiO2−ch), amorphous silica (SiO2−am), moganite (SiO2−m), cristobalite (SiO2−c), etc. This is one of the reasons, why there are several calibrations known for silica geothermometers based on experimental, theoretical or field studies. They have been summarized, for example, by Gunnarsson and Arnórsson (2000) or Arnórsson (2000). However, the silica phases of interest for hydrogeochemical studies of thermal waters are first of all quartz, chalcedony and amorphous silica (Arnórsoon 2000).
In the majority of known geothermal systems, the silica minerals with which the aqueous silica [SiO2(aq)] attains equilibrium are precipitated from solution, forming secondary minerals. Secondary silica mineral constitutes the controlling phase for SiO2(aq) concentrations, which depends on the rate of two counteracting processes: (1) dissolution of primary silicate minerals in the rock, and (2) precipitation of secondary silica minerals (Arnórsson 2000). The rate of dissolution of the primary rock-forming silicate minerals depends first on their thermodynamic properties and the reactivity of water in the system, which is largely controlled by its pH. Thus, roughly speaking, the water pH dependent rate of dissolution/precipitation of the silica minerals finally may control attaining the equilibrium of SiO2(aq) with secondary quartz, chalcedony or amorphous silica.
The dissolution of silica minerals (for example, quartz) in the pH up to 7.8 can be expressed as (for instance: Langmuir 1997):
and the solubility product (K osp ) for this reaction is:
where a, m and γ refer to the activity of a chemical compound, its molality and activity coefficient, respectively.
The most soluble form is amorphous silica and the least soluble is quartz. Generally, the solubilities of quartz and its polymorphs are fixed and pH-independent in most natural waters (Langmuir 1997). However, the dissociation of silicic acid (i.e. H4SiO4 which is equal to SiO2(aq) in our considerations) at alkaline pH leads to substantial increases in their solubilities. As a weak acid, H4SiO4 dissociates in two steps, according to the reactions (Langmuir 1997):
Finally, the dissolution of quartz at alkaline pH (between 9 and 10), can be expressed as:
H4SiO4 o concentration exceeds that of the anionic silica species up to pH = 9.82, whereas up to pH = 7.82 the concentration of anionic silica species is less than 1% (Langmuir 1997). Such behavior of the silicic acid in alkaline water is surely one of the most important causes of serious errors in application of silica geothermometers. The case in point is the understanding that the concentration of uncharged aqueous silica controls the equilibrium with secondary silica minerals and its concentration must be corrected according to the pH of water. The thermal waters of the Sudetes Mts are a good example. The total silica concentrations measured in these thermal waters are presented in Table 4, along with saturation indices (SI) with respect to quartz, chalcedony and amorphous silica. As can be seen, the measured (m m) total concentrations of silica expressed as aqueous silica, with assumption that SiO2(aq) = H4SiO4, are equal to real (m r) concentrations of SiO2(aq) in the solution only in low-pH thermal waters of Duszniki area; in all other cases corrections are needed to obtain true SiO2(aq) values.
Then, measured (and corrected if needed) SiO2(aq) is inserted into the respective silica geothermometer equation. For the low-enthalpy Sudetic thermal waters, the temperature–solubility equations for quartz (Fournier 1977), chalcedony (Fournier 1977, Arnórssson et al. 1983) and amorphous silica (Gunnarsson and Arnórsson 2000) were tested (Table 3).
Generally, the low pH water, such as CO2 rich waters of Duszniki hydrogeothermal system, tends to dissolve silicate minerals rather rapidly, and neither secondary quartz and chalcedony nor even amorphous silica reaches equilibrium with aqueous silica in the solution due to kinetic reasons. Moreover, very low temperatures of these waters (below 20°C in all springs and wells studied) and possible mixing with shallow waters of the modern hydrologic cycle, indicate that there are no favorable hydrogeological settings to approach any equilibrium in the water–rock system. Therefore, the application of chemical geothermometry in such geothermal system is baseless and gives unreal temperature estimations (Table 3). This agrees with some conclusions derived by Dowgiałło (2002) who shows on Giggenbach diagram (Giggenbach 1988) that these waters belong to the immature ones.
The application of quartz geothermometer to waters of Lądek and Cieplice generally gives overestimated temperature values except the water from deepest well C-1 (Table 3). As can be seen in Table 4, the water from well C-1 has the highest temperature at the outflow, i.e. 86.7°C and is almost in equilibrium with quartz. Taking into account the corrected (m r) concentration of uncharged SiO2(aq) the estimated temperature will be about 81°C, which is very close to the one measured at the outflow. However, the bottom temperature in well C-1 was about 96°C. In the case where reservoir temperature exceeds 100°C (due to the elevated water column pressure) boiling of water occurs. It causes the concentration of aqueous solutes to increase in proportion to the steam formation. It also causes the pH of water to increase due to the transfer of weak acids dissolved in water into the steam phase (Arnórsson 2000). Therefore, in this case, it seems to be more appropriate to use a totally analyzed SiO2(aq) (i.e., m m ) instead of the calculated unionized one (m r) to estimate the quartz equilibrium temperature. Such approximation gives the reservoir temperature about 94°C, which is very close to that obtained by bottom-hole temperature survey (Table 3). For other waters of the Cieplice geothermal system the best reservoir temperature estimations are obtained by inserting the calculated unionized SiO2(aq) (m r) concentrations (Table 4) to chalcedony equilibrium equations. These waters belong to the same hydrogeothermal system as the water from C-1 well. However, they must be cooled down considerably in the upflow zone gaining oversaturation with respect to chalcedony.
The thermal waters of Lądek are obviously colder and belong to a shallower circulation system than those of Cieplice area. In that case, the best actual temperature in the reservoir may also be obtained by applying the chalcedony equilibrium equations (Table 3).
Na–K and Na–K–Ca geothermometers
Many empirical calibrations have been proposed for the Na–Ka geothermometer (Arnórsson 2000). The newest one by Arnórssson et al. (1983), tested in this study, is based on the assumption that Na+ and K+ in thermal waters are in simultaneous equilibrium with quite pure albite and K-feldspar (Arnórsson 2000). The reaction involved can be expressed as:
Although albite and K-feldspar are widespread as secondary minerals in the rock environment of geothermal systems, they do not form in all types of rocks subjected to hydrothermal alteration (Arnórsson 2000). That is why there is no universal Na–K geothermometer. From the thermodynamic point of view, the equilibrium in the water–rock system is attained when secondary albite and K-feldspar tend to precipitate from solution; it means that thermal waters must be somewhat supersaturated with respect to these minerals. However, the supersaturation is caused by the excessive aluminum in solution (Langmuir 1997). The solubilities of both albite and K-feldspar decrease with decreasing temperature, which causes the feldspars’ tendency to precipitate in upflow zones. In the Sudetic thermal waters the concentrations of Al are very small, below the detection limits of ICP-AES method. Therefore, saturation indices (SI) with respect to albite and feldspar or other aluminosilicates have not been tracked in this study. Nevertheless, application of Na–K geothermometer to Sudetic thermal waters gives a reasonable range of reservoir temperature estimations (i.e. 98–113°C) only for the Cieplice geothermal system. In the case of thermal waters of Lądek, calculated reservoir temperatures are somewhat inflated (Table 3).
A very promising and consistent with silica geothermometers range of reservoir temperatures was obtained applying Na–K–Ca geothermometer calibrated empirically by Fournier and Truesdell (1973). The main advantage of this temperature indicator is that it does not give high and misleading results especially for low-temperature thermal waters. Generally, there is no agreement among scientists, which minerals are particularly involved in controlling the abundance of Na, K and Ca in thermal waters; usually proposed are clay minerals, micas and feldspars. Arnórsson (2000) suggests following reactions, which may be involved:
The application of Na–K–Ca geothermometer to waters of the Cieplice geothermal system gives reservoir temperatures between about 94–104°C, whereas when applied to colder waters of Lądek area it gives reservoir temperatures between about 30–51°C (Table 3).
An important first geothermometric study based on aluminosilicate equilibria in the Sudetic thermal waters was published by Lesniak and Nowak (1993). However, the Al3+ concentrations, they presented in chemical records for most thermal waters seem to be unreliable (i.e., too high) and do not agree completely with our studies. In consequence, they suggest rather lower range of possible reservoir temperatures in the Cieplice and Lądek areas, i.e., 60–70°C and higher in the Duszniki area: 50–95°C.
Oxygen isotope geothermometer in SO4–H2O system (Δ18O SO4–H2O)
This geothermometer is based on the fact that the exchange of oxygen isotopes between SO4 2− and H2O is a function of temperature. It is counted among the most useful ones for water-dominated geothermal fields and was successfully applied in many geothermal systems in the range of temperatures 100–350°C (Mizutani and Rafter 1969; Mizutani 1972; Cortecci 1974; Cortecci and Dowgiałło 1975; McKenzie and Truesdell 1977; Fouillac et al. 1990).
A few experimentally determined fractionation factors (α) of oxygen isotopes in the SO4–H2O system are in use up to now (Table 5). Such variety of α value is due to the fact that the oxygen isotope exchange rate is strongly dependent on temperature and also on pH of the solution. Chiba and Sakai (1985) deduced that the isotope exchange reaction between dissolved sulfates and water proceeds through collision between H2SO4 0 and H2O at low pH, and between HSO4 − and H2O at intermediate pH. The rate of the oxygen isotopic exchange between sulfate and water in acid and neutral thermal waters of temperature above 100°C is sufficiently fast to expect that sulfates be in isotopic equilibrium with the reservoir waters. Below 100°C, the rate of this exchange is much slower. For instance, according to the equation proposed by Lloyd (1968) for half time of the exchange:
where t 1/2, T and b refer to the half-time of the exchange in hours, temperature in °K, and to coefficient equals −1.17 at pH 7, respectively; the time for 99.9% isotopic exchange to equilibrium is about 18 years at 200°C and about 500 years at 100°C. However, in some groundwaters, the residence time of SO4 2− ion may be long enough to attain the isotopic equilibrium with water at temperatures even much below 100°C. The evidence of such equilibrium was found, for example, by Halas et al. (1993) in the study of postglacial waters from deep horizons under the Baltic Sea bottom, eastern Pomerania, Poland, or by Fouillac et al. (1990) in thermal waters of the Dogger geothermal aquifer in the Paris Basin, where the average temperature measured in situ was about 80°C. Taking into account the intermediate pH in majority of the waters studied, sulfates must exist in a mixture of SO4 2− and HSO4 − (Sakai 1977; Chiba and Sakai 1985). Therefore, the temperature scale should be between the scales for HSO4 − and SO4 2− (Table 3). It means, that the application of fractionation factors of Mizutani and Rafter (1969) and Lloyd (1968) should give the most appropriate range of water temperatures at depth.
The results of application of the SO4–H2O oxygen isotope geothermometer to thermal waters studied are summarized in Table 3. As it can be seen, this geothermometer gives reliable temperature values only in the Cieplice geothermal system. Therefore, we suggest that the equilibrium of oxygen isotopes in the sulfate–water system is reached in these thermal waters. The range of calculated temperatures between 99 and 116°C sufficiently well agrees with the temperature measured at the bottom of the deepest well C-1, i.e. 96.1°C. Moreover, the results are generally consistent with temperatures obtained while applying other chemical geothermometers. Unfortunately, the well C-1 is closed since its construction and testing in the year 2000 and sampling of water was not possible. Other studied wells and springs in the Cieplice area extract water from the same circulation system but are much shallower and the water is cooled down considerably due to ascension in drainage areas. Thermal waters in the Duszniki and Lądek areas are colder than those at Cieplice. Evidently, they represent shallower circulation systems where the temperature of rock environment is much lower and the residence time of water is much shorter. Moreover, thermal waters at Duszniki contain considerable amounts of CO2, which affects the oxygen isotopic composition of water and accelerates its cooling. The waters in Lądek contain H2S, which is an evidence of bacterial reduction of sulfates thus disqualifying the application of SO4–H2O geothermometer.
Conclusions
In this study, the applicability of selected chemical and isotopic geothermometers to assess reservoir temperatures in low-enthalpy geothermal systems of the Sudetes Mts has been demonstrated. The results show that in favorable hydrogeological settings the best temperature estimations are obtained from silica (mainly chalcedony) and alkaline (Na–K–Ca, partly Na–K) geothermometers. Moreover, the ranges of calculated temperatures are quite consistent (Table 3). The rule of thumb in application of silica geothermometry is thermodynamic modeling of silica speciations and further interpretation of unionized SiO2(aq) concentration, which controls the equilibrium attainment with secondary silica minerals.
Reliable reservoir temperature estimations were obtained while applying the oxygen isotope geothermometer in SO4–H2O system. However, the attention must be paid to secondary processes in geothermal systems, which may disqualify the use of this temperature indicator, for example: bacterial reduction of sulfates (waters from Lądek) or considerable CO2 content (carbonated waters of Duszniki).
There are some hydrogeothermal systems which are quite shallow, slightly thermal and CO2 rich, where the application of known chemical or isotopic geothremometers gives misleading and unreal temperature data, as for example in the Duszniki area.
Based on geothermometric survey it is suggested, that the Cieplice region is the most prospective one with respect to finding thermal water with temperatures close or slightly exceeding 100°C. Waters in this geothermal system contain the least amounts of 14C (Table 2), which suggests the longest residence time in the aquifer from infiltration to discharge. Consequently, the chemical equilibrium between some rock-forming minerals and water or isotopic equilibrium between oxygen and hydrogen of water and its selected chemical compounds is most probably reached here.
References
Arnórssson S, Gunnlaugsson E, Svavarsson H (1983) The chemistry of geothermal waters in Iceland. III. Chemical geothermometry in geothermal investigations. Geochim Cosmochim Acta 47:547–566
Arnórsson S (2000) Isotopic and chemical techniques in geothermal exploration development and use. IAEA, Vienna
Bruszewska B (2000) The geothermal conditions in lower silesia (SW Poland). Geolog Rev 48:639–643 [in Polish]
Chiba H, Sakai H (1985) Oxygen isotope exchange rate between dissolved sulfate and water at hydrothermal temperatures. Geochim Cosmochim Acta 49:993–1000
Ciężkowski W (1990) A study on the hydrogeochemistry of mineral and thermal waters in the Polish Sudets Mtn. Prace Naukowe Instytutu Geotechniki Politechniki Wrocławskiej 60:1–33 [in Polish]
Ciężkowski W, Groning M, Leśniak PM, Weise SM, Zuber A (1992) Origin and age of thermal waters in Cieplice Spa, Sudeten, Poland, inferred from isotope, chemical and noble gas data. J Hydrol 140:89–117
Ciężkowski W, Doktór S, Graniczny M, Kabat T, Liber-Madziarz E, Przylibski T, Teisseyre B, Wiśniewska M, Zuber A (1996) Determination of recharge areas for curative water of meteoric origin in Poland basing on isotopic study: annex 3, 6, and 20. Zakład Badawczo-Usługowy ‘Zdroje’, Wrocław. [in Polish, unpublished]
Cortecci G (1974) Oxygen isotopic ratios of sulfate ions-water pairs as a possible geothermometer. Geothermics 3:60–64
Cortecci G, Dowgiałło J (1975) Oxygen and sulfur isotopic composition of the sulfate ions from mineral and thermal groundwaters of Poland. J Hydrol 24:271–282
Dowgiałło J (1976) Thermal waters of the Sudetes Mts. Acta Geol Pol 26:617–643 [in Polish]
Dowgiałło J (2000) Thermal water prospecting results at Jelenia Góra - Cieplice (Sudetes, Poland) versus geothermometric forecasts. Env Geol 39:433–436
Dowgiałło J (2002) The Sudetic Geothermal Region of Poland. Geothermics 31:343–359
Dowgiałło J, Florkowski T, Grabczak J (1974) Tritium and 14C dating of Sudetic thermal waters. Bulletin of the Polish Academy of Sciences-Earth Sciences 22:101–109
Dowgiałło J, Fistek J (1995) The Jelenia Góra geothermal system (Western Sudetes, Poland). Bulletin of the Polish Academy of Sciences - Earth Sciences 43:243–252
Dowgiałło J, Fistek J (2003) New findings in the Wałbrzych–Kłodzko geothermal sub–region (Sudetes, Poland). Geothermics 32:689–699
Dowgiałło J, Halas S, Porowski A (2005) Isotope temperature indicators of thermal waters in south-western Poland. Proceedings World Geothermal Congress 2005, Antalya, Turkey, 24–29 April, 2005: 1–8
Fouillac C, Fouillac AM, Criaud A (1990) Sulphur and oxygen isotopes of dissolved sulphur species in formation waters from the Dogger geothermal aquifer, Paris Basin, France. Appl Geochem 5:415–427
Fournier RO (1977) Chemical geothermometers and mixing models for geothermal systems. Geothermics 5:41–50
Fournier RO, Truesdell AH (1973) An empirical Na–K-Ca geothermometer for natural waters. Geochim Cosmochim Acta 37:1255–1275
Giggenbach WF (1988) Geothermal solute equilibria. Derivation of Na–K-Mg–Ca geoindicators. Geochim Cosmochim Acta 52:2749–2765
Gunnarsson I, Arnórsson S (2000) Amorphous silica solubility and the thermodynamic properties of H4SiO4 o in the range of 0o to 350 °C. Geochim Cosmochim Acta 64:2295–2307
Halas S, Pluta I (2000) Empirical calibration of isotope thermometer δ18O (SO4 2−) − δ18O (H2O) for low temperature brines. Book of Abstracts: V Isotope Workshop. European Society for Isotope Research, Kraków, Poland, pp 68–71
Halas S, Trembaczowski A, Sołtyk W, Walendziak J (1993) Sulphur and oxygen isotopes in sulphates in natural waters: (2) deep-waters from horizons below Baltic sea floor. Isotopenpraxis 28:229–235
Kusakabe D, Robinson BW (1977) Oxygen isotopic fractionation factor between barite and H2O at hydrothermal temperature. Oxygen and sulfur isotope equilibria in the BaSO4-HSO4–H2O system from 110 to 350 °C and its applications. Geochim Cosmochim Acta 44:1033–1040
Langmuir D (1997) Aqueous environmental geochemistry. Prentice Hall, Upper Saddle River, NJ, p 598
Leśniak PM, Nowak D (1993) Water-rock interaction In some mineral waters In the Sudetes, Poland: implications for chemical geothermometry. Ann Soc Geol Pol 63:101–118
Lloyd RM (1968) Oxygen isotopic behavior in the sulfate-water system. J Geophys Res 73:6099–6110
McKenzie WF, Truesdell AH (1977) Geothermal reservoir temperatures estimated from the oxygen isotope compositions of dissolved sulfate and water from hot springs and shallow drillholes. Geothermics 5:51–61
Mizutani Y, Rafter TA (1969) Oxygen isotopic composition of sulfates–3. Oxygen isotopic fractionation in the bisulphate ion-water system. New Zeeland Journal of Science 12:54–59
Mizutani Y (1972) Isotopic composition and underground temperature of the Otake geothermal water, Kyushu, Japan. Geochemistry Journal 6:67–73
Porowski A, Dowgialło J (2008) Application of selected chemical and isotopic geothermometers to low enthalpy thermal waters in Poland. In: Future of Hydrogeology: Modern trends and perspectives. St. Petersburg University Press [in print]
Różański K, Araguas-Araguas L, Gonfiantini R (1993) Isotopic patterns in modern global precipitation. In: Stewart PK, Lohmann KC, McKenzie J, Savin S (eds) Climate change in continental isotopic records. Geophysical Monograph 76: 1–36
Sakai H (1977) Sulfate-water isotope thermometry applied to geothermal systems. Geothermics 5:67–74
Zuber A, Ciężkowski W, Kryza J, Grabczak J (1989) Origin of thermal waters of Cieplice Zdrój Spa and their anomal chemical and isotopic composition. Prace Naukowe Instytutu Politechniki Wrocławskiej 58:389–400 [in Polish]
Zuber A, Weise SM, Grabczak J, Ciężkowski W (1995) Age and recharge area of thermal waters in Ladek Spa (Sudeten, Poland), deduced from environmental isotope and noble gas data. J Hydrol 167:327–349
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s12665-009-0350-8
Rights and permissions
About this article
Cite this article
Adam, P., Jan, D. Application of selected geothermometers to exploration of low-enthalpy thermal water: the Sudetic Geothermal Region in Poland. Environ Geol 58, 1629–1638 (2009). https://doi.org/10.1007/s00254-008-1409-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00254-008-1409-7