
Abstract
The biocatalytic production of fuels and chemicals from plant biomass represents an attractive alternative to fossil fuel-based refineries. In this context, the mining and characterization of novel biocatalysts can promote disruptive innovation opportunities in the field of lignocellulose conversion and valorization. In the present work, we conducted the biochemical and structural characterization of two novel hydroxycinnamic acid catabolic enzymes, isolated from a lignin-degrading microbial consortium, a feruloyl-CoA synthetase, and a feruloyl-CoA hydratase-lyase, named LM-FCS2 and LM-FCHL2, respectively. Besides establishing the homology model structures for novel FCS and FCHL members with unique characteristics, the enzymes presented interesting biochemical features: LM-FCS2 showed stability in alkaline pHs and was able to convert a wide array of p-hydroxycinnamic acids to their respective CoA-thioesters, including sinapic acid; LM-FCHL2 efficiently converted feruloyl-CoA and p-coumaroyl-CoA into vanillin and 4-hydroxybenzaldehyde, respectively, and could produce vanillin directly from ferulic acid. The coupled reaction of LM-FCS2 and LM-FCHL2 produced vanillin, not only from commercial ferulic acid but also from a crude lignocellulosic hydrolysate. Collectively, this work illuminates the structure and function of two critical enzymes involved in converting ferulic acid into high-value molecules, thus providing valuable concepts applied to the development of plant biomass biorefineries.
Key points
• Comprehensive characterization of feruloyl-CoA synthetase from metagenomic origin.
• Novel low-resolution structures of hydroxycinnamate catabolic enzymes.
• Production of vanillin via enzymatic reaction using lignocellulosic hydrolysates.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Global climate and environmental change, intrinsically associated with an overdependence on fossil resources such as oil and coal, have driven scientists and governments to seek alternative sustainable sources for fuels and chemicals (Souza et al. 2017). In a biobased economy, microbial fermentation, enzyme-based processes, and green chemistry may be used to generate liquid biofuels and high-value chemicals from renewable feedstocks such as lignocellulosic biomass (Lange et al. 2021). The latter is comprised of three main components, cellulose, hemicellulose, and lignin, presenting a wide array of C5 and C6 sugars and aromatic molecules that could be converted into numerous products (Supplemental Fig. S1). However, overcoming the inherent recalcitrance of lignocellulose — and, particularly, of lignin, a highly hydrophobic and heterogenous phenolic polymer — to exploit these resources for advanced material applications display numerous challenges (Irmak 2019).
As degradation of lignocellulose is a primordial step of natural carbon cycling, it is logical to look in nature for solutions to this industrial challenge. Thus, metagenomic approaches can provide important insights on the often-untapped reservoirs of naturally occurring biocatalysts, which can be applied to lignocellulose conversion and valorization in an industrial context (Pandey and Singhal 2021). In a previous study, the metagenomic profiling of a lignin-degrading microbial consortium, established using soil from a sugarcane field as inoculum, disclosed ferulic acid (FA) catabolism pathways helpful for lignin valorization strategies (Moraes et al. 2018). This pathway employs feruloyl-CoA synthetases (EC 6.2.1.34, FCS) and feruloyl-CoA hydratase-lyases (EC 4.2.1.101, FCHL), and subsequent reactions that result in intermediates of potential biotechnological interest.
FA can be obtained from the hydrolysis of lignocellulosic feedstocks, such as rice bran and sugarcane bagasse, and lignin streams derived from biomass pretreatment (Rosazza et al. 1995; Brenelli et al. 2016). The use of lignin waste for the bioproduction of fine chemicals such as vanillin is particularly interesting, representing the dual benefit of using non-edible, renewable feedstocks as starting material instead of petroleum, and adding value to the production chain of lignocellulosic biofuels and pulp/paper industries (Chen and Wan 2017; Becker and Wittmann 2019; Sharma et al. 2020).
Vanillin (3-methoxy-4-hydroxybenzaldehyde; C8H8O3) is an aromatic aldehyde with highly desired organoleptic properties for food, cosmetics, and pharmaceutical industries. Natural vanillin is traditionally extracted from the cured pods of the orchid Vanilla planifolia. This labor-intensive and slow process cannot meet an ever-increasing global demand for natural products (Ciriminna et al. 2019). Thus, global demand has been met mainly through vanillin produced from non-renewable sources, such as guaiacol, or waste sulfite liquor lignin; however, these processes are not environment-friendly, and the vanillin produced must be labeled as “artificial” or “synthetic,” which does not appeal to many consumer markets (Fache et al. 2016; Paul et al. 2021). On the other hand, vanillin produced by biotechnological routes, such as microbial fermentation or enzymatic catalysis, is considered natural and may be sold as such (Banerjee and Chattopadhyay 2019).
The plant orthologues of FCSs — 4-coumarate:CoA ligases — are better characterized, and several studies have focused on the structural elucidation of these enzymes, especially regarding their substrate binding capacity (Lozoya et al. 1988; Lee and Douglas 1996; Schneider et al. 2003; Lindermayr et al. 2002; Costa et al. 2005). On the other hand, there are scarce biophysical and biochemical characterizations of prokaryotic FCSs, since most published works, while important, mainly focus on their direct application for vanillin production (Ghosh et al. 2007; Gräf and Altenbüchner 2014; Yang et al. 2013; Kaur et al. 2014; Chakraborty et al. 2016, 2017; Fleige et al. 2016; Chen et al. 2022), or the genetic characterization of their endogenous expression and role in FA catabolism (Overhage et al. 1999; Achterholt et al. 2000; Masai et al. 2002; Otani et al. 2014). Similarly, there are only two published structures of prokaryotic FCHLs (Liberato et al. 2020; Bennett et al. 2008). However, the thorough biochemical and biophysical characterization of prokaryotic FCSs and FCHLs are important to drive industrial applications for these enzymes.
Specifically, as shown in several studies recently reviewed (Galadima et al. 2020), FCSs and FCHLs are auspicious candidates for the biocatalytic production of vanillin from FA (Supplemental Fig. S1) via a sequential two-step coenzyme-A (CoA)-dependent non-β-oxidative reaction (Mathew and Abraham 2006). In the first step, FA is activated via CoA-thiosterification to feruloyl-CoA, with ATP consumption; secondly, feruloyl-CoA is hydrated and cleaved, yielding vanillin and acetyl-CoA (Graf and Altenbuchner 2014).
In the present work, we describe the heterologous expression, purification, and characterization of an FCS and an FCHL isolated from a lignin-degrading microbial consortium established using soil from a sugarcane plantation in Brazil (Moraes et al. 2018), named LM-FCS2 and LM-FCHL2, respectively. We provide the biochemical and biophysical characterization of these enzymes and demonstrate their combined use to produce vanillin from lignin-rich lignocellulosic hydrolysates.
Materials and methods
Phylogenetic analysis
The protein sequences sharing at least 60% similarity to LM-FCS2 or LM-FCHL2 found in the UniProt database (https://www.uniprot.org/) were retrieved. The multiple protein alignments were carried out using MUSCLE (Edgar 2004a, b), and ambiguously aligned positions were removed using Gblock (Talavera and Castresana 2007). The evolutionary relationships among sequences were inferred using RAxML (Stamatakis 2014) with a PROTGAMMAAUTO model, and 1000 bootstrap replicates. The phylogenetic trees generated were manually edited for improved visualization using the iTOL web tool (https://itol.embl.de).
Expression and purification of LM-FCS2 and LM-FCHL2 enzymes
The coding sequences corresponding to LM-FCS2 and LM-FCHL2 (GenBank accession numbers MZ983551 and MZ983550, respectively) were retrieved from metagenomic data of a lignin-degrading microbial consortium, codon-optimized (GenBank accession number OM677839 and OM677840, respectively), and cloned into pET-28a( +) as previously described (Moraes et al. 2018). The vectors pET-28a-LM-FCS2 and pET-28a-LM-FCHL2 were transformed in Escherichia coli BL21(DE3) (New England Biolabs, Ipswich, MA). A single colony of each transformation was sterilely picked and inoculated in LB medium supplemented with kanamycin (50 μg/mL) and incubated at 37 °C, 200 rpm, for 16 h. The overnight cultures were added to a 1% final concentration to fresh LB medium supplemented with kanamycin (50 μg/mL) and incubated at 37 °C, 200 until the optical density at 600 nm (OD600) reached 0.6, followed by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). For LM-FCHL2 expression, cultures were kept at 37 °C for 4 h, as previously described (Liberato et al. 2020). For LM-FCS2 expression, cultures were kept at 30 °C for 4 h. After induction, the cells were harvested by centrifugation at 4000 × g, and LM-FCS2 and LM-FCHL2 were purified by affinity (HiTrap Chelating HP column, GE Healthcare, Chicago, IL; Buffer A: 20 mM sodium phosphate buffer pH 7.0, 100 mM NaCl, 5 mM imidazole; Buffer B: 20 mM sodium phosphate buffer pH 7.0, 100 mM NaCl, 500 mM imidazole) and size-exclusion chromatography (Superdex 200 HiLoad 16/600 GL column, GE Healthcare, Chicago, IL; Buffer C: 20 mM sodium phosphate buffer pH 7.4, 100 mM NaCl), as previously described (Sodré et al. 2019). The purified protein fractions were analyzed by 12% SDS-PAGE.
LM-FCS2 optimal temperature, pH, and substrates
Enzymatic reactions were prepared and monitored by detecting the formation of feruloyl-CoA at 345 nm, as previously described (Sodré et al. 2019). The reactions were analyzed spectroscopically after adding 50 µL of 2 M sodium phosphate buffer. The specific activity was determined as µmols of product/minute/mg of protein. To determine LM-FCS2 optimal temperature, a range from 20 to 60 °C was used. The optimal pH was determined in either 100 mM potassium phosphate buffer (pH 6.2–8.0) or 20 mM acetate-borate-phosphate (ABF) buffer (pH 6.2–9.8), and different substrates were evaluated (ferulic acid, p-coumaric acid, caffeic acid, and sinapic acid). The absorbance and molar extinction coefficient used for each substrate are detailed in Supplemental Table S1. The formation of products was also analyzed by capillary electrophoresis after reaction in 100 mM phosphate buffer, as described previously (Sodré et al. 2019).
Generation of alkaline treatment hydrolysates from sugarcane bagasse and straw
Sugar cane bagasse was collected from the Granelli mill, Charqueada, SP, Brazil. Before experimental use, the bagasse was manually chopped for size reduction of the larger particles and ground in a cross-beater mill SK300 (Retsch, Haan, Germany) with a 1.0-mm control sieve. The resulting material was dried at room temperature until humidity reached ∼10% moisture content, and stored.
The condition chosen for pretreatment of the bagasse was 0.7% w/v NaOH, 85 °C, 5 h, based on Lima and collaborators (Lima et al. 2018), and performed in a 0.5-L batch reactor in triplicate. The reactor was heated in a glycerin thermostatic bath during the reaction residence time and then cooled down for 10 min in an ice bath. The content of the reactor was poured into a polypropylene mesh for filtration, resulting in hydrolysate named SBPT, which was used as the substrate for vanillin production.
The coupled reaction of LM-FCS2 and LM-FCHL2 using ferulic acid and lignocellulosic hydrolysate as substrates and analysis by LC–MS/MS
The vanillin production by LM-FCS2 and LM-FCHL2 was initially evaluated using ferulic acid as substrate, and product formation was detected by capillary electrophoresis. Sugarcane bagasse liquid streams derived from diluted alkali pretreatment (SBPT) were evaluated as a source of ferulic acid utilized as substrate for LM-FCS2/LM-FCHL2 coupled reaction, generating vanillin as a bioproduct. The reaction mixture was assembled as described above; however, the volume correspondent to ferulic acid was substituted by SBPT.
Following incubation of 15 h at 25 °C, the reactions with LM-FCS2 and LM-FCHL2 were prepared for liquid chromatography coupled to mass spectrometry analysis. Reactions (500 µL) were mixed with 1 mL of ethyl acetate, vortexed vigorously, and left standing at least for 5 min to allow phase separation. The organic phase (800 µL) were then collected and dried using a vacuum centrifuge (Eppendorf Concentrator plus™, Eppendorf, Hamburg, Germany). Finally, the dried samples were resuspended in 1 mL methanol 100% and filtered using 0.22 µm polytetrafluorethylene (PTFE) syringe filters.
For liquid chromatography, an UltiMate 3000 UHPLC with a Thermo Scientific Accucore C18 column (2.6 μm pore size, 2.1 mm for 100 mm) was used at 45 °C. To establish the elution gradient, 0.1% formic acid in water (solvent A) and acetonitrile (solvent B) were used. The elution profile (A:B) proceeded as follows: 0–10 min 5% solvent B, 10–21.2 min 98% solvent B, and 21.2–30 min 5% solvent B. The flow was set to 0.3 mL min−1, and the injection volume was 3 μL. The liquid chromatography was coupled to an Orbitrap Q-Exactive (Thermo Fisher Scientific, Bremen, Germany) mass spectrometer, using an electrospray ionization source set to 3500 V. The analysis parameters for mass spectrometry analysis were set in positive mode [M + H]+, with evaluated m/z = 100 to 1500. All spectra analyses and operations were processed using Xcalibur software version 3.0.63 (Thermo Fisher Scientific, Bremen, Germany).
Structural analysis
As previously described, circular dichroism (CD) spectra and fluorescence emission measurements of LM-FCHL2 and LM-FCS2 were collected (Sodré et al. 2019; Liberato et al. 2020). Small-angle X-ray scattering (SAXS) measurements were performed at the SAXS beamline of National Synchrotron Light Laboratory (LNLS), Campinas, SP, Brazil. LM-FCHL2 (1 and 29 mg/mL) and LM-FCS2 (1 and 10 mg/mL) were measured at 20 °C in 20 mM ABF buffer pH 7. The X-ray beam wavelength was 1.48 Å, and the sample-to-detector distance was set at 0.95 m. Buffer scattering was measured and subtracted from the total measured intensities. The X-ray patterns were measured using a CCD detector (MarXperts, Norderstedt, Germany). The integration of SAXS patterns was performed with the FIT2D program (www.esrf.eu/computing/scientific/FIT2D). The radii of gyration were determined using Guinier approximation and the GNOM program (Svergun et al. 2001). CRYSOL program was used to calculate the theoretical X-ray scattering curve from the crystal structures and homology models (Kozin and Svergun 2001). The remote homology detection server HHPred (Söding et al. 2005) was used to search for homologs of the LM-FCHL2 and LM-FCS2 in the Protein Data Bank (PDB) with default parameters.
Molecular modeling
The tertiary structure of LM-FCS2 was predicted using the molecular homology modeling approach implemented in MODELLER (Webb and Sali 2016). The amino acid sequence was obtained from the NCBI data bank (accession WP_067990405) and then compared using HHpred against Protein Data Bank (PDB) to select the templates. The crystal structures used as templates are a fatty acyl-CoA synthetase from Mycobacterium tuberculosis (PDB ID: 3R44), acyl-adenylate synthetase from Methanosarcina acetivorans (PDB ID: 3ETC), acetyl-CoA synthetase from Salmonella enterica (PDB ID: 5JRH), and long-chain-fatty-acid-CoA ligase from Archaeoglobus fulgidus (PDB ID: 3G7S). All sequences were aligned using the Multiple Sequence Alignment software MUSCLE (Edgar 2004a, b). MODELLER was able to predict the 3D structure of LM-FCS2 through homology modeling based on its FASTA sequence. The best model was selected based on DOPE (Discrete Optimized Protein Energy) value (Shen and Sali 2006), and Molprobity for the structure validation (Chen et al. 2010). In this work, we selected the model with the following specifications: the Ramachandran plot with more than 90% residues in the favorable region and no outliers and with lower DOPE energy values. The protein model constructed using MODELLER was validated to identify possible errors and estimate the quality of its tertiary structure. The correct stereochemistry of the protein is important since it is critical for its interpretation. The stereochemical quality of the model is evaluated through Molprobity (Chen et al. 2010) to evaluate bond lengths, plane angles, chirality, side-chain conformations, and the steric clashes between atoms guaranteeing the best evaluation of the quality of the theoretical model.
Results
Sequence analysis, expression, and purification of LM-FCS2 and LM-FCHL2
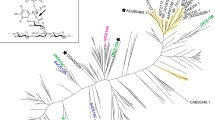
To evaluate the evolutionary relationships of LM-FCS2 and LM-FCHL2 with other FCSs and FCHLs, protein amino acid sequences from Uniprot displaying at least 60% amino acid identity to our sequences were retrieved, aligned, and used in phylogenetic analysis (Fig. 1). All the close protein sequences of LM-FCS2 were assigned to the Actinobacteria phylum, mainly to the Mycolicibacterium genus. The LM-FCS2 shared 85.5% amino acid identity to Mycolicibacterium litorale (A0A6S6NXL9), followed by Mycolicibacterium agri (85.4%, A0A2A7N4Y6) and Mycobacterium sp. (81.2%, A0A1A3T202). Nonetheless, although the phylogenetic analysis showed LM-FCS2 formed a unique cluster with M. litorale, the low bootstrap value suggests a divergence among sequences (Fig. 1). The LM-FCHL2 protein sequence shared similarities with different Proteobacteria phylum families, including Sphingomonadaceae (Sphingobium and Sphingorhabdus genera) and Parvularculaceae (Marinicaulis genus). Moreover, LM-FCHL2 formed a unique branch with Sphingobium sp. NBRC 103,272 (G2IJ08) and Sphingobium sp. B10D3C (A0A7W0BP28) with higher bootstrap, sharing 95% and 93% amino acid identity, respectively (Fig. 1).

Phylogenetic trees of LM-FCS2 (A) and LM-FCHL2 (B) and other homologous protein sequences. The trees were constructed using maximum likelihood, as implemented in RaxML. Each protein is labelled by its UniProt accession number, followed by the species name. LM-FCS2 and LM-FCHL2 are highlighted in bold
LM-FCS2 and LM-FCHL2 coding sequences were cloned into a pET-28a( +) bacterial vector, with a sequence for an N-terminal His6-tag, for purification by affinity chromatography. Afterward, the obtained fractions were further purified by size-exclusion chromatography (SEC). Figure 2 depicts the pattern of expression and the purified enzymes after SEC, with LM-FCS2 and LM-FCHL2 presenting approximately 60 kDa and 30 kDa, respectively, which is in close agreement with the predicted molecular weight without the His6-tag (59.6 kDa and 32.4 kDa, respectively).

Heterologous expression and chromatography purification of LM-FCS2 and LM-FCHL2. A and B Affinity chromatography of LM-FCS2 and LM-FCHL2, respectively. C and D Size-exclusion chromatography (SEC) of LM-FCS2 and LM-FCHL2, respectively. 12% SDS-PAGE. M: PageRuler™ Unstained Protein Ladder (Thermo Scientific™), T0: BL21 lysated cells before induction, 4 h: BL21 lysated cell after 4 h of IPTG induction
Biochemical characterization of LM-FCS2
Biochemical characterization of LM-FCS2 followed the experimental conditions previously established for FCS1 (Sodré et al. 2019). The optimum pH determination was performed in two buffers: 20 mM acetate-borate-phosphate (ABF) buffer (pH 6.2–9.8) and 100 mM potassium phosphate (pH 6.2–8.0). Using ABF as a buffer, the LM-FCS2 presented maximum specific activity on FA (0.36 U/mg) at pH 8.6 for FA (Fig. 3A). While for potassium phosphate buffer, the maximum specific activity on FA was 0.29 U/mg at pH 7.8 (Fig. 3B). In addition to FA, optimal pH was determined using p-coumaric, caffeic, and sinapic acids as substrates (Fig. 3A and B). As with FA, the optimal pH in the ABF buffer was 8.6 for all substrates and varied from 7.8 to 8.0 in potassium phosphate buffer. The optimal temperature was determined using FA as substrate and 100 mM potassium phosphate buffer pH 7.8. As shown in Fig. 3C, LM-FCS2 presented maximum activity at 40 °C.

Determination of LM-FCS2 optimal pH, temperature and substrate. A Optimal pH observed in ABF buffer using different hydroxycinnamic acids as substrates. B Optimal pH observed in potassium phosphate buffer using different hydroxycinnamic acids as substrates. C Optimal temperature determined using FA and potassium phosphate buffer pH 7.8. D LM-FCS2 relative activity observed in the optimal pH and buffer conditions for each substrate
Furthermore, the relative activity in each substrate was evaluated in the respective optimal pH of each buffer system (Fig. 3D). While the highest activity was detected with FA as substrate in both buffers, the activity profiles for the other hydroxycinnamic acids were buffer-dependent. In the ABF buffer, after FA, the highest activities were observed for p-coumaric acid, followed by sinapic acid. In contrast, caffeic acid showed excessively high and unstable absorbance in this buffer, not allowing evaluation by the spectroscopic method. Conversely, caffeic acid presented the second highest activity in potassium phosphate buffer, followed closely by sinapic and p-coumaric acids.
To further confirm the enzymatic activity observed using spectroscopy, reactions with hydroxycinnamic acids in potassium phosphate buffer were evaluated by capillary electrophoresis, which denoted the formation of the respective CoA-thioesters from FA, p-coumaric acid, sinapic acid, and caffeic acid (Supplementary Material Fig. S2). In addition, LM-FCS2 activity in ABF buffer using FA as substrate was also confirmed by capillary electrophoresis, as shown in Supplemental Fig. S2.
Biophysical characterization of LM-FCS2 and LM-FCHL2
The effects of pH on the secondary and tertiary structures of the LM-FCHL2 and LM-FCS2 enzymes were analyzed using circular dichroism (CD) spectroscopy and intrinsic fluorescence spectroscopy, respectively. Figure 4 shows the CD spectra for LM-FCHL2 and LM-FCS2 at different pH values. In both cases, the CD spectra at pH 7 were characterized by two minima around 208 nm and 222 nm, indicating the presence of α-helical structures. At pH values higher than 7, no significant alterations were observed in the spectra profiles, which means that the secondary structures of both enzymes are stable under alkaline conditions. However, at acidic pH values (in particular at pH 3 and 4), changes were detected in the spectra profiles for both enzymes, with a decrease of the minima characteristic of α-helix and, therefore, indicative of secondary structure loss.

CD spectra of LM-FCHL2 at pHs 3–7 (A) and pHs 7–10 (B). CD spectra of LM-FCS2 at pHs 3–7 (C) and pHs 7–10 (D). All measurements were performed at 20 °C
Figure 5 shows the fluorescence emission spectra for LM-FCHL2 and LM-FCS2, where the eight tryptophan residues present in both structures were used to monitor likely conformational alterations caused by pH variation. At pH 7, the fluorescence emission spectra of LM-FCHL2 and LM-FCS2 are characterized by maximum emissions at 348 nm (typical, on average, of partially solvent-exposed tryptophan residues) and 334 nm (typical, on average, of tryptophan residues buried inside the protein core), respectively (Souza et al. 2016).

Fluorescence emission spectra of LM-FCHL2 at pHs 3–7 (A) and pHs 7–10 (B). Fluorescence emission spectra of LM-FCS2 at pHs 3–7 (C) and pHs 7–10 (D). All measurements were performed at 20 °C
At pH values higher than 7, no significant alterations were observed in the fluorescence emission spectra, suggesting an absence of changes in the enzyme structures and the positions of their tryptophan residues under alkaline conditions. However, at pH values lower than 7, a reduction in quantum yield and a shift in the spectral center of mass were observed, indicating that the enzyme structures and their tryptophan residues positions changed under acidic conditions. Taken together, the alterations revealed on the secondary and tertiary structures at acidic conditions can be correlated with the absence of activity, as described above, for both enzymes in non-alkaline conditions.
SAXS method was used to obtain information about the quaternary structures of LM-FCHL2 and LM-FCS2 in solution. Figure 6 shows the X-ray scattering curve measured for LM-FCS2 and the associated Guinier plot. The Guinier plot was linear, and the radius of gyration Rg calculated was 34 ± 1 Å. The Rg and the maximum dimension (Dmax) of the molecule, calculated using the GNOM program, were 35.84 ± 0.03 Å and 115 ± 5 Å, respectively. The remote homology detection program HHpred found that LM-FCS2 presents, in general, a low amino acid sequence identity (35.0%; 23.8%; 22.9%; 27.0%) when compared to homologous structures (PDB ID: 3R44, 3ETC, 5JRH, and 3G7S, respectively), despite a high probability of having the same folding. Thus, LM-FCS2 was modeled as a homodimer using an ensemble of the four crystallographic structures mentioned above as a template. The homology model built for LM-FCS2 monomer showed an intermediate theoretical model compared to homologous crystallographic structures, with a mean RMSD (root-mean-square-deviation) value of 3.11 Å, which corresponds to an average structure, attenuating possible biases eventually found in models obtained through homology modeling. The theoretical X-ray scattering curve calculated from the homodimeric LM-FCS2 homology model resulted in an excellent fit to SAXS data. The homodimeric LM-FCS2 homology model has Rg and Dmax of 33.50 Å and 106.3 Å, respectively, values in agreement with the results obtained by SAXS analysis. Therefore, the results are most consistent with a homodimeric molecule for LM-FCS2 in solution under native conditions. A summary of the main SAXS results for LM-FCS2 is given in Supplemental Table S2.

SAXS data measured for LM-FCS2 at 20 °C. Experimental X-ray scattering curve of the LM-FCS2 at pH 7 (open black circles) superimposed on the computed scattering curves based on the homodimeric LM-FCS2 homology model (black line). Left inset: Guinier plot. Right inset: homodimeric LM-FCS2 homology model
Figure 7 shows the X-ray scattering curve measured for LM-FCHL2 and the associated Guinier plot. The Guinier plot was linear, and the Rg calculated was 37 ± 1 Å. The Rg and the Dmax of the molecule, calculated using the GNOM program, were 35.53 ± 0.01 Å and 100 ± 5 Å, respectively. The HHPred program (Söding et al. 2005) found that LM-FCHL2 presents 49% sequential identity and 100% probability to have the same folding when compared with feruloyl-CoA hydratase-lyase (FCHL) from a lignin-degrading consortium (Liberato et al. 2020). As recently reported, FCHL is a hexameric protein formed of six identical monomers, and each monomer is folded in two domains: the N-terminal domain, composed of one layer of six-stranded β-sheets and one layer of α-helices; and the C-terminal domain, which has three α-helices (Liberato et al. 2020).

SAXS data measured for LM-FCHL2 at 20 °C. Experimental X-ray scattering curve of the LM-FCHL2 at pH 7 (open black circles) superimposed on the computed scattering curves based on the hexameric feruloyl-CoA hydratase-lyase (FCHL) crystal structure (black line). Left inset: Guinier plot. Right inset: hexameric FCHL crystal structure (PDB 6P5U)
As shown in Fig. 7, the experimental X-ray scattering curve for LM-FCHL2, superimposed with the theoretical X-ray scattering curve calculated from the hexameric crystal structure of FCHL (PDB 6P5U), presented a good fit. The FCHL crystal structure has Rg and Dmax of 32.29 Å and 94.2 Å, respectively. However, in the FCHL crystal structure, eleven N-terminal amino acid residues and twenty-nine C-terminal amino acid residues are not present, indicating that both regions are possibly flexible (Bennett et al. 2008). Thus, the flexibility of the terminal regions of the LM-FCHL2 in solution may explain the differences observed in the superimposition mainly to higher values of the scattering vector. Therefore, the results are most consistent with a hexameric molecule for LM-FCHL2 in solution, presenting some degree of flexibility under native conditions. A summary of the main SAXS results for LM-FCS2 and LM-FCHL2 is given in Supplemental Table S2. The homology model (monomer) for the two enzymes, depicting the motifs involved in catalytic mechanism, is shown in Supplemental Fig. S3.
Biocatalytic production of vanillin via LM-FCS2/LM-FCHL2 coupled reaction
As proof-of-concept, vanillin production via LM-FCS2/LM-FCHL was first attempted using pure FA as substrate (Fig. 8a; Supplemental Fig. S4). The chromatogram shows that the coupled reaction produced vanillin from FA, while the reaction containing only LM-FCS2 did not (Fig. 8a; Supplemental Fig. S4). Interestingly, LM-FCHL2 could produce vanillin directly from FA in the absence of LM-FCS2, albeit in smaller quantities than observed in the coupled reaction.

LC–MS chromatograms depicting the biocatalytic production of vanillin from ferulic acid (A) and SBPT (B) using LM-FCS2 and FCHL2 coupled reaction. The control refers to the reaction medium without any added enzymes. LM-FCHL2 and LM-FCS2 refer to the reaction medium containing either LM-FCHL2 and LM-FCS2, respectively. LM-FCS2 + LM-FCHL2 refers to the reaction medium containing both enzymes
To evaluate the biocatalytic conversion of lignin-rich liquors to vanillin, the liquid fraction of alkaline pretreated sugarcane bagasse hydrolysate (SBPT) was used as a substitute to FA in the standard enzymatic reaction of LM-FCS2/LM-FCHL2. As shown in Fig. 8b, SBPT contains a mixture of biomass-derived hydroxycinnamic acids, which were used as substrates for LM-FCS2 and LM-FCHL2, resulting in the formation of 4-hydroxybenzaldehyde and vanillin from p-coumaric and ferulic acids, respectively. We further confirmed these biocatalytic conversions from FA and SBPT by filtering the LC–MS data to detect FA, p-coumaric acid, vanillin, or 4-hydroxybenzaldehyde, and display the peak area for each compound (Supplemental Fig. S5). A high-resolution analysis was performed, allowing the determination of the experimental m/z ratio presenting minimal error compared to the theoretical m/z ratio described in the literature. The m/z ratio and the retention times and product ions for all the compounds are shown in Supplemental Table S3. Residual vanillin was detected in the control reaction of SBPT, which agrees with previous studies that reported this molecule as a common inhibitor found in lignocellulosic hydrolysates (Sodré et al. 2021). Nevertheless, the samples containing SBPT and both enzymes (LM-FCS2 and LM-FCHL2) showed a tenfold increase in the normalization level (NL) of vanillin compared to the control, demonstrating the formation of this compound via biocatalysis. To our knowledge, this is the first report of vanillin bioproduction from alkaline pretreated sugarcane bagasse hydrolysates using feruloyl-CoA synthetase/feruloyl-CoA hydratase-lyase coupled reaction.
Discussion
LM-FCS2 was biochemically and biophysically characterized according to a previous work that characterized another feruloyl-CoA synthetase — FCS1 — derived from a lignin-degrading consortium. LM-FCS2 presented maximum activity at 40 °C, slightly higher than other characterized prokaryotic feruloyl-CoA synthetases, which presented optimal temperature between 30–37 °C (Zenk et al. 1980; Yang et al. 2013; Sodré et al. 2019). Interestingly, the enzyme presented more than 70% of its activity in a wide range of temperatures, from 25 to 50 °C.
LM-FCS2 presented higher specific activity in ABF buffer than in potassium phosphate buffer for all evaluated hydroxycinnamic acids. Furthermore, ABF buffer provided optimal pHs in more alkaline values than potassium phosphate buffer, suggesting that the enzymatic activity depends not only on the pH but also on the ion used to establish the buffer system. Interestingly, while this shift in optimal pH due to buffer was also observed for FCS1 (Sodré et al. 2019), the latter presented higher specific activity values in potassium phosphate buffer instead of ABF.
The reasons for this buffer-induced change in activity are unknown. However, this observation has been reported for other similar enzymes; for instance, Pseudomonas putida 4-cinnamoyl CoA ligase activity reached its maximum at pH 6.7 in potassium phosphate buffer but showed 30% less activity in Tris–HCl buffer 0.5 M in this same pH (Zenk et al. 1980). This was also observed in 4-coumarate CoA ligase from spruce, which displayed maximum activity in potassium phosphate buffer pH 7.5 and roughly half the activity in Tris–HCl buffer in the same pH (Lüderitz et al. 1982). Sarni and colleagues researched buffer and pH-related changes in cinnamoyl-CoA reductase and cinnamyl alcohol dehydrogenase activities. They found different activity profiles in 100 mM KH2PO4/NaOH, Tris maleate/Na2OH, KH2PO4/Na2HPO4, KH2PO4/Na2HPO4, and citrate/NaOH buffers (Sarni et al. 1984). The buffer system used for purification can also exert influence in activity, as reported by Sofeo and colleagues when purifying an acetyl-CoA synthetase using HEPES or Tris–HCl buffer (higher activity on the former) (Sofeo et al. 2019).
The LM-FCS2 structural data presented in Figs. 4 and 5 corroborate with the optimal pH results from the biochemical characterization, in which little to no activity is detected at acidic pH values. Interestingly, poor activity and stability in low pHs were also observed for FCS1 (Sodré et al. 2019), which was retrieved from the same microbial community (Moraes et al. 2018). LM-FCS2 shares the same highly conserved histidine residue as FCS1 (FCS1: H267; LM-FCS2: H205) and another nucleoside diphosphate (NDP)-forming acyl-CoA synthetases homologs (Supplemental Fig. S6a), such as E. coli succinyl-CoA synthetase (SCS; PDB: 1SCU_A) (Wolodko et al. 1994), Candidatus Korachaeum cryptofilum NDP-forming acetyl-CoA synthetase (PDB: 4XYL_A) (Weiße et al. 2016), and Pyrococcus furiosus ADP-forming acetyl-CoA synthetase (ACS; PDB: 2CSU) (Bräsen et al. 2008). In addition, 4-coumarate: CoA-ligases from Arabidopsis thaliana (At4CL1, At4CL2, At4CL3, At4CL5) and Populus tremuloides (Pt4CL1 and Pt4CL2) also present this conserved histidine residue (Hu et al. 1998; Schneider et al. 2003). It has been shown in E. coli SCS and P. furiosus ACS that this residue is transiently phosphorylated during catalysis through phosphoramidite bonds, which are more unstable and prone to degradation in acidic pHs than phosphodiester bonds presented by other amino acids (Wolodko et al. 1994; Klumpp and Krieglstein 2002; Bräsen et al. 2008). Similarly, LM-FCHL2 presents higher structural stability at alkaline pHs, which agrees with its capacity of converting feruloyl-CoA to vanillin in reaction media at pH 8.6 or even using lignin-rich alkaline lignocellulosic hydrolysate as substrate.
Remarkably, LM-FCS2 can utilize sinapic acid as a substrate. Previous studies investigating A. thaliana At4CL2 mutants disclosed a 12-amino acid motif on the substrate-binding pocket that grants specificity to different hydroxycinnamic acids (Schneider et al. 2003). The binding of larger molecules, such as ferulic and sinapic acids, was conditioned to the presence of small amino acids in the positions correspondent to At4CL2’s Met-293 or Lys-320 (Supplemental Fig. S6b). In addition, the binding of sinapic acid in At4CL2 was prevented by the amino acid residues Val-355 and Leu-356, and deletion of either promoted conversion of sinapic acid in 4CL2 ferulic acid-converting mutants (however, deletion of both residues completely obliterated activity). Indeed, previously characterized homologous 4CLs from A. thaliana (At4CL5; At3g21030) and Glycine max (Gm4CL1), capable of converting sinapic acid, harbored deletions on position correspondent to Leu-356 (Lindermayr et al. 2002; Schneider et al. 2003). Interestingly, LM-FCS2 converts sinapic acid but does not present deletions in this region; instead, the residue corresponding to At4CL2’s Leu-356 is a threonine (Supplemental Fig. S6b). It is unclear whether substituting non-polar leucine for polar and slightly smaller threonine is the only factor to grant sinapic acid-binding capability in LM-FCS2. Conversely, Rhodococcus jostii RHA1 hydroxycinnamoyl-CoA synthetase CouL, which does not bind sinapic acid, contains substitutions in this region. The residues correspond to At4CL2’s Val-355 and Leu-356 are serine and valine, respectively (Otani et al. 2014).
Interestingly, LM-FCHL2 was able to produce vanillin directly from FA without the presence of LM-FCS2 to produce the feruloyl-CoA intermediate. The CoA-independent deacetylation of FA, yielding vanillin and acetic acid, has been suggested for some microorganisms; in this case, the double bond of trans-ferulic acid is hydrated to 4-hydroxy-3-methoxyphenyl-β-hydroxy propionic acid and subsequently cleaved in an aldolase reaction (Toms and Wood 1970; Priefert et al. 2001). However, clear evidence of possible genes and enzymes involved in this pathway is lacking, in part due to the difficulty in producing the unstable transient intermediate 4-hydroxy-3-methoxyphenyl-β-hydroxy propionic acid.
In conclusion, the biochemical and biophysical characterization of two novel biocatalysts active in hydroxycinnamic acids has been presented. Both enzymes presented interesting features: LM-FCS2 showed stability in alkaline pHs and was able to convert a wide array of p-hydroxycinnamic acids to their respective CoA-thioesters, including bulky sinapic acid; LM-FCHL2 efficiently converted feruloyl-CoA and p-coumaroyl-CoA into vanillin and 4-hydroxybenzaldehyde, respectively, and could produce vanillin directly from FA — a remarkable feature indicative of a possible role in CoA-independent deacetylation of FA. The coupled reaction of LM-FCS2 and LM-FCHL2 produced vanillin, not only from commercial FA but also from a crude lignocellulosic hydrolysate.
This work builds upon previous studies in the valorization of biomass and lignin, offering further options for the biocatalytic production of high-value molecules from these feedstocks. We showed the feasibility of employing enzymatic strategies to detoxify and convert lignocellulosic hydrolysates into compounds of commercial interest, confirming that the FCS and FCHL are relevant enzymes to be applied in the context of lignocellulose biorefineries.
Data availability
The data supporting this study’s findings are available from the corresponding author upon reasonable request.
References
Achterholt S, Priefert H, Steinbüchel A (2000) Identification of Amycolatopsis sp. strain HR167 genes, involved in the bioconversion of ferulic acid to vanillin. Appl Microbiol Biotechnol 54(6):799–807
Banerjee G, Chattopadhyay P (2019) Vanillin biotechnology: the perspectives and future. J Sci Food Agric 99:499–506. https://doi.org/10.1002/jsfa.9303
Becker J, Wittmann C (2019) A field of dreams: lignin valorization into chemicals, materials, fuels, and health-care products. Biotechnol Adv 37(6):107360. https://doi.org/10.1016/j.biotechadv.2019.02.016
Bennett JP, Bertin L, Moulton B, Fairlamb IJS, Brzozowski AM, Walton NJ, Grogan G (2008) A ternary complex of hydroxycinnamoyl-CoA hydratase-lyase (HCHL) with acetyl-CoA and vanillin gives insights into substrate specificity and mechanism. Biochem J 414:281–289. https://doi.org/10.1042/BJ20080714
Bräsen C, Schmidt M, Grötzinger J, Schönheit P (2008) Reaction mechanism and structural model of ADP-forming acetyl-CoA synthetase from the hyperthermophilic archaeon Pyrococcus furiosus: evidence for a second active site histidine residue. J Biol Chem 283:15409–15418. https://doi.org/10.1074/jbc.M710218200
Brenelli LB, Mandelli F, Mercadante AZ, Rocha GJ de M, Rocco SA, Craievich AF, Gonçalves AR, Centeno D da C, de Oliveira Neto M, Squina FM (2016) Acidification treatment of lignin from sugarcane bagasse results in fractions of reduced polydispersity and high free-radical scavenging capacity. Ind Crops Prod 83:94–103. https://doi.org/10.1016/j.indcrop.2015.12.013
Chakraborty D, Gupta G, Kaur B (2016) Metabolic engineering of E. coli TOP10 for production of vanillin through FA catabolic pathway and bioprocess optimization using RSM. Protein Expr Purif 128:123–133. https://doi.org/10.1016/j.pep.2016.08.015
Chakraborty D, Selvam A, Kaur B, Wong JWC, Karthikeyan OP (2017) Application of recombinant Pediococcus acidilactici BD16 (fcs +/ech +) for bioconversion of agrowaste to vanillin. Appl Microbiol Biotechnol 101:5615–5626. https://doi.org/10.1007/s00253-017-8283-8
Chen Z, Wan C (2017) Biological valorization strategies for converting lignin into fuels and chemicals. Renew Sustain Energy Rev 73:610–621. https://doi.org/10.1016/j.rser.2017.01.166
Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr 66:12–21. https://doi.org/10.1107/S0907444909042073
Chen QH, Xie DT, Qiang S, Hu CY, Meng YH (2022) Developing efficient vanillin biosynthesis system by regulating feruloyl-CoA synthetase and enoyl-CoA hydratase enzymes. Appl Microbiol Biotechnol 106:247–259. https://doi.org/10.1007/s00253-021-11709-w
Ciriminna R, Fidalgo A, Meneguzzo F, Parrino F, Ilharco LM, Pagliaro M (2019) Vanillin: the case for greener production driven by sustainability megatrend. ChemistryOpen 8:660–667. https://doi.org/10.1002/open.201900083
Costa MA, Bedgar DL, Moinuddin SGA, Kim K-W, Cardenas CL, Cochrane FC, Shockey JM, Helms GL, Amakura Y, Takahashi H, Milhollan JK, Davin LB, Browse J, Lewis NG (2005) Characterization in vitro and in vivo of the putative multigene 4-coumarate:CoA ligase network in Arabidopsis: syringyl lignin and sinapate/sinapyl alcohol derivative formation. Phytochemistry 66(17):2072–2091. https://doi.org/10.1016/j.phytochem.2005.06.022
Edgar RC (2004a) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Edgar RC (2004b) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:1–19. https://doi.org/10.1186/1471-2105-5-113
Fache M, Boutevin B, Caillol S (2016) Vanillin production from lignin and its use as a renewable chemical. ACS Sustain Chem Eng 4:35–46. https://doi.org/10.1021/acssuschemeng.5b01344
Fleige C, Meyer F, Steinbüchel A (2016) Metabolic engineering of the actinomycete Amycolatopsis sp. strain ATCC 39116 towards enhanced production of natural vanillin. Appl Environ Microb 82(11):3410–3419. https://doi.org/10.1128/AEM.00802-16
Galadima AI, Salleh MM, Hussin H, Chong CS, Yahya A, Mohamad SE, Abd-Aziz S, Yusof NNM, Naser MA, Al-Junid AFM (2020) Biovanillin: production concepts and prevention of side product formation. Biomass Convers Biorefinery 10:589–609. https://doi.org/10.1007/s13399-019-00418-0
Ghosh S, Sachan A, Sen SK, Mitra A (2007) Microbial transformation of ferulic acid to vanillic acid by Streptomyces sannanensis MTCC 6637. J Ind Microbiol Biotechnol 34(2):131–138. https://doi.org/10.1007/s10295-006-0177-1
Graf N, Altenbuchner J (2014) Genetic engineering of Pseudomonas putida KT2440 for rapid and high-yield production of vanillin from ferulic acid. Appl Microbiol Biotechnol 98:137–149. https://doi.org/10.1007/s00253-013-5303-1
Hu WJ, Kawaoka A, Tsai CJ, Lung J, Osakabe K, Ebinuma H, Chiang VL (1998) Compartmentalized expression of two structurally and functionally distinct 4-coumarate:CoA ligase genes in aspen (Populus tremuloides). Proc Natl Acad Sci U S A 95:5407–5412. https://doi.org/10.1073/pnas.95.9.5407
Irmak S (2019) Challenges of biomass utilization for biofuels. In: Abomohra AE (ed) Biomass for Bioenergy - Recent Trends and Future Challenges. IntechOpen. https://doi.org/10.5772/intechopen.83752
Kaur B, Chakraborty D, Kumar B (2014) Metabolic engineering of Pediococcus acidilactici BD16 for production of vanillin through ferulic acid catabolic pathway and process optimization using response surface methodology. Appl Microbiol Biotechnol 98:8539–8551. https://doi.org/10.1007/s00253-014-5950-x
Klumpp S, Krieglstein J (2002) Phosphorylation and dephosphorylation of histidine residues in proteins. Eur J Biochem 269:1067–1071
Kozin MB, Svergun DI (2001) Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34:33–41
Lange L, Connor KO, Arason S, Bundgård-Jørgensen U, Canalis A, Carrez D, Gallagher J, Gøtke N, Huyghe C, Jarry B, Llorente P, Marinova M, Martins LO, Mengal P, Paiano P, Panoutsou C, Rodrigues L, Stengel DB, van der Meer Y, Vieira H (2021) Developing a sustainable and circular bio-based economy in eu: by partnering across sectors, upscaling and using new knowledge faster, and for the benefit of climate, environment & biodiversity, and people & business. Front Bioeng Biotechnol 8:619066. https://doi.org/10.3389/FBIOE.2020.619066
Lee D, Douglas CJ (1996) Two divergent members of a tobacco 4-coumarate:coenzyme A Ligase (4CL) gene family (cDNA structure, gene inheritance and expression, and properties of recombinant proteins). Plant Physiol 112(1):193–205. https://doi.org/10.1104/pp.112.1.193
Liberato MV, Araújo JN, Sodré V, Gonçalves TA, Vilela N, Moraes EC, Garcia W, Squina FM (2020) The structure of a prokaryotic feruloyl-CoA hydratase-lyase from a lignin-degrading consortium with high oligomerization stability under extreme pHs. Biochim Biophys Acta Proteins Proteomics 1868:140344. https://doi.org/10.1016/j.bbapap.2019.140344
Lima CS, Rabelo SC, Ciesielski PN, Roberto IC, Rocha GJM, Driemeier C (2018) Multiscale alterations in sugar cane bagasse and straw submitted to alkaline deacetylation. ACS Sustain Chem Eng 6:3796–3804. https://doi.org/10.1021/acssuschemeng.7b04158
Lindermayr C, Möllers B, Fliegmann J, Uhlmann A, Lottspeich F, Meimberg H, Ebel J (2002) Divergent members of a soybean (Glycine max L.) 4-coumarate:coenzyme A ligase gene family: primary structures, catalytic properties, and differential expression. Eur J Biochem 269:1304–1315. https://doi.org/10.1046/j.1432-1033.2002.02775.x
Lozoya E, Hoffmann H, Douglas C, Schulz W, Scheel D, Hahlbrock K (1988) Primary structures and catalytic properties of isoenzymes encoded by the two 4-coumarate: CoA ligase genes in parsley. Eur J Biochem 176:661–667. https://doi.org/10.1111/j.1432-1033.1988.tb14328.x
Lüderitz T, Schatz G, Grisebach H (1982) Enzymic synthesis of lignin precursors purification and properties of 4-coumarate:CoA ligase from cambial sap of spruce (Picea dies L.). Eur J Biochem 123:583–586. https://doi.org/10.1111/j.1432-1033.1982.tb06572.x
Masai E, Harada K, Peng X, Kitayama H, Katayama Y, Fukuda M (2002) Cloning and characterization of the ferulic acid catabolic genes of Sphingomonas paucimobilis SYK-6. Appl Environ Microbiol 68:4416–4424. https://doi.org/10.1128/aem.68.9.4416-4424.2002
Mathew S, Abraham TE (2006) Bioconversions of ferulic acid, an hydroxycinnamic acid. Crit Rev Microbiol 32:115–125. https://doi.org/10.1080/10408410600709628
Moraes EC, Alvarez TM, Persinoti GF, Tomazetto G, Brenelli LB, Paixão DAA, Ematsu GC, Aricetti JA, Caldana C, Dixon N, Bugg TDH, Squina FM (2018) Lignolytic-consortium omics analyses reveal novel genomes and pathways involved in lignin modification and valorization. Biotechnol Biofuels 11:75. https://doi.org/10.1186/s13068-018-1073-4
Otani H, Lee Y, Eltis LD (2014) Characterization of p-hydroxycinnamate catabolism in a soil actinobacterium. J Bacteriol 196:4293–4303. https://doi.org/10.1128/JB.02247-14
Overhage J, Priefert H, Steinbüchel A (1999) Biochemical and genetic analyses of ferulic acid catabolism in Pseudomonas sp. Strain HR199. Appl Environ Microbiol 65(11):4837–47
Pandey M, Singhal B (2021) Metagenomics: adding new dimensions in bioeconomy. Biomass Convers Biorefinery:1–20. https://doi.org/10.1007/S13399-021-01585-9
Paul V, Rai DC, Ramyaa RL, Srivastava SK, Tripathi AD (2021) A comprehensive review on vanillin: its microbial synthesis, isolation and recovery. Food Biotechnol 35:22–49. https://doi.org/10.1080/08905436.2020.1869039
Priefert H, Rabenhorst J, Steinbüchel A (2001) Biotechnological production of vanillin. Appl Microbiol Biotechnol 56:296–314. https://doi.org/10.1007/s002530100687
Rosazza JPN, Huang Z, Dostal L, Volm T, Rousseau B (1995) Biocatalytic transformations of ferulic acid: an abundant aromatic natural product. J Ind Microbiol 15:457–471
Sarni F, Grand C, Am B (1984) Purification and properties of cinnamoyl-CoA reductase and cinnamyl alcohol dehydrogenase from poplar stems (Populus X euramericana). Eur J Biochem 139:259–265. https://doi.org/10.1111/j.1432-1033.1984.tb08002.x
Schneider K, Hövel K, Witzel K, Hamberger B, Schomburg D, Kombrink E, Stuible HP (2003) The substrate specificity-determining amino acid code of 4-coumarate:CoA ligase. Proc Natl Acad Sci U S A 100:8601–8606. https://doi.org/10.1073/pnas.1430550100
Sharma A, Sharma P, Singh J, Singh S, Nain L (2020) Prospecting the potential of agroresidues as substrate for microbial flavor production. Front Sustain Food Syst 4:1–11. https://doi.org/10.3389/fsufs.2020.00018
Shen M, Sali A (2006) Statistical potential for assessment and prediction of protein structures. Protein Sci 15:2507–2524. https://doi.org/10.1110/ps.062416606
Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244-248
Sodré V, Araujo JN, Gonçalves TA, Vilela N, Braz ASK, Franco TT, de Oliveira NM, Damasio AR de L, Garcia W, Squina FM (2019) An alkaline active feruloyl-CoA synthetase from soil metagenome as a potential key enzyme for lignin valorization strategies. PLoS One 14:e0212629. https://doi.org/10.1371/journal.pone.0212629
Sodré V, Vilela N, Tramontina R, Squina FM (2021) Microorganisms as bioabatement agents in biomass to bioproducts applications. Biomass Bioenergy 151:106161. https://doi.org/10.1016/j.biombioe.2021.106161
Sofeo N, Hart JH, Butler B, Oliver DJ, Yandeau-Nelson MD, Nikolau BJ (2019) Altering the substrate specificity of acetyl-CoA synthetase by rational mutagenesis of the carboxylate binding pocket. ACS Synth Biol 8:1325–1336. https://doi.org/10.1021/acssynbio.9b00008
Souza GM, Ballester MVR, de Brito Cruz CH, Chum H, Dale B, Dale VH, Fernandes ECM, Foust T, Karp A, Lynd L, Maciel Filho R, Milanez A, Nigro F, Osseweijer P, Verdade LM, Victoria RL, Van der Wielen L (2017) The role of bioenergy in a climate-changing world. Environ Dev 23:57–64. https://doi.org/10.1016/J.ENVDEV.2017.02.008
Souza TV, Nascimento JA, da Silva VM, Liberato MV, Pimentel AC, Alvarez TM, Squina FM, Garcia W (2016) Chemical stability of a cold-active cellulase with high tolerance toward surfactants and chaotropic agent. Biotechnol Rep 9:1–8
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Svergun DI, Petoukhov MV, Koch MH (2001) Determination of domain structure of proteins from X-ray solution scattering. Biophys J 80:2946–2953. https://doi.org/10.1016/S0006-3495(01)76260-1
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. https://doi.org/10.1080/10635150701472164
Toms A, Wood JM (1970) Degradation of trans-ferulic acid by Pseudomonas acidovorans. Biochemistry 9:337–343. https://doi.org/10.1021/bi00804a021
Webb B, Sali A (2016) Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 54:5.6.1-5.6.37. https://doi.org/10.1002/cpbi.3
Weiße RHJ, Faust A, Schmidt M, Schönheit P, Scheidig AJ (2016) Structure of NDP-forming acetyl-CoA synthetase ACD1 reveals a large rearrangement for phosphoryl transfer. Proc Natl Acad Sci U S A 113:E519–E528. https://doi.org/10.1073/pnas.1518614113
Wolodko WT, Fraser ME, James MN, Bridger WA (1994) The crystal structure of succinyl-CoA synthetase from Escherichia coli at 2.5-A resolution. J Biol Chem 269:10883–10890
Yang W, Tang H, Ni J, Wu Q, Hua D, Tao F, Xu P (2013) Characterization of two Streptomyces enzymes that convert ferulic acid to vanillin. PLoS One 8:e67339. https://doi.org/10.1371/journal.pone.0067339
Zenk MH, Ulbrich B, Busse J, Stöckigt A (1980) Procedure for the enzymatic synthesis and isolation of cinnamoyl-CoA thiolesters using a bacterial system. Anal Biochem 101:182–187
Acknowledgements
The authors would like to acknowledge the facilities Beamline SAXS-2 (CNPEM) and the Process Development Laboratories (CNPEM) for structural support.
Funding
The study was funded by FAPESP (2015/50590–4, 20/05784–3, 2015/50612–8, 2017/22669–0, 15/23279–6, 17/16089–1, 2018/18101–1, 2017/17275–3, 2017/16976–8) and CNPq (428527/2018–3, 306279/2020–7, 404654/2018–5, 304816/2017–5, 305740/2017–2, 422132/2018–7, 305816/2020–9).
Author information
Authors and Affiliations
Contributions
TAG: conceptualization, methodology, investigation, writing — reviewing and editing; VS: conceptualization, investigation, original draft preparation, writing — reviewing and editing; SNS: methodology, investigation, writing — reviewing and editing; NV: investigation, reviewing and editing; GT: original draft preparation, methodology, investigation, writing — reviewing and editing; JNA: methodology, investigation, reviewing and editing; JRCM: methodology, investigation, data curation, reviewing and editing; TPF: investigation, data curation, reviewing and editing; AD: investigation, data curation, writing — reviewing and editing; WG: conceptualization, original draft preparation, data curation, writing — reviewing and editing; FMS: conceptualization, data curation, writing—reviewing and editing.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies involving human participants or animals.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gonçalves, T.A., Sodré, V., da Silva, S.N. et al. Applying biochemical and structural characterization of hydroxycinnamate catabolic enzymes from soil metagenome for lignin valorization strategies. Appl Microbiol Biotechnol 106, 2503–2516 (2022). https://doi.org/10.1007/s00253-022-11885-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-11885-3