
Abstract
Mitogen-activated protein kinase (MAPK) cascades are broadly conserved and play essential roles in multiple cellular processes, including fungal development, pathogenicity, and secondary metabolism. Their function, however, also exhibits species and strain specificity. Penicillium oxalicum secretes plant-biomass-degrading enzymes (PBDEs) that contribute to the carbon cycle in the natural environment and to utilization of lignocellulose in industrial processes. However, knowledge of the MAPK pathway in P. oxalicum has been relatively limited. In this study, comparative transcriptomic analysis of P. oxalicum, cultured on different carbon sources, found ten putative kinase genes with significantly modified transcriptional levels. Six of these putative kinase genes were knocked out in the parental strain ∆PoxKu70, and deletion of the gene, Fus3/Kss1-like PoxMK1 (POX00158), resulted in the largest reduction (91.1%) in filter paper cellulase production. Further tests revealed that the mutant ∆PoxMK1 lost 37.1 to 92.2% of PBDE production, under both submerged- and solid-state fermentation conditions, compared with ∆PoxKu70. In addition, the mutant ∆PoxMK1 had reduced vegetative growth and increased pigment biosynthesis. Comparative transcriptomic analysis showed that PoxMK1 deletion from P. oxalicum downregulated the expression of major PBDE genes and known regulatory genes such as PoxClrB and PoxCxrB, whereas the transcription of pigment biosynthesis-related genes was upregulated. Comparative phosphoproteomic analysis revealed that PoxMK1 deletion considerably modified phosphorylation of key transcription- and signal transduction-associated proteins, including transcription factors Mcm1 and Atf1, RNA polymerase II subunits Rpb1 and Rpb9, MAPK-associated Hog1 and Ste7, and cyclin-dependent kinase Kin28. These findings provide novel insights into understanding signal transduction and regulation of PBDE gene expression in fungi.
Key points
• PoxMK1 is involved in expression of PBDE- and pigment synthesis-related genes.
• PoxMK1 is required for vegetative growth of P. oxalicum.
• PoxMK1 is involved in phosphorylation of key TFs, kinases, and RNA polymerase II.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is a fundamental property of living organisms to sense and respond appropriately to external environmental stimuli, through signal transduction pathways, in which protein kinases play central roles by mediating protein phosphorylation. The mitogen-activated protein kinase (MAPK) signaling pathway is evolutionarily conserved in all eukaryotes, from yeast to mammals, with diverse regulatory functions in cell differentiation, proliferation, stress response, aging, and apoptosis (Hagiwara et al. 2016). The MAPK pathway is composed of three sequentially activated kinases, MAPK kinase kinase (MAPKKK), MAPK kinase (MAPKK), and MAPK. The activated form of MAPK phosphorylates, and in turn activates, downstream target proteins (Martínez-Soto and Ruiz-Herrera 2017).
Three MAPK signaling pathways have been identified in many filamentous fungi, which are orthologous to Hog1, Slt2, and Fus3/Kss1 in Saccharomyces cerevisiae (Segorbe et al. 2017; Tong and Feng 2019). Of these, the Fus3/Kss1-like pathway is involved in multiple physiological and developmental processes, such as sexual and asexual sporulation, vegetative growth, and production of secondary metabolites (Martínez-Soto and Ruiz-Herrera 2017). For example, deletion of MpkB, or AnFus3 from Aspergillus, resulted in uncompleted sexual development, aberrant conidiophores, slowed hyphal growth, and low aflatoxin production (Priegnitz et al. 2015). In contrast, deletion of Fus3-like Tmk1 from Trichoderma reesei accelerated vegetative growth on several carbon sources, such as Avicel (Wang et al. 2017), whereas deletion of Tmk1 had no effect on growth and sporulation, in the presence of sugarcane bagasse (de Paula et al. 2018). Wang et al. (2017) reported that Tmk1 negatively affected cellulase production in T. reesei, but had no effect on the expression of cellulase genes in response to Avicel plus wheat bran. Conversely, de Paula et al. (2018) found that deletion of Tmk1 from T. reesei cultured on sugarcane bagasse reduced cellulase production, by repressing the expression of major cellulase genes. In addition, Tmk1 mediates the expression of xylanase genes. These findings suggest that the functions of Tmk1 vary between different fungal species, different strains of the same species, and different carbon sources.
The filamentous fungus, Penicillium oxalicum, is one of the most important industrial workhorses for cellulase production in China, because of its high β-glucosidase (BGL) activity and superior hydrolytic performance (Yan et al. 2017). In P. oxalicum, the expression of genes encoding plant-biomass-degrading enzymes (PBDEs) such as cellulase, xylanase, and amylase is regulated by their corresponding signal transduction pathways and downstream transcription factors (TFs). P. oxalicum can produce large amounts of PBDEs during both submerged-state (SmF) and solid-state fermentation (SSF) (Zhao et al. 2019a). Most previous studies, however, were carried out under SmF conditions.
In this study, we identified a Fus3/Fss1-like PoxMK1 that was induced by wheat bran at the transcriptional level. The effects of PoxMK1 on PBDE production during both SmF and SSF, vegetative growth, and pigment biosynthesis were characterized. Subsequently, transcriptomic and phosphoproteomic analysis were employed to elucidate the mechanism of PoxMK1 function.
Materials and methods
Fungal strains
P. oxalicum wild-type strain HP7-1 (China General Microbiological Culture Collection [CGMCC], #10781) and the parental strain ΔPoxKu70 (CGMCC 3.15650) were described previously (Zhao et al. 2016). The strain ΔPoxKu70 was generated by deleting the gene PoxKu70, which is involved in a non-homologous end-joining pathway, in the wild-type strain HP7-1. The deletion mutants of six putative kinase genes (Table 1) and the complementation strain CPoxMK1 were constructed by homologous recombination techniques, in the parental strains ΔPoxKu70 and ΔPoxMK1, respectively. All these P. oxalicum strains were maintained on potato dextrose agar (PDA; Sigma-Aldrich, Darmstadt, Germany) plates and stored at 4 °C for up to 1 month. For long-term storage, fungal strains were grown on PDA plates at 28 °C for 6 days, and the asexual spores produced were harvested with sterile H2O containing 0.1% (w/v) Tween-80 (Sango, Shanghai, China). The final concentration of spores was adjusted to 1 × 108 per milliliter. Fungal spores were stored in 25% (v/v) glycerol (Sango) at − 80 °C.
Mutant generation and complementation
Deletion mutants of putative kinase-encoding genes and their corresponding complementation strains were constructed by homologous recombination, as described previously (Yan et al. 2017). A knockout cassette was constructed for gene deletion, by fusion PCR with specific primer pairs (Supplementary Table S1), to replace each target gene in the parental strain ΔPoxKu70. The knockout cassette consisted of the G418-resistance gene fragment and each of the upstream and downstream DNA fragments flanking the target gene (Supplementary Fig. S1). Similarly, for the complementation strains of ΔPoxMK1, a complementary cassette was generated by fusion PCR of four DNA fragments to replace an aspartic protease gene PoxPepA, consisting of the complete PoxMK1 coding region, along with its native promoter and terminator, the bleomycin resistance gene fragment, and the left- and right-flanking sequences of the PoxPepA gene (Wang et al. 2018). The cassette fragments for knockout, or complementation, were generated and then directly transformed into fresh protoplasts of ΔPoxKu70 and ΔPoxMK1. The transformants were selected on a PDA medium supplemented with the antibiotics G418 (0.5 mg/mL; Biotopped, Beijing, China) and bleomycin (0.08 mg/mL; Roche, Basel, Switzerland), as selective markers for the deletion mutants and complementation strains, respectively.
Enzymatic activity assays
Sample preparation
For preliminary screening of mutants in putative kinase genes, 1 × 108 spores of each P. oxalicum mutant were directly inoculated into modified minimal medium (MMM; 4.0 g/L KH2PO4, 4.0 g/L (NH4)2SO4, 0.6 g/L MgSO4·7H2O, 0.6 g/L CaCl2, 1.0 mL/L Tween-80; 0.005 g/L FeSO4·7H2O, 0.0016 g/L MnSO4, 0.0017 g/L ZnCl2 and 0.002 g/L CoCl2; pH 5.0; Yan et al. 2017) with Avicel (2.0% w/v) and cultured for 6 days at 28 °C, with shaking at 180 rpm. Subsequently, the culture was centrifuged at 11,300 × g, for 10 min at 4 °C, then the supernatant (crude enzyme solution) was used directly to assay enzymatic activity.
For transfer experiments from glucose, 1.0 × 108 fresh spores of P. oxalicum mutants were inoculated into MMM with 1% (w/v) glucose and pre-cultured at 28 °C with shaking, at 180 rpm for 24 h. Hyphae (~ 1.0 g) were harvested and transferred, for SmF, into liquid medium (MMM with 2% w/v Avicel or 1.0% w/v soluble corn starch [SCS]). For SSF, hyphae (~ 1.0 g) were harvested and transferred onto solid medium (6.0 g rice straw, 4.0 g wheat bran, and 20 mL mineral salt solution [2.5 g/L MgSO4·7H2O, 2.5 g/L KH2PO4, 5.0 mg/L FeSO4·7H2O, 1.6 mg/L MnSO4·H2O, 1.4 mg/L ZnSO4·7H2O, 2.0 mg/L CoCl2, 5.0 g/L yeast extract, and 1.0 mL/L Tween-80, pH 5.0]) and cultured for 2–4 days at 28 °C. Crude enzyme solution was extracted from liquid media as above and from solid media as described previously (Su et al. 2017).
Enzymatic assay procedures
The activity assays for cellulase and xylanase activities were as reported previously (Zhao et al. 2016), and that for amylase activity was as described previously (Xu et al. 2016). The activity unit (U) for filter paper cellulase (PFase), carboxymethylcellulase (CMCase), xylanase, soluble starch-degrading enzyme (SSDE), and raw starch-degrading enzyme (RSDE) was defined as the amount of enzyme required to produce 1 μmol of glucose or xylose per min from the substrate under optimal conditions; the unit (U) for p-nitrophenyl-β-cellobiosidase (pNPCase) and p-nitrophenyl-β-glucopyranosidase (pNPGase) activity was defined as the amount of enzyme required to produce 1 μmol of pNP per min from the substrates.
Total DNA extraction and Southern hybridization analysis
Genomic DNA extraction from P. oxalicum, using a modified phenol-chloroform method, was carried out as described previously (Zhao et al. 2016). Briefly, 1 × 108 spores were inoculated into liquid complete medium (CM; 10.0 g/L d-glucose, 2.0 g/L peptone, 1.0 g/L yeast extract, 1.0 g/L acid hydrolyzed casein, and 50 mL/L 20 × nitrate solution [120.0 g/L NaNO3, 10.40 g/L KCl, 10.40 g/L MgSO4·7H2O, 30.40 g/L KH2PO4; pH 6.5]) at 28 °C for 24 h, with shaking at 180 rpm. The mycelia were collected and used for DNA extraction.
Total DNAs were digested with PstI (TaKaRa Bio Inc. Dalian, China). The resulting DNA fragments were fractionated on a 0.8% (w/v) agarose gel and transferred onto Hybond N+ (GE Healthcare Limited, Amersham, UK). Southern hybridization was performed following the manufacturer’s protocols. The probe for Southern hybridization was amplified by PCR with a primer pair (Supplementary Table S1).
Determination of biomass and intracellular protein
To compare vegetative growth of the different P. oxalicum strains, equal numbers of spores were inoculated into MMM (100 mL), supplemented with different carbon sources and incubated with shaking, at 180 rpm and 28 °C. For glucose (1.0% w/v) or SCS medium, the hyphae were collected every 12 h until 72 h and dried at 50 °C for 48 h, to determine dry weight. For Avicel (2.0% w/v) medium, the growth profile was evaluated by the content of intracellular protein in the mycelia. Protein extraction was carried out as described by Zhao et al. (2016), and total protein concentration was quantified with a Bradford Assay Kit (Pierce Biotechnology, Rockford, IL) according to the manufacturer’s instructions.
Total RNA extraction, real-time quantitative PCR, and RNA-sequencing analysis
For total RNA extraction, spores (1 × 108) were pre-cultured in MMM with glucose (1% w/v) for 24 h, then transferred to liquid medium and solid medium with 2.0% (w/v) Avicel, or 1.0% SCS, respectively. The hyphae were harvested after a shift from glucose at 4, 12, 24, and 48 h for real-time quantitative PCR (RT-qPCR), as well as 24 h for RNA sequencing. Total RNA of fungal strains was extracted using TaKaRa RNAiso Plus (TaKaRa Bio Inc.) in accordance with the manufacturer’s instructions and stored at − 80 °C until needed.
For RT-qPCR analysis, total RNA (1.0 μg) was used as the template to synthesize first-strand cDNA with TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix (Transgen, Beijing, China). Reaction mixtures (20 μL) were prepared using SYBR Premix Ex Taq II (10 μL; TaKaRa Bio Inc.), first-strand cDNA (0.2 μL), and special primers (0.8 μL; 10 μM). Subsequently, RT-qPCR was performed on an ABI 7500 Real-time PCR system (Thermo Fisher Scientific, Waltham, MA). The actin gene (POX09248) of P. oxalicum was used as the internal reference, and the relative gene expression was normalized against that of the parental strain of ∆PoxKu70 and calculated by the 2−∆∆CT method (Livak and Schmittgen 2001). All RT-qPCR assays were performed in triplicate for each sample.
RNA-sequencing (RNA-seq) was carried out at BGI, Shenzhen, China, on an Illumina HiSeq2000 platform. The generated clean reads were mapped onto the referred genome of P. oxalicum strain HP7-1 for functional annotation by BWA software, version 0.7.10-r789, and Bowtie2, version 2.1.0 (Langmead and Salzberg 2012). The relative gene expression abundances were represented by FPKM (fragments per kilobase of exon per million mapped reads) and calculated with RSEM software, version 1.2.12 (Li and Dewey 2011). Differentially expressed genes (DEGs) were screened for by DESeq2 tools (Love et al. 2014) with |log2 fold change| ≥ 1 and probability ≥ 0.8. The reliability of different biological replicates from each sample was assessed by Pearson’s correlation coefficient.
Phosphoproteomic analysis
Mycelia from P. oxalicum mutants ΔPoxKu70 and ΔPoxMK1 were harvested after a shift from glucose to MMM containing Avicel and incubation for 4 h. Total protein was extracted as described previously (Xiong et al. 2014). Total protein samples were sent to BGI (Shenzhen) for quantitative phosphoproteomic sequencing. Briefly, protein (100 μg from each sample) was digested with trypsin (Promega, Madison, WI; 25 μg/g total protein) at 37 °C for 4 h, then the same amount of trypsin was added for a second digestion, for a further 8 h. Subsequently, the resulting peptides were desalted with a Strata X C18 SPE column (Phenomenex, Guangzhou, China) and then labeled with 4-plex iTRAQ reagents (AB Sciex, Foster City, CA) according to the manufacturer’s instructions. After labeling, the tryptic peptides were desalted and enriched using TiO2. The enriched peptides were fractionated by high pH reversed-phase HPLC using a Betasil C18 column (5-μm particle size, 10 × 250 mm; Thermo Fisher).
For LC-MS/MS analysis, a nano-flow LC system (Shimadzu, Kyoto, Japan) coupled with a Q Exactive HF mass spectrometer (Thermo Fisher) was employed. The fractionated peptides, as described above, were dissolved in phase A (2.0% acetonitrile and 0.1% formic acid) and centrifuged at 13000 × g at 4 °C for 15 min. The supernatant was loaded onto a Trap column (Thermo Fisher), to concentrate and desalt the sample, then transferred to a reversed-phase analytical column (Thermo Fisher) to separate the peptides at a flow rate of 0.3 μL/min. The linear gradient elution program started at 5% (v/v) phase B (98% acetonitrile and 0.1% formic acid) for 8 min, increased to 21% at 76 min, 32% at 82 min, and 80% at 90 min. The eluted peptides were detected by tandem mass spectrometry (MS/MS), in the data-dependent acquisition mode.
The resulting phosphoproteomic data were analyzed quantitatively with Proteome Discoverer 1.4 (Thermo Scientific) and Mascot 2.3 (Matrix Science, Boston, MA). The LC-MS/MS spectra were matched against all the proteins of P. oxalicum strain HP7-1. Pearson’s correlation coefficient was used to assess the reliability of three biological replicates from ΔPoxKu70, or ΔPoxMK1, according to the peptide signal intensity (PSI) of each sample. The phosphorylation site location confidence was determined by phosphoRS 3.1 (Taus et al. 2011) with a phosphoRS probability ≥ 0.75. Differentially expressed phosphopeptides were screened for and selected with a threshold of an absolute fold-change of ≥ 1.5 and P < 0.05.
For the analysis of differential phosphopeptide motifs, the MEME software version 5.0.4 (https://meme-suite.org/tools/momo) was used to enrich the kinase phosphorylation motifs with the following settings: verbosity 1, motif width 15, elimination repeats 15, minimum occurrences 20 and score threshold 1.0E-6, central T/P residues with the same modification mass combined, and expansion of modified peptides extracted from protein database of P. oxalicum.
Sequence analysis and deposition
The conserved domain of PoxMK1 (POX00158) was analyzed by SMART online (http://smart.embl.de/). Multiple alignments were carried out using MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/). Construction of the phylogenetic tree was performed with MEGA 7.0, using the neighbor-joining method and 1000 replicates were used for calculating the bootstrap values (Kumar et al. 2016).
The sequence of PoxMK1 and transcriptomic data were submitted to GenBank and SRA at NCBI with accession number MT468552 and GSE154636, respectively. Phosphoproteomic data were deposited in ProteomeXchange with accession number PXD020729.
Results
Deletion of POX00158 in P. oxalicum significantly reduces the production of filter paper cellulase
Comparative transcriptomic data from P. oxalicum HP7-1 grown in MMM containing glucose, wheat bran, or wheat bran plus Avicel, for 72 h (Yan et al. 2017), were re-analyzed; expression of ten genes encoding putative protein kinases showed significant alteration, with a threshold of probability ≥ 0.8 (Table 1). Most of these candidate genes were annotated to encode serine/threonine protein kinases, in particular, genes POX00158 and POX05156 encoded putative MpkB/Fus3 and protein kinase C, respectively. Comparative transcriptional analysis indicated that the transcriptional levels of all the genes, except for POX00158, were downregulated in P. oxalicum strain HP7-1, with a log2 fold change between − 2.68 and − 1.15, when cultured in MMM containing wheat bran, compared with MMM containing glucose. Gene POX00158, however, was upregulated by 95.9% on wheat bran. Five genes, POX01286, POX02455, POX02629, POX04065, and POX07454, were downregulated with a log2 fold change between − 2.28 and − 1.18, when cultured on wheat bran plus Avicel, compared with glucose (Table 1).
To investigate the effects of these putative kinase genes on cellulase production, we tried to knock them out in the P. oxalicum parental strain ∆PoxKu70. A total of six deletion mutants (∆POX00158, ∆POX01286, ∆POX02455, ∆POX02629, ∆POX02702, and ∆POX07454) were successfully constructed and confirmed by PCR amplification (Supplementary Fig. S1A) with specific primers (Supplementary Table S1). All six deletion mutants were directly inoculated into MMM containing Avicel as the sole carbon source and cultured for 6 days; crude enzyme extracts were collected for measurement of filter paper cellulase (FPase) activity. The parental strain ∆PoxKu70 was used as a control. Four of the deletion mutants, i.e., ∆POX00158, ∆POX01286, ∆POX02455, and ∆POX02702, had significantly reduced FPase production (by 9.2–91.1%), compared with ΔPoxKu70. The greatest reduction in FPase production was in ∆POX00158 (Table 1); hence, we selected POX00158 for further investigation.
Southern hybridization analysis was employed to confirm that no additional recombination events had occurred in the ∆POX00158 chromosome. The probe used was generated by PCR, using a special primer pair (Supplementary Table S1). Two bands of approximately 4.5 kb and 2.9 kb appeared in ∆PoxKu70 and ∆POX00158, respectively, which was as expected (Supplementary Fig. S1B).
The complementation strain of ∆POX00158, CPOX00158, was constructed simultaneously, via homologous recombination and confirmed by PCR amplification (Supplementary Fig. S2) using specific primers (Supplementary Table S1).
Gene POX00158 encodes a Fus3/Kss1-like MAPK in P. oxalicum
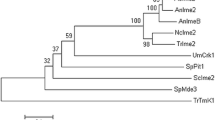
In the genome of P. oxalicum strain HP7-1, POX00158 was 1754 bp in length, contained two introns, and encoded a protein of 478 amino acids. The POX00158 protein contained a conserved serine/threonine protein kinase catalytic (S_TKc) domain (amino acid residues 140–428; E-value = 1.03e-98; Fig. 1a), as determined by Simple Modular Architecture Research Tool (SMART) analysis. NCBI BlastP alignment analyses revealed that POX00158 shared 100%, 97.5%, 96.9%, and 93.1% identity in amino acid sequence with PDE08196 (GenBank accession number EPS33234.1) in P. oxalicum 114-2, MpkB (XP_014534839.1) in Penicillium digitatum Pd1, MpkB (AAF12815) in Aspergillus nidulans FGSC4, and Tmk1 (EGR49073.1) in T. reesei QM6a, respectively. However, POX00158 had a lower identity with Kss1 (60.7%) and Fus3 (59.7%) in S. cerevisiae S288c. Surprisingly, multiple alignment analysis of POX00158 from P. oxalicum strain HP7-1 (Fig. 1a) revealed that it was over 100 amino acids longer than other homologous proteins at the N-terminus. Phylogenetic analysis indicated that POX00158 was closely grouped into the Fus3/Kss1 branch, and clearly separated from MAP kinases Slt2 and Hog1 (Fig. 1b). For the purpose of further study, POX00158 was re-designated as PoxMK1.

Sequence analyses of PoxMK1 (POX00158) from P. oxalicum. a Mutiple alignment analysis. Blue frame represents S_TKc domain. CAA84835.1: KSS1 of Saccharomyces cerevisiae S288C; CAA97038.1: FUS3 of S. cerevisiae S288C. EKV07229.1: PdMpkB of Penicillium digitatum PHI26; EHA18509.1: MpkB of Aspergillus niger ATCC 1015; b Unrooted phylogenetic tree of POX00158 and its orthologs. The phylogenetic tree was constructed using MEGA 7.0 with the neighbor-joining method and a Poisson model. The bootstrap values at the nodes were calculated with 1,000 replicates. S_TKc: serine/threonine protein kinase catalytic domain
PoxMK1 is required for PBDE production in P. oxalicum
The effects of PoxMK1 on the production of PBDEs including cellulase, xylanase, and amylase were investigated in detail under both SSF and SmF conditions. P. oxalicum mutant ∆PoxMK1, complementation strain CPoxMK1, and the parental strain ∆PoxKu70 were pre-cultured in MMM containing glucose for 24 h, then transferred to SmF in MMM containing Avicel or SCS, as well as SSF on wheat bran plus rice straw, for 2–4 days. Enzymatic activity assays determined that cellulase and xylanase production by ∆PoxMK1 decreased by 52.8–82.4% in SmF, except for pNPGase production on day 2, which increased by 174.2% (P < 0.01; Fig. 2(a–e)), compared with ∆PoxKu70. As expected, the production of cellulase and xylanase from SSF of ∆PoxMK1 decreased by 37.1–92.2% (P < 0.01; Fig. 2(h–l)). Surprisingly, the mutant ∆PoxMK1 had reduced production of amylases, including SSDE and RSDE, until 4 days of SCS induction (Fig. 2(f–g)). The complementation strain CPoxMK1 partially restored production of all the assayed PBDEs, compared with that of ∆PoxMK1 (P < 0.01; Fig. 2), suggesting that the changes in PBDE production by ∆PoxMK1 resulted from the deletion of PoxMK1.

PBDE production by P. oxalicum strains. (a–d) Cellulase production and (e) xylanase production, when P. oxalicum strains were cultured in MMM containing Avicel as the sole carbon source by SmF, for 2–4 days at 28 °C, after a transfer from glucose. (a) FPase; (b) CMCase; (c) pNPCase; (d) pNPGase. (f–g) Amylase production when cultured in MMM containing SCS for for 2–4 days by SmF at 28 °C, after a transfer from glucose. (f) SSDE with SCS as substrate; (g) RSDE with raw cassva flour as the substrate. (h–k) Cellulase production and (l) xylanase production when cultured in MMM containing wheat bran plus rice straw as the carbon source for 2–4 days by SSF at 28 °C, after a transfer from glucose. (h) FPase; (i) CMCase; (j) pNPCase; (k) pNPGase. FPase, filter paper cellulase; CMCase, carboxymethylcellulase; pNPCase, p-nitrophenyl-β-cellobiosidase; pNPGase, p-nitrophenyl-β-glucopyranosidase. MMM, modified minimal medium; SCS, soluble corn strach; SSDE, soluble starch-degrading enzyme; RSDE, raw cassava starch-degrading enzyme; SmF, submerged-state fermentation; SSF, solid-state fermentation. ** and * represent P ≤ 0.01 and P ≤ 0.05 by Student’s t test, which show significant differences between the deletion mutant and the parental strain ∆PoxKu70, and between the deletion mutant and the complementation strain CPoxMK1, respectively
PoxMK1 is involved in vegetative growth and pigment biosynthesis in P. oxalicum
To explore the effects of PoxMK1 on vegetative growth of P. oxalicum, fresh spores of both the mutant ∆PoxMK1 and parental strain ∆PoxKu70 were directly inoculated into MMM containing glucose, SCS, or Avicel and CM, cultured for 72 h, and the accumulated mycelial biomass, or mycelial intracellular protein were measured. ∆PoxMK1 had reduced vegetative growth during the later period of culture in MMM, containing either glucose or Avicel, compared with ∆PoxKu70 (Fig. 3(a and b)), whereas there was no significant difference in CM, or MMM containing SCS (Fig. 3(c and d)).

Growth and pigment production of P. oxalicum mutant strains. (a) Growth curves in MMM containing glucose; (b) growth curves in MMM containing Avicel; (c) growth curves in MMM containing SCS; (d) growth curves in complete medium. (e) Pigment biosynthesis by the mutant ΔPoxMK1, the complementation strain CPoxMK1, and the parental strain ΔPoxKu70. All P. oxalicum strains were directly inoculated into the corresponding medium and incubated for 72 h for growth measurement and six days for culture color determination at 28 °C, with shaking at 180 rpm. MMM, modified minimal medium; SCS, soluble corn starch
In addition, when ∆PoxMK1, ∆PoxKu70, and CPoxMK1 were cultured in MMM containing glucose, Avicel, or SCS for 2 days, considerable differences were observed in the colors of the cultures. ∆PoxMK1 cultures were light gray/orange to yellow, whereas ∆PoxKu70 and CPoxMK1 were white to pale orange (Fig. 3(e)). These data suggested that PoxMK1 in P. oxalicum influences vegetative growth and pigment biosynthesis.
Transcriptomic analyses showed that PoxMK1 is involved in global regulation in P. oxalicum
To gain more insights into the effects of PoxMK1 on genome-wide transcriptional profiling, RNA-Seq was employed. Total RNAs were extracted from P. oxalicum strains ∆PoxMK1 and ∆PoxKu70, cultured in MMM containing Avicel as the sole carbon source, for 24 h, after a transfer from glucose. The reproducibility of three biological repeats of each P. oxalicum strain was verified by Pearson’s correlation coefficient (R2 > 0.98) (Supplementary Fig. S3). About 24 million clean reads with a length of 100 bp were obtained from each sample, over 90% of which were mapped onto the HP7-1 genome (Supplementary Table S2). A total of 1037 differentially expressed genes (DEGs) were selected, using a threshold of |log2 fold change| ≥ 1 and probability ≥ 0.8, including 521 upregulated genes (1.0 < log2 fold change < 14.3) and 516 downregulated genes (− 6.9 < log2 fold change < − 1.0) in ∆PoxMK1, relative to ∆PoxKu70 (Table S3). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated that most of these selected DEGs were involved in metabolism (accounting for 80.0%), including carbon metabolism (23.2%), amino acid metabolism (14.6%), and lipid metabolism (6.9%) (Fig. 4a).

Comparative analysis of the transcriptomes from P. oxalicum mutant ΔPoxMK1 and the parental strain ΔPoxKu70 cultured in medium containing Avicel by SmF. a Differentially expressed genes (DEGs) in ΔPoxMK1 annotated by KEGG database. DEGs were selected with a threthod of |log2 fold change| ≥ 1 and probability ≥ 0.8. b DEGs encoding plant cell wall–degrading enzymes (CWDEs). c DEGs encoding putative transcriptional factors. d DEGs encoding putative sugar transporters. FPKM, fragments per kilobase of exon per million mapped reads
Of the 1037 DEGs, 113 encoded carbohydrate-active enzymes (CAZymes), including 69 downregulated genes (− 5.1 < log2 fold change < − 1.0) and 44 upregulated genes (1.0 < log2 fold change < 13.2). Of these, 43 DEGs were annotated to encode plant cell wall–degrading enzymes (CWDEs) and almost all had decreased expression in ∆PoxMK1 compared with that in ∆PoxKu70. In particular, the key cellulase and xylanase genes were included, such as cellobiohydrolase gene POX05587/Cel7A-2, endo-β-1,4-glucanase genes POX05571/Cel7B, POX07535/Cel12A, β-1,4-glucosidase gene POX06835/Bgl3A, and endo-xylanase genes POX00063/Xyn10A and POX06783/Xyn11A (Fig. 4b).
Expression of fungal cellulase and xylanase genes was tightly regulated by transcription factors (TFs). A total of 38 putative TF-encoded DGEs was detected, most of which were annotated as zinc finger proteins (Fig. 4c). Remarkably, there were 12 (POX07025/AbaA, POX06534/BrlA, POX05726, POX01118, POX5692, POX09469, POX01960/ClrB, POX06337, POX04860/HtfA, POX00972/ClrC, POX06759, and POX04420/CxrB) reported as participating in the regulation of fungal PBDE production (Li et al. 2017; Li et al. 2020; Liao et al. 2018; Yan et al. 2017; Zhao et al. 2019a, b), five (POX01257/Ste12, POX06534/BrlA, POX04860/HtfA, POX07025/AbaA, and POX07099/FlbD) contribute to fungal development and conidiation, and two DGEs (POX06534/BrlA and POX07025/AbaA) are related to synthesis of secondary metabolites (Park and Yu 2012).
Carbohydrate uptake by fungi from the extracellular environment, which is also necessary for PBDE induction, requires sugar transporters (Li et al. 2013). Screening for DEGs encoding putative sugar transporters detected 27 genes with a log2 fold change from − 4.4 to 8.4. Of those, 19 genes showed downregulated transcripts. Notably, two genes POX05915 and POX06051 encoding cellodextrin transporters CdtC and CdtD had transcription reduced by 60.5% and 72.5%, respectively, whereas gene POX07576 encoding the homologous protein of glucose transporter RCO-3 in N. crassa (Madi et al. 1997) was upregulated by 4151.8% (Fig. 4d).
Previous experiments confirmed that PoxMK1 influences pigment synthesis in P. oxalicum (Fig. 3(e)), which may result from altered expression of the corresponding genes. Six DEGs were involved in dihydroxynaphthalene melanin (DHN-melanin) biosynthesis (Perez-Cuesta et al. 2020), i.e., POX01390/ayg1, POX01391/abr1, POX01430/alb1/wA, POX01431/abr2/yA, POX03412/arp1, and POX03413/arp2. Their expression was upregulated with a log2 fold change of 5.1–9.7 in ∆PoxMK1, compared with ∆PoxKu70 (Fig. 4e).
RT-qPCR analysis indicated that PoxMK1 is dynamically involved in expression of genes related to PBDE production and pigment synthesis in P. oxalicum
To confirm the findings from transcriptomic analysis and dynamic regulation of PoxMK1, RT-qPCR was employed. The expression of all tested genes decreased by 15.1–97.7% in ∆PoxMK1, compared with ∆PoxKu70 (P < 0.05; Fig. 5(a)). In addition to cellulase genes, the transcription of tested amylase genes first increased considerably, by 410.7–917.6% at 4 h, then decreased sharply by 56.3–94.6% at 12 and 24 h (P < 0.01), during induction by SCS (Fig. 5(b)).

Transcription analyses of selected genes in P. oxalicum mutant strain ΔPoxMK1 by real-time quantitative reverse transcription-PCR. (a) Expression of major cellulase and xylanase genes in the presence of Avicel under SmF conditions, for 4–24 h. (b) Expression of major amylase genes in the presence of soluble corn starch under SmF conditions, for 4–24 h. (c) Expression of major cellulase and xylanase genes in the presence of wheat bran plus rice straw under SSF conditions, for 12–48 h. (d) Expression of the known regulatory genes in the presence of Avicel under SmF conditions, for 4–24 h. POX01960/ClrB and POX00972/ClrC regulate the expression of cellulase and xylanase genes in P. oxalicum, whereas POX06534/BrlA and POX07099/FlbD are involved in asexual development. (e) Expression of the known regulatory genes in the presence of soluble corn starch under SmF conditions, for 4–24 h. POX03890/AmyR positivaly regulates the expression of amylase genes in P. oxalicum. (f) Expression of the known regulatory genes in the presence of wheat bran plus rice straw under SSF conditions, for 12–48 h. (g) Expression of pigment biosynthesis-relative genes. Gene expression levels in the mutant ∆PoxMK1 were normalized against those in the parental strain ∆PoxKu70. ** and * represent P ≤ 0.01 and P ≤ 0.05 by Student’s t test, which show significant differences between the deletion mutant and the parental strain ∆PoxKu70
Similarly, the expression of cellulase and xylanase genes in SSF was reduced to various degrees in ∆PoxMK1, compared with ∆PoxKu70. Four genes, POX04786/Cel6A, POX05587/Cel7A-2, POX01166/Cel5B, and POX06835/Bgl3A, were downregulated by 13.6–91.2% at 12–48 h. Two genes, POX06079 and POX08484/Xyn11A, decreased before 24 h, while both POX05571/Cel7B and POX00063/Xyn10A decreased at 12 h and 48 h, respectively (P < 0.05; Fig. 5(c)).
The expression of several known regulatory genes, related to the expression of PBDE genes, exhibited various changes in ∆PoxMK1. In SmF using Avicel as the sole carbon source, POX01960/ClrB significantly decreased by 21.8–26.0% at 4–24 h. By contrast, POX00972/ClrC increased its expression by 260% and 448% at 4 and 12 h, respectively, while decreased by 74.6% at 24 h (Fig. 5(d)). When cultured by SmF with SCS, the expression of POX01960/ClrB, POX00972/ClrC, and POX03890/AmyR decreased by 31.7–78.5% (Fig. 5(e)). In SSF with wheat bran plus rice straw, the transcriptional levels of both POX01960/ClrB and POX00972/ClrC were downregulated by 21.7–77.0% during the whole induction period, except for POX01960/ClrB at 48 h, which increased by 24.4% (P < 0.05; Fig. 5(f)).
The changes in pigment biosynthesis-related genes, resulting from deletion of PoxMK1, cultured by SmF with Avicel, was further investigated by RT-qPCR. Transcriptional levels of all six above mentioned genes involved in DHN-melanin biosynthesis increased considerably, up to a thousand-fold, during the whole induction period (P < 0.01; Fig. 5(g)). Taken together, these findings indicate that PoxMK1 is involved in both PBDE production and pigment biosynthesis, by regulating transcriptional levels of the corresponding genes.
Phosphoproteomic analysis revealed putative targets of PoxMK1 in P. oxalicum
To further characterize PoxMK1-dependent phosphorylation, comparative phosphoproteomic analysis was performed, between P. oxalicum mutant ∆PoxMK1 and the parental strain ∆PoxKu70, cultured in MMM containing Avicel, for 4 h, after a transfer from glucose. The resulting phosphoproteomic data from three biological replicates of each strain were analyzed by Pearson’s correlation coefficient, which determined that they were reliable, with R > 0.91 (Supplementary Fig. S4). A total of 1649 phosphopeptides, with lengths of 9-45 amino acids, was identified (Supplementary Fig. S5a). Of these, 1409 phosphosites, belonging to 887 phosphoproteins, were further characterized using phosphoRS, with probability ≥ 0.75 as a threshold (Supplementary Fig. S5b), in which the relative frequency of serine/threonine/tyrosine was 79/20/0.8 (Supplementary Fig. S5c), comparable with the reported protein phosphorylation in T. reesei (Nguyen et al. 2016). Most phosphosites were detected in peptides carrying a single phosphate (Supplementary Fig. S5d). Further analysis indicated that the phosphosites identified by PoxMK1 were predicted as shown in Fig. Supplementary S5e.
Using an absolute fold change of ≥ 1.5 and p < 0.05 as thresholds, 770 phosphopeptides were detected, representing 462 unique proteins that were differentially phosphorylated in the mutant ∆PoxMK1, compared with the parental strain ∆PoxKu70 (Supplementary Table S4). These included 121 phosphopeptides from 84 unique proteins with reduced phosphorylation and 649 phosphopeptides from 378 unique proteins with increased phosphorylation. KEGG pathway analysis revealed that these phosphorylated proteins were mainly involved in metabolism and genetic information processing, specifically carbohydrate metabolism and translation (Supplementary Fig. S5f).
Among the 462 phosphorylated proteins, 22 were annotated to bind DNA (Table 2) and most of them were found to have one phosphosite. Only two of these proteins showed reduced phosphorylation, including POX03695 with phospho-Ser at position 4 (S4) and POX04493 with phospho-Ser at position 12 (S12). POX03695 and POX04493 were annotated as RNA polymerase II (RNA Pol II) subunit Rpb9 and SRF-MADS protein Mcm1, respectively. The remaining 20 proteins had increased phosphorylation, suggesting that these resulted indirectly from PoxMK1 depletion (Table 2). One of these, POX03016, a bZIP protein Atf1, with a phosphorylation site at amino acid T136, negatively regulated the production of cellulase and xylanase in P. oxalicum (Zhao et al. 2019b).
In addition to TFs, 11 putative kinase proteins showed altered phosphorylation to various degrees (Table 3). However, only two kinase proteins had decreased phosphorylation, POX07703 (at amino acids S10, S17 and T222) and POX07948 (at S461). POX07703 and POX07948 were annotated as cyclin-dependent kinase 7 (CDK7, or Kin28) and MAPK kinase MAPKK Ste7, respectively. MAPK Hog1 protein, POX06496, had increased phosphorylation, at amino acids T171 and Y173.
Discussion
In this study, we found through comparative transcriptomic analysis that the Fus3/Fss1-like gene, PoxMK1, is induced by culture of P. oxalicum on wheat bran as sole carbon source. Furthermore, PoxMK1 was required for plant-biomass-degrading enzyme (PBDE; cellulase, xylanase and amylase) production under both submerged (SmF)- and solid-state (SSF) fermentation conditions, as well as pigment biosynthesis, vegetative growth, and the expression of their corresponding genes. Comparative transcriptomic and phosphoproteomic analyses indicated that PoxMK1 mediated phosphorylation of key kinases, RNA polymerase II, and transcription factors (TFs), as well as the transcriptional levels of genes encoding essential TFs and sugar transporters (Fig. 6). These findings have expanded knowledge of the functional diversity of Fus3/Fss1-like proteins in fungi and provide novel insights into understanding the mechanism of signal transduction and expressional regulation of fungal PBDE genes.

Proposed schematic model illustrating PoxMK1-dependent functions in P. oxalicum. Under induction of Avicel, PoxMK1 is involved in regulation of PBDE genes through complex pathways. PoxMK1 promotes the phosphorylation of transcription factor POX04493/Mcm1, kinase POX07703/Kin28, and RNA polymerase II subunit POX03695/Rpb9, as well as the expression of essential regulatory genes POX01960/ClrB and POX04420/CxrB, and cellodextrin transporter-encoding genes POX06051/CdtC and POX05915/CdtD. Mcm1 and ClrB synergistically co-regulate expression of cellulase and xylanase genes, and Mcm1 can assist in ClrB recruitment to the promoter regions of target genes. CxrB positively regulates the expression of cellulase and xylanase genes, as well as amylase genes and ClrB (Zhao et al., unpublished). Both CdtC and CdtD are required for cellobiose consumption and induce the expression of cellulase genes. Kinase Kin28 can phosphorylate the largest subunit of RNA polymerase II POX06706/Rpb1, which is involved in DNA transcription, as well as another subunit, Rpb9. In addition, PoxMK1 enables repression of the phosphorylation of the MAPK kinases POX06496/Hog1 and Rpb9, as well as the expression of glucose transporter–encoding gene POX07576/RCO-3. Hog1 phosphorylates and activates the transcription factor POX03016/Atf1, which negatively regulates the production of cellulase and xylanase. RCO-3 facilitates glucose uptake, resulting in carbon catabolite repression. PoxMK1 negatively regulates pigment biosynthesis through repressing the expression of the corresponding genes. Dashed lines indicate that the relationship has not yet been confirmed experimentally
Previous accumulated evidence showed that the regulatory functions of Fus3/Kss1-like kinase depended on both the carbon source and the fungal species/strain. For example, in Fusarium oxysporum, deletion of Fmk1 impacted hyphal growth on both nutrient-poor minimal medium and nutrient-rich medium (Segorbe et al. 2017), whereas in T. reesei, Tmk1 had no effect on fungal vegetative growth, when grown on sugarcane bagasse, but negatively regulated vegetative growth when grown on Avicel. Similarly, the effects of PoxMK1 on vegetative growth in P. oxalicum varied depending on carbon source. There was no significant difference in CM and MMM with SCS, while a decrease in MMM containing either glucose or Avicel (Fig. 3). Regarding the regulation of genes encoding PBDEs, TmK1 positively regulated cellulase genes in the presence of sugarcane bagasse, but did not on wheat bran plus Avicel (de Paula et al. 2018; Wang et al. 2017). However, our findings suggested that PoxMK1 is involved in regulation of cellulase, xylanase, and amylase gene expression in P. oxalicum, independent of both carbon source and fermentation mode.
SSF, a fungal fermentation mode that uses moist solid substrates with low water activity, has been used industrially for three decades to produce a number of extracellular fungal enzymes, including PBDEs and is a potential technology for developing lignocellulosic biomass-based biorefineries (Bentil et al. 2018; Diaz et al. 2016; Marone et al. 2019). SSF offers several advantages over SmF, such as greater enzyme yields (resulting from the reduced catabolite repression), enhanced enzyme stability, high volumetric productivities, and low manufacturing costs, resulting from simulation of a natural habitat and favoring the growth of filamentous fungi over other micro-organisms (Diaz et al. 2016; Su et al. 2017). Therefore, it is important to improve understanding of the regulatory mechanisms controlling enzyme production during SSF with P. oxalicum. An important finding from this study was that PoxMK1 positively regulates PBDE production during SSF with P. oxalicum, which will contribute to improving the production of industrial enzymes.
Deletion of PoxMK1 resulted in changes to the color of P. oxalicum cultures and upregulation of DHN-melanin biosynthesis-cluster gene expression, suggesting that PoxMK1 participates in signal transduction of DHN-melanin biosynthesis. This is consistent with a report that depletion of the Fus3 ortholog, MpkB, in Aspergillus fumigatus induced a strong enhancement of DHN-melanin production, thereby resulting in a more strongly colored culture (Perez-Cuesta et al. 2020). The DHN-melanin biosynthesis-cluster includes six genes, ayg1, abr1, alb1/wA, abr2/yA, arp1, and arp2. The polyketide synthase Alb1/wA catalyzes β-ketoacyl condensation of malonyl-CoA and acetyl-CoA to generate the heptaketide napthopyrone YWA1, which is yellow. The heptaketide hydrolyase Ayg1 hydrolyzes YWA1 to generate 1,3,6,8-tetrahydroxynaphthalene flaviolin (1,3,6,8-THN), which is transformed to scytalone by 1,3,6,8-THN reductase Arp2. Scytalone is converted to 1,3,8-THN after dehydrogenation by scytalone dehydratase Arp1, and, in turn, to vermelon by Arp2. Subsequently, the multicopper oxidase Abr1 converts vermelon to 1,8-DHN, followed by polymerization to form DHN-melanin by laccase Abr2. These precursors of melanin are yellowish to brown (Perez-Cuesta et al. 2020). The color of a particular culture is related to the relative amounts of the various intermediates during melanin biosynthesis. The signal-transduction pathway that activates the DHN-melanin biosynthesis cluster remains unknown, but may be associated with G protein–mediated signaling (Perez-Cuesta et al. 2020). The MAPK pathway generally functions downstream of G protein–mediated signaling (Hagiwara et al. 2016).
Comparative phosphoproteomic analysis identified three potential targets for PoxMK1, with reduced phosphorylation in the mutant ∆PoxMK1, i.e., POX03695/Rpb9, POX04493/Mcm1, and POX07703/Kin28, but this finding needs confirmation. The SRF-MADS protein Mcm1/McmA is involved in the regulation of asexual development and extracellular enzyme production, including that of cellulase, hemi-cellulase, and protease (Li et al. 2016a). McmA and ClrB synergistically co-regulate expression of cellulase and hemi-cellulase genes in A. nidulans, and McmA assists recruitment of ClrB to the promoter regions of target genes, not vice versa (Li et al. 2016b).
Mcm1 is N-terminally phosphorylated at amino acids S2 and T8, and putatively recognized by casein kinase II and I in S. cerevisiae, under high salt stress (Kuo et al. 1997). However, alignment analysis indicated that POX04493/Mcm1 has 74.2% sequence-identity with S. cerevisiae Mcm1 (accession number NP_013757.1), but with a different N-terminal sequence (data not shown). This study identified the phosphosite of POX04493/Mcm1 at amino acid S12, in agreement with that recognized by Fus3 (Mok et al. 2010).
Rpb9, as a nonessential subunit of RNA Pol II, facilitates transcription-coupled RNA repair in S. cerevisiae (Li and Li 2017). Rpb9 participates in transcriptional proofreading, can slow down RNA Pol II chain-elongation after a misincorporation event, and enhances TFIIS-mediated error excision, thereby ensuring transcriptional fidelity (Knippa and Peterson 2013). Deletion of rpb9 resulted in change off Rad2 chromatin binding and increased Rad2 occupancy on Mediator complex–bound upstream activating sequences (Georges et al. 2019).
Cyclin-dependent kinase Kin28 phosphorylates Rpb1, the largest subunit of RNA Pol II, at amino acid S5 of the sequence YSPTSPS, in the carboxyl-terminal domain, which facilitates the association of RNA Pol II with factors involved in transcriptional elongation, including the Mediator and SAGA complexes (Suh et al. 2013). Kin28 depletion, or inactivation, inhibits dissociation of RNA Pol II from its promoter and stabilizes Mediator complex association (Knoll et al. 2020). Kin28 is activated by phosphorylation of CDK-activated kinase Cak1 (Espinoza et al. 1998). Surprisingly, POX06076/Rpb1 was found to increase its phosphorylation at amino acids S1536 and S1553 that seemed to be different from that recognized by Kin28.
Surprisingly, MAPKK POX07948/Ste7 reduced its phosphorylation level in ∆PoxMK1. It is known to localize upstream of PoxMK1 and phosphorylate MAPK Fus3/Kss1. Therefore, it needs to be further studied. Moreover, the known target protein Ste12 of Fus3/Kss1, or its orthologs, was not detected by comparative phosphoproteomic analysis. Ste12 and Ste12-like proteins share conserved functions involved in the regulation of fungal development and pathogenicity (Wong and Dumas 2010). POX01257, which encodes Ste12, was detected through comparative transcriptomic analysis of P. oxalicum cultured on Avicel, because its expression was reduced in the mutant ∆PoxMK1. In addition, deletion of POX01257 significantly affected cellulase and xylanase production of P. oxalicum (Zhao et al. unpublished).
It is notable that POX03016, which codes for Atf1, a bZIP protein with a phosphorylation site at amino acid T136, negatively regulated the production of cellulase and xylanase in P. oxalicum (Zhao et al. 2019b). Atf1 is phosphorylated by MAPK Styl/Hog1 in Schizosaccharomyces pombe, which is required for its functions under oxidative stress (Salat-Canela et al. 2017). Screening for kinase proteins with altered phosphorylation found a MAPK Hog1 protein, POX06496, with increased phosphorylation at amino acids T171 and Y173 (Table 2). Depletion of PoxMK1 weakened and/or eliminated secretion of PBDEs, thereby leading to the changes in osmolarity that occurred as a result of the presence or absence of free sugars in the extracellular environment (Huberman et al. 2017). However, the mechanism for PoxMK1 depletion resulting in increased phosphorylation of POX06496/Hog1 needs to be further studied.
To sum up, this work characterized PoxMK1 in P. oxalicum as an ortholog of the Fus3/Kss1 MAP kinase and revealed that it positively regulates PBDE production of the fungus in both SSF and SmF, represses pigment biosynthesis, and affects fungal vegetative growth. Moreover, transcriptomic and phosphoproteomic analysis provided novel insights into understanding signal transduction and regulation of PBDE gene expression in the filamentous fungi.
References
Bentil JA, Thygesen A, Mensah M, Lange L, Meyer AS (2018) Cellulase production by white-rot basidiomycetous fungi: solid-state versus submerged cultivation. Appl Microbiol Biotechnol 102(14):5827–5839
de Paula RG, Antoniêto ACC, Carraro CB, Lopes DCB, Persinoti GF, Peres NTA, Martinez-Rossi NM, Silva-Rocha R, Silva RN (2018) The duality of the MAPK signaling pathway in the control of metabolic processes and cellulase production in Trichoderma reesei. Sci Rep 8:14931
Diaz AB, Blandino A, Webb C, Caro I (2016) Modelling of different enzyme productions by solid-state fermentation on several agro-industrial residues. Appl Microbiol Biotechnol 100(22):9555–9566
Espinoza FH, Farrell A, Nourse JL, Chamberlin HM, Gileadi O, Morgan DO (1998) Cak1 is required for Kin28 phosphorylation and activation in vivo. Mol Cell Biol 18:6365–6373
Georges A, Gopaul D, Wilkes CD, Aiach NG, Novikova E, Barrault MB, Alibert O, Soutourina J (2019) Functional interplay between Mediator and RNA polymerase II in Rad2/XPG loading to the chromatin. Nucleic Acids Res 47:8988–9004
Hagiwara D, Sakamoto K, Abe K, Gomi K (2016) Signaling pathways for stress responses and adaptation in Aspergillus species: stress biology in the post-genomic era. Biosci Biotech Bioch 80:1667–1680
Huberman LB, Coradetti ST, Glass NL (2017) Network of nutrient-sensing pathways and a conserved kinase cascade integrate osmolarity and carbon sensing in Neurospora crassa. P Natl Acad Sci USA 114:E8665–E8674
Knippa K, Peterson DO (2013) Fidelity of RNA polymerase II transcription: role of Rpb9 in error detection and proofreading. Biochemistry 52:7807–7817
Knoll ER, Zhu ZL, Sarkar D, Landsman D, Morse RH (2020) Kin28 depletion increases association of TFIID subunits Taf1 and Taf4 with promoters in Saccharomyces cerevisiae. Nucleic Acids Res 48:4244–4255
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 7:1870
Kuo MH, Nadeau ET, Grayhack EJ (1997) Multiple phosphorylated forms of the Saccharomyces cerevisiae Mcm1 protein include an isoform induced in response to high salt concentrations. Mol Cell Biol. 17:819–832
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323
Li WT, Li SS (2017) Facilitators and repressors of transcription-coupled DNA repair in Saccharomyces cerevisiae. Photochem Phoyobiol 93:259–267
Li J, Liu GD, Chen M, Li ZH, Qin YQ, Qu YB (2013) Cellodextrin transporters play important roles in cellulase induction in the cellulolytic fungus Penicillium oxalicum. Appl Microbiol Biotechnol 97:10479–10488
Li N, Kunitake E, Endo Y, Aoyama M, Kanamaru K, Kimura M, Kato M, Kobayashi T (2016a) Involvement of an SRF-MADS protein McmA in regulation of extracellular enzyme production and asexual/sexual development in Aspergillus nidulans. Biosci Biotech Bioch 80:1820–1828
Li N, Kunitake E, Endo Y, Aoyama M, Kimura M, Koyama Y, Kobayashi T (2016b) McmA-dependent and -independent regulatory systems governing expression of ClrB-regulated cellulase and hemicellulase genes in Aspergillus nidulans. Mol Microbiol 102:810–826
Li ZH, Liu GD, Qu YB (2017) Improvement of cellulolytic enzyme production and performance by rational designing expression regulatory network and enzyme system composition. Bioresource Technol 245:1718–1726
Li CX, Zhao S, Luo XM, Feng JX (2020) Weighted gene co-expression network analysis identifies critical genes for the production of cellulase and xylanase in Penicillium oxalicum. Front Microbiol 11:520–520
Liao GY, Zhao S, Zhang T, Li CX, Liao LS, Zhang FF, Luo XM, Feng JX (2018) The transcription factor TpRfx1 is an essential regulator of amylase and cellulase gene expression in Talaromyces pinophilus. Biotechnol Biofuels 11:276
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆CT method. Methods 25:402–408
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Madi L, McBride SA, Bailey LA, Ebbole DJ (1997) rco-3, a gene involved in glucose transport and conidiation in Neurospora crassa. Genetics 146:499
Marone A, Trably E, Carrère H, Prompsy P, Guillon F, Joseph-Aimé M, Barakat A, Fayoud N, Bernet N, Escudié R (2019) Enhancement of corn stover conversion to carboxylates by extrusion and biotic triggers in solid-state fermentation. Appl Microbiol Biotechnol 103(1):489–503
Martínez-Soto D, Ruiz-Herrera J (2017) Functional analysis of the MAPK pathways in fungi. Revista Iberoamericana de Micología 34:192–202
Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M, Cameroni E, Niu H, Good M, Remenyi A, Ma JL, Sheu YJ, Sassi HE, Sopko R, Chan CS, De Virgilio C, Hollingsworth NM, Lim WA, Stern DF, Stillman B, Andrews BJ, Gerstein MB, Snyder M, Turk BE (2010) Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci Signal 3:ra12
Nguyen EV, Imanishi SY, Haapaniemi P, Yadav A, Saloheimo M, Corthals GL, Pakula TM (2016) Quantitative site-specific phosphoproteomics of Trichoderma reesei signaling pathways upon induction of hydrolytic enzyme production. J Proteome Res 15:457–467
Park HS, Yu JH (2012) Genetic control of asexual sporulation in filamentous fungi. Curr Opin Microbiol 15:669–677
Perez-Cuesta U, Aparicio-Fernandez L, Guruceaga X, Martin-Souto L, Abad-Diaz-de-Cerio A, Antoran A, Buldain I, Hernando FL, Ramirez-Garcia A, Rementeria A (2020) Melanin and pyomelanin in Aspergillus fumigatus: from its genetics to host interaction. Int Microbiol 23:55–63
Priegnitz BE, Brandt U, Pahirulzaman KA, Dickschat JS, Fleißner A (2015) The AngFus3 mitogen-activated protein kinase controls hyphal differentiation and secondary metabolism in Aspergillus niger. Eukaryot Cell 14:602–615
Salat-Canela C, Paulo E, Sánchez-Mir L, Carmona M, Ayté J, Oliva B, Hidalgo E (2017) Deciphering the role of the signal- and Sty1 kinase-dependent phosphorylation of the stress-responsive transcription factor Atf1 on gene activation. J Biol Chem 292:13635–13644
Segorbe D, Pietro AD, Pérez-Nadales E, Turrà D (2017) Three Fusarium oxysporum mitogen-activated protein kinases (MAPKs) have distinct and complementary roles in stress adaptation and cross-kingdom pathogenicity. Mol Plant Pathol 18:912–924
Su LH, Zhao S, Jiang SX, Liao XZ, Duan CJ, Feng JX (2017) Cellulase with high β-glucosidase activity by Penicillium oxalicum under solid state fermentation and its use in hydrolysis of cassava residue. World J Microbiol Biotechnol 33:37
Suh H, Hazelbaker DZ, Soares LM, Buratowski S (2013) The C-terminal domain of RPB1 functions on other RNA polymerase II subunits. Mol Cell 51:850–858
Taus T, Köcher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K (2011) Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10:5354–5362
Tong SM, Feng MG (2019) Insights into regulatory roles of MAPK-cascaded pathways in multiple stress responses and life cycles of insect and nematode mycopathogens. Appl Microbiol Biotechnol 103:577–587
Wang MY, Zhang ML, Li L, Dong Y, Jiang YM, Liu KM, Zhang RQ, Jiang BJ, Niu KL, Fang X (2017) Role of Trichoderma reesei mitogen-activated protein kinases (MAPKs) in cellulase formation. Biotechnol Biofuels 10:99
Wang L, Zhao S, Chen XX, Deng QP, Li CX, Feng JX (2018) Secretory overproduction of a raw starch-degrading glucoamylase in Penicillium oxalicum using strong promoter and signal peptide. Appl Microbiol Biotechnol 102:9291–9301
Wong SHJ, Dumas B (2010) Ste12 and ste12-like proteins, fungal transcription factors regulating development and pathogenicity. Eukaryot Cell 9:480–485
Xiong Y, Coradetti ST, Li X, Gritsenko MA, Clauss T, Petyuk V, Camp D, Smith R, Cate JHD, Yang F, Glass NL (2014) The proteome and phosphoproteome of Neurospora crassa in response to cellulose, sucrose and carbon starvation. Fungal Genet Biol 72:21–33
Xu QS, Yan YS, Feng JX (2016) Efficient hydrolysis of raw starch and ethanol fermentation: a novel raw starch-digesting glucoamylase from Penicillium oxalicum. Biotechnol Biofuels 9:216
Yan YS, Zhao S, Liao LS, He QP, Xiong YR, Wang L, Li CX, Feng JX (2017) Transcriptomic profiling and genetic analyses reveal novel key regulators of cellulase and xylanase gene expression in Penicillium oxalicum. Biotechnol Biofuels 10:279
Zhao S, Yan YS, He QP, Yang L, Yin X, Li CX, Mao LC, Liao LS, Huang JQ, Xie SB, Nong QD, Zhang Z, Jing L, Xiong YR, Duan CJ, Liu JL, Feng JX (2016) Comparative genomic, transcriptomic and secretomic profiling of Penicillium oxalicum HP7-1 and its cellulase and xylanase hyper-producing mutant EU2106, and identification of two novel regulatory genes of cellulase and xylanase gene expression. Biotechnol Biofuels 9:203
Zhao S, Liao XZ, Wang JX, Ning YN, Li CX, Liao LS, Liu Q, Jiang Q, Gu LS, Fu LH, Yan YS, Xiong YR, He QP, Su LH, Duan CJ, Luo XM, Feng JX (2019a) Transcription factor Atf1 regulates expression of cellulase and xylanase genes during solid-sate fermentation of ascomycetes. Appl Environ Microbiol 85:e01226–e01219
Zhao S, Liu Q, Wang JX, Liao XZ, Guo H, Li CX, Zhang FF, Liao LS, Luo XM, Feng JX (2019b) Differential transcriptomic profiling of filamentous fungus during solid-state and submerged fermentation and identification of an essential regulatory gene PoxMBF1 that directly regulated cellulase and xylanase gene expression. Biotechnol Biofuels 12:103
Funding
This work was financially supported by the National Natural Science Foundation of China (grants 31760023 and 31660305), the Autonomous Research Project of State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources (SKLCUSA-a201902 and SKLCUSA-a201923), the Training Program for 1000 Young and Middle-aged Key Teachers in Guangxi at 2019, and the One Hundred Person Project of Guangxi.
Author information
Authors and Affiliations
Contributions
JXF supervised this study and revised the manuscript. SZ co-supervised and directed all experiments and revised the manuscript. BM conducted construction of deletion mutants, measurement of enzymatic activities, transcriptomes, and phosphoproteomes, and wrote the manuscript. YNN performed construction of complementary strain and enzymatic activity assay. CXL conducted bioinformatic analysis. DT and HG conducted RT-qPCR analysis. XMP performed phenotypic analyses. XML was involved in preparation of experimental materials.
Corresponding authors
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 5280 kb)
Rights and permissions
About this article

Cite this article
Ma, B., Ning, YN., Li, CX. et al. A mitogen-activated protein kinase PoxMK1 mediates regulation of the production of plant-biomass-degrading enzymes, vegetative growth, and pigment biosynthesis in Penicillium oxalicum. Appl Microbiol Biotechnol 105, 661–678 (2021). https://doi.org/10.1007/s00253-020-11020-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-11020-0