
Abstract
Aspergillus aculeatus produces cellulolytic enzymes in the presence of their substrates. We screened a library of 12,000 A. aculeatus T-DNA-inserted mutants to identify a regulatory factor involved in the expression of their enzyme genes in response to inducers. We found one mutant that reduced the expression of FIII-avicelase (chbI) in response to cellulose. T-DNA was inserted into a putative protein kinase gene similar to AN10082 in A. nidulans, serine–arginine protein kinase F, SrpkF. Fold increases in srpkF gene expression in response to various carbon sources were 2.3 (d-xylose), 44 (Avicel®), 59 (Bacto™ Tryptone), and 98 (no carbon) compared with d-glucose. Deletion of srpkF in A. aculeatus resulted in a significant reduction in cellulose-responsive expression of chbI, hydrocellulase (cel7b), and FIb-xylanase (xynIb) genes at an early induction phase. Further, the srpkF-overexpressing strain showed upregulation of the srpkF gene from four- to nine-fold higher than in the control strain. srpkF overexpression upregulated cbhI and cel7b in response to cellobiose and the FI-carboxymethyl cellulase gene (cmc1) and xynIb in response to d-xylose. However, the srpkF deletion did not affect the expression of xynIb in response to d-xylose due to the less expression of srpkF under the d-xylose condition. Our data demonstrate that SrpkF is primarily involved in cellulose-responsive expression, though it has a potential to stimulate gene expression in response to both cellobiose and d-xylose in A. aculeatus.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lignocellulosic biomass has long been recognized as a potential sustainable source of mixed sugars for fermentation to biofuels and chemicals (Himmel et al. 2007). A key step for bioconversion of lignocellulose is enzymatic hydrolysis of pretreated lignocellulose to fermentable sugars. Filamentous fungi are prominent producers of enzymes that degrade lignocellulose (Payne et al. 2015). Trichoderma reesei is a well-known species that produces copious amounts of cellulolytic enzymes (Bischof et al. 2016). T. reesei glycoside hydrolases are being continuously improved to utilize lignocellulose as a feedstock for the generation of bio-based products. For example, Aspergillus aculeatus no. F-50 [NBRC 108789] was isolated from soil as a host for production of carbohydrate-active enzymes that cooperatively hydrolyze pulp in combination with Trichoderma reesei (Murao et al. 1979). β-Glucosidase from A. aculeatus no. F-50 was introduced into T. reesei, which accelerated cellulose hydrolysis (Baba et al. 2015; Nakazawa et al. 2012). This β-glucosidase showed high compatibility with T. reesei, suggesting that A. aculeatus produces promising enzymes for liberating fermentable sugars from lignocellulose. However, cellulolytic and xylanolytic enzymes of A. aculeatus are not utilized in industry because of low production levels. We aimed to understand the regulatory mechanisms of the associated genes and apply this knowledge to improving enzyme production in A. aculeatus.
Cellulolytic and xylanolytic enzyme production in Aspergillus is regulated at the transcriptional level. The first identified regulator of cellulolytic and xylanolytic genes was XlnR, a Zn(II)2Cys6-type transcriptional activator that coordinates xylanolytic expression in Aspergillus niger (van Peij et al. 1998). Genetic analysis indicates that XlnR controls the expression of xylanolytic and cellulolytic enzyme genes in A. aculeatus (Kunitake and Kobayashi 2017; Tani et al. 2014). XlnR regulates expression of FIb-xylanase (xynIb) and FI-carboxymethyl cellulase (cmc1) genes in response to cellulose and d-xylose, respectively. In contrast, FIII-avicelase (cbhI), FII-carboxymethyl cellulase (cmc2), and hydrocellulase (cel7b) are induced in response to cellulose via an XlnR-independent signaling pathway in A. aculeatus (Tani et al. 2012). Clr-2 in Neurospora crassa and ClrB, a Clr-2 homolog in Aspergillus nidulans, were identified as Zn(II)2Cys6-type transcriptional activators that control expression of cellulolytic enzyme genes in response to cellulosic carbon sources (Coradetti et al. 2012). ManR, a ClrB ortholog, participates in the XlnR-independent signaling pathway in Aspergillus oryzae (Ogawa et al. 2013). We confirmed that ManR controls expression of cbhI, cmc2, and cel7b in response to cellulose, which is controlled by the XlnR-independent signaling pathway in A. aculeatus (Tsumura et al. 2021).
Various factors are involved in a complex regulatory network to maintain the precise balance of carbon sources required for growth and hydrolytic enzyme production. Cellodextrin transporters function as transceptors that recognize cellobiose and initiate induction of cellulase gene expression in N. crassa (Znameroski, et al. 2014). Trichoderma mitogen-activated protein kinase, Tmk2, is involved in cell wall integrity, sporulation, and cellulase production (Wang et al. 2014). Cyclic AMP-dependent protein kinase A (PKA) affects cellulase gene expression in response to light in T. reesei (Schuster et al. 2012). PKA in A. nidulans is also involved in the repression of cellulolytic enzyme genes in response to carbon sources (Kunitake et al. 2019). However, molecular mechanisms underlying cooperative control by these factors of cellulolytic and xylanolytic enzyme genes remain unknown.
We previously established a positive screening method to identify regulators involved in the cellulose-responsive induction in A. aculeatus. We constructed a random insertional mutagenesis library using Agrobacterium tumefaciens-mediated transformation of A. aculeatus NCP2, which harbors a transcriptional fusion between the cbhI promoter (PCBHI) and the orotidine 5′-phosphate decarboxylase gene (pyrG). Cellulose-responsive expression-deficient mutants could then be isolated by screening for 5-fluoroorotic acid (5-FOA)-resistant mutants on minimal medium (MM) with wheat bran as a sole carbon source (Kunitake et al. 2011, 2013). We identified ClbR, dipeptidyl peptidase IV, and SepM as regulators that stimulate the expression of cellulolytic enzyme genes in response to cellobiose (Kunitake et al. 2015, 2013; Tani et al. 2017; Tsumura et al. 2021). ClbR is a Zn(II)2Cys6-type transcriptional activator that controls the expression of cellulolytic enzyme genes in response to cellobiose (Kunitake, et al. 2013). SepM interacts with SepL, a putative kinase in the septation initiation network complex, which participates in septa formation and regulation of ManR-dependent signaling (Tsumura et al. 2021).
We further screened for a new regulator to better understand the regulatory mechanisms underlying ManR- and XlnR-dependent signaling pathways that modulate gene expression in response to cellulose. We identified a putative protein kinase, SrpkF, which increased expression of cellulolytic and xylanolytic genes in response to cellobiose and d-xylose under control of both ManR-dependent and XlnR-dependent signaling in A. aculeatus.
Materials and methods
Strains, transformation, marker recycling, and T-DNA insertion
All A. aculeatus strains used in this study were derived from wild-type A. aculeatus no. F-50 [NBRC 108789]. Unless otherwise stated, all strains were propagated at 30 ℃ in an appropriately supplemented MM (Adachi et al. 2009). A. aculeatus NCP2 (niaD1::niaD::PCHB1-pyrG; pyrG1) was used to construct A. aculeatus strains for T-DNA insertion via Agrobacterium tumefaciens-mediated transformation. Counterselection on 5-FOA and marker recycling were performed as described previously (Kunitake et al. 2011, 2013). A. aculeatus MR12 (pyrG1; ∆ku80) was used for the disruption and complementation of the srpkF gene (Tani et al. 2013). Escherichia coli DH5αF′ was used for plasmid construction.
Disruption and complementation of srpkF
The A. aculeatus srpkF-deficient mutant (pyrG1; ∆ku80; ∆srpkF) was created by replacing srpkF with the A. nidulans orotidine 5′-phosphate decarboxylase gene (AnpyrG) followed by marker recycling (Tani et al. 2013). The srpkF deletion cassette was constructed with the 5′ and 3′ regions of srpkF, which are key in homologous recombination to replace srpkF with AnpyrG) were amplified from A. aculeatus genomic DNA using primer pairs 2.6 k-5′srpkF/2.6 k-5′srpkR and L-kinaseF/L-kinaseR, respectively. The AnpyrG gene was amplified from A. nidulans genomic DNA using the primer pair AnpyrG-KF/AnpyrG-KR. The 3′ flanking region of sepM was amplified using the primer pair 2.6 k-MsrpkF/M-kinaseR to eliminate AnpyrG by intramolecular homologous recombination at the srpkF locus. The 5′ region of AnpyrG (responsible for marker recycling) and the 3′ region were fused via PCR using the primer pair 2.6 k-5′srpkF/L-kinaseR and subcloned into the EcoRV site of pBluescriptIIKS( +) to yield pDsrpkF. The srpkF deletion cassette was amplified via PCR using the primer pair 2.6 k-5′srpkF/L-kinaseR from pDsrpkF and introduced into MR12 (pyrG1; ∆ku80) using the protoplast-PEG method to yield the A. aculeatus ∆srpkF plus pyrG strain (pyrG1; ∆ku80; ∆srpkF::AnpyrG). Marker recycling used 1 × 104 transformant spores spread onto MM supplemented with 0.01% uridine and 1 mM 5-FOA. A. aculeatus ∆srpkF (pyrG1; ∆ku80; ∆srpkF) was obtained by monospore isolation (Supplementary Fig. S1). Supplementary Table S1 summarizes the primers used in the study.
The srpkF promoter, open reading frame, and 3′-untranslated region (UTR) were first amplified using the primer pair 2.6 k-5′srpkF/M-kinaseR to complement A. aculeatus ∆srpkF. AnpyrG and the 3′ region required for homologous recombination at the srpkF locus were subsequently amplified with primer pairs AnpyrG-KF/AnpyrG-CR and C-kinaseF/C-kinaseR, respectively. The three DNA fragments were fused via PCR using the primer pair 2.6 k-5′srpkF/C-kinaseR. Finally, A. aculeatus ∆srpkF was transformed with the amplified DNA fragments to yield the srpkF-complemented strain (pyrG1; ∆ku80; ∆srpkF::srpkF::AnpyrG) (Supplementary Fig. S1).
The A. aculeatus SrpkF C-terminus deletion mutant was generated by introducing a stop codon at 1129 nt. The ORF with its 5′ flanking region (− 23–1,121 bp) and 3′-UTR of srpkF was amplified using primer pairs pK-F/srpkFstopMR-R and 3′UTRF/3′UTRR, respectively. Products were fused using PCR with primer pair pK-F/3′UTRR. The region of AnpyrG for marker recycling and the 3′ region required for homologous recombination at the srpkF locus were subsequently amplified with primer pair MRstop-srpkF-F/L-kinaseR using pDsrpkF as a template. These fragments were fused via PCR using the primer pair pK-F/L-kinaseR. Fused fragments were introduced into MR12 (Supplementary Fig. S2). A. aculeatus ∆CsrpkF (pyrG1; ∆ku80; ∆CsrpkF1-327) was obtained after marker recycling. The ∆CsrpkF strain was complemented by the same fragments used to complement ∆srpkF (Supplementary Fig. S2).
The A. aculeatus srpkF-overexpressing strain (pyrG1::Ptef-srpkF::AapyrG; Δku80; ΔsrpkF) was generated by inserting an srpkF-overexpressing cassette into the A. aculeatus pyrG locus in ∆sprkF. The AapyrG ORF with its promoter and terminator region and the AapyrG 3′ region were amplified using primer pairs AapyrGORFF/AapyrGORFR and AapyrG3′F/AapyrG3′R, respectively, to introduce the expression cassette at the AapyrG locus. The promoter region of translation elongation factor 1α gene (Ptef) was amplified using primer set AaPtefF/AaPtefR and subsequently fused to the AapyrG 3′ region via PCR using primer set AapyrG3′F/AaPtefR, followed by digestion with NotI and SacI. The amplified AapyrG ORF fragment was digested with KpnI and XbaI and subcloned into pBS KpnI and SalI sites. The plasmid was digested with NotI and SacI and ligated with digested AapyrG 3′ and Ptef fragments. The srpkF-overexpressing cassette was amplified using primer set 5′AapyrGF/3′AapyrGR. Finally, A. aculeatus ∆srpkF was transformed with amplified DNA fragments to yield the srpkF-overexpressing strain (Supplementary Fig. S3).
Gene expression analysis by quantitative RT-PCR
Quantitative RT-PCR (qRT-PCR) was used to quantify the expression of cellulase and hemicellulase genes as previously described (Tani et al. 2017). 0.1% Bacto™ Tryptone (Thermo Fisher Scientific, Tokyo, Japan) was used as a neutral carbon source (noninducing condition). Indicated carbon sources were added to media supplemented with 0.1% Bacto™ Tryptone to investigate the expression of test genes. Total RNA (500 ng) was used to amplify cDNA with ReverTra Ace™ qPCR RT-Master Mix (Toyobo, Tokyo, Japan). qRT-PCR was performed in a Thermal Cycler Dice™ Real-Time System (Takara, Kyoto, Japan). For amplification, a SYBR® Green I assay using THUNDERBIRD™ SYBR® qPCR Mix (Toyobo) was performed in a 20 μl reaction. Primers used for qRT-PCR are listed in Supplementary Table S1. Expression of the glyceraldehyde-3-phosphate dehydrogenase A gene (gpdA) was used as an internal control. The specificity of the PCR amplification was confirmed by melting curve analysis. The expression profile of each gene was analyzed with the delta–deltaCT method. More than three biological replicates were performed for each experiment, and each replicate was evaluated in triplicate.
Additional methods
Genomic DNA preparation and Southern blotting were performed as described previously (Kunitake et al. 2013). An in-house A. aculeatus draft genome database was used to obtain the genomic sequence of the srpkF gene. Two independently amplified cDNA fragments were analyzed to determine the srpkF cDNA sequence. Conidia from the A. aculeatus strains were collected in a 0.1% Tween® 80/0.8% NaCl solution and counted using a hemocytometer. The number of conidia was normalized by colony area.
Nucleotide sequence data
Nucleotide sequence data were deposited in Japan’s DNA Data Bank (DDBJ) Nucleotide Sequence Data Libraries. The accession number of srpkF in A. aculeatus is DDBJ Acc. no. LC638744.
Results
Isolation of a cellulose-responsive induction-deficient mutant from an A. aculeatus T-DNA insertion mutant library
We previously screened an A. aculeatus T-DNA insertion mutant library of approximately 12,000 transformants for strains that were 5-FOA-resistant and cellulose-responsive induction-deficient. We isolated five 5-FOA-resistant strains that showed reduced growth on medium supplemented with 1% Avicel® but showed normal growth on media supplemented with 1% glucose, 1% beechwood xylan, or 1% d-xylose (Tani et al. 2017). We further analyzed one 5-FOA-resistant strain (Q3) that showed a strong correlation between the function of the gene disrupted by T-DNA and the cellulose-responsive induction-deficient phenotype.
We first investigated cbhI expression profiles in response to Avicel® in Q3 because the cbhI promoter was fused to the pyrG gene. This reporter gene was used as bait to screen for factors involved in the cellulose-induced signaling pathway with the 5-FOA-resistant phenotype (Kunitake et al. 2013). Expression of cbhI was induced in response to Avicel® at 9 h postinduction in the control strain (NCP2) but significantly reduced in Q3 to approximately 20% of NCP2 expression levels (p < 0.05, Student’s t test) (Fig. 1). Expression of xynIb in response to Avicel® was also significantly reduced in Q3 to approximately 30% of NCP2 expression levels (p < 0.05, Student’s t test) (Fig. 1). These data suggest that the T-DNA insertion in Q3 disrupted a gene required for the induction of test genes in response to Avicel®.

Identification of a cellulose-responsive induction-deficient mutant. qRT-PCR analysis of cbhI and xynIb expression at 9 h postinduction with 1% (w/v) Avicel® was performed for control (NCP2) and Q3 strains. Relative expression corresponds to the ratio of the mean expression levels of cbhI divided by mean expression of gpdA, the reference gene. Relative expression levels are means of three independent experiments, and error bars indicate the standard deviations. Letters indicate significant differences between groups (p < 0.05, Student’s t test)
Serine–arginine protein kinase F participates in the early phase of cellulose induction
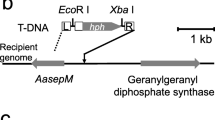
T-DNA integration into Q3 was analyzed by Southern blotting since the recovery of T-DNA flanking sequences by inverse PCR differs depending on integration pattern. Genomic DNA isolated from Q3 was digested with EcoRI, XbaI and SpeI, HindIII, and PstI. Single digestion by EcoRI (unique site in the T-DNA left flanking region), HindIII (absent from the vector), and PstI (unique site in the vector backbone) and double digestion with XbaI and SpeI (unique site in the T-DNA right flanking region and absent from the vector, respectively) all yielded a single band (Fig. 2a). These data demonstrate that one copy of T-DNA was integrated into the Q3 genome. We amplified the T-DNA flanking sequences via inverse PCR using Q3 genomic DNA digested with EcoRI and XbaI/SpeI to recover the right and left flanking sequences, respectively (Fig. 2b). We sequenced DNA fragments amplified via inverse PCR, which showed that the T-DNA integrated into the ORF encoding the putative serine–threonine protein kinase (Fig. 2b). The right and left flanking sequences of the T-DNA were inserted at 1122 and 1137 nt from the translation start site of the gene with a 15 bp deletion in the recipient genome. The putative protein kinase gene was composed of 1443 bp with five exons and encoded a 416-amino acid protein. Based on a homology search in FungiDB (https://fungidb.org/fungidb/), the putative protein kinase was most similar to ACLA_003920 in Aspergillus clavatus (E value = 0.0; Identities = 81%). The gene was also similar to AN10082 in A. nidulans, SrpkF (E value = 7e–100; Identities = 38%). This protein is a member of a family of serine–threonine protein kinases that includes an expanded group of seven serine–arginine protein kinases (SRPK) in A. nidulans (de Souza et al. 2013). Therefore, this gene was designated srpkF in A. aculeatus. The T-DNA insertion at 1122 nt from the translation start site caused production of a truncated protein of 327 amino acids in the original protein and seven additional amino acids (SNTDSLN) and a stop codon derived from the T-DNA fragment. Thus, srpkF is the candidate gene causing the cellulose-responsive induction-deficient phenotype of Q3.

Determination of the T-DNA integration pattern and identification of the disrupted gene. Deduced T-DNA integration via Southern blotting (a) and a schematic representation of the SrpkF locus (b)
The srpkF gene expressed under the carbon starvation condition
Expression profiles of srpkF were assessed under inducing (1% Avicel® or 1% d-xylose), repressing (1% d-glucose), carbon neutral (0.1% Bacto™ Tryptone), and carbon starvation conditions of the cellulolytic and xylanolytic enzyme genes. Transcripts of srpkF were quantified using RNA prepared from A. aculeatus wild-type strain grown under the following conditions: 1% d-glucose medium for 3 h, 1% d-xylose medium for 3 h, 1% Avicel® medium for 9 h, 0.1% Bacto™ Tryptone medium for 3 h, and no carbon source for 3 h. Fold increases in srpkF gene expression compared with d-glucose medium were 2.3 (d-xylose), 44 (Avicel®), 59 (Bacto™ Tryptone), and 98 (no carbon) (Fig. 3). A. aculeatus grew poorly in 1% Avicel® medium, 0.1% Bacto™ Tryptone medium, and under the no carbon condition, whereas it grew well on 1% glucose and 1% d-xylose media, indicating that A. aculeatus did not acquire sufficient carbon from 1% Avicel® and 0.1% Bacto™ Tryptone media. These data indicate that srpkF is expressed under conditions in which available carbon is limited.

qRT-PCR analysis of srpkF expression in wild-type A. aculeatus under the following conditions: 1% d-glucose for 3 h (G), 1% d-xylose for 3 h (X), 1% Avicel® for 9 h (A), 0.1% Bacto™ Tryptone for 3 h (B), and no carbon source for 3 h (N). Relative expression corresponds to the ratio of srpkF divided by mean expression levels of gpdA. Relative expression levels are means of three independent experiments, and error bars indicate the standard deviations. Letters indicate significant differences between groups (p < 0.05, one-way ANOVA)
Functional analysis of SrpkF in A. aculeatus
To genetically analyze the function of srpkF, the entire srpkF gene was deleted by homologous recombination (ΔsrpkF, pyrG+) followed by marker recycling to yield an A. aculeatus srpkF deletion mutant (ΔsrpkF, pyrG−). A srpkF deletion mutant was never isolated using the DNA fragment corresponding to the 1522 nt region upstream from the translation start site that was used for homologous recombination. Thus the DNA fragment from 1523 to 2613 nt upstream was targeted. Further, the 3′ flanking region of srpkF was deleted after marker recycling in ΔsrpkF. Thus, the srpkF deletion mutant included elimination of 1.5 kb upstream and 1.3 kb downstream flanking sequences. ΔsrpkF was transformed with srpkF DNA fragments yielding a complemented strain (srpkF+) (Supplementary Fig. S1). A C-terminal deletion mutant of SrpkF composed of 327 amino acids (ΔCsrpkF) in Q3 was generated by introducing a stop codon at 1129 nt from the translation start site of the ORF, followed by marker recycling of pyrG to yield ΔCsrpkF (SrpkF1-327, pyrG−). ΔCsrpkF was also complemented using the srpkF DNA fragment. We confirmed that gene replacement and complementation occurred as expected by Southern blotting (Supplementary Fig. S2). MR12, ΔsrpkF, ΔCsrpkF, and srpkF+ grew equally well on MM supplemented with 1% d-glucose, 1% d-xylose, and 1%Avicel® (data not shown). Expression profiles of test genes in the complemented ΔCsrpkF strain were not different from profiles from MR12 and srpkF+, and we thus used srpkF+ as a control strain (data not shown).
We investigated the effect of srpkF deletion on gene expression in response to cellulosic carbon sources. Transcripts of cbhI and xynIb were quantified at 6 and 9 h postinduction in MR12, ΔCsrpkF, ΔsrpkF, and srpkF+ strains. Expression of cbhI and xynIb was reduced in ΔCsrpkF and ΔsrpkF only after induction. Fold induction of cbhI in response to Avicel® decreased significantly to 50% in ΔsrpkF and 31% in ΔCsrpkF of the response in MR12 at 9 h postinduction (p < 0.05, Student’s t test). Similarly, fold induction of xynIb significantly decreased to 26% in ΔsrpkF and 32% in ΔCsrpkF (p < 0.05, Student’s t test) (Supplementary Fig. S4 and Fig. 4). SrpkF participates in the expression of both genes in response to Avicel®. Expression levels decreased similarly in both ΔCsrpkF and ΔsrpkF and were restored by srpkF complementation. Hence, we further analyzed the SrpkF function using only ΔsrpkF. Expression of cel7b in ΔsrpkF decreased significantly to 55% of expression in MR12 at 9 h postinduction (p < 0.05, Student’s t test), which was restored in srpkF+ (Fig. 4). The expression of cmc2, cmc1, and xynIa, decreased to approximately 60%, but these reductions were not statistically significant (Fig. 4).

Effect of srpkF deletion on expression of cellulase and hemicellulase genes. qRT-PCR results of each gene in MR12 (M, black bars), ΔsrpkF (Δ, white bars), and srpkF+ (+ , gray bars) incubated for 6 and 9 h under the noninduced condition ( −) and the 1% Avicel®-inducing condition (Avi). The relative expression corresponds to the ratio of the mean expression levels of each gene divided by that of gpdA. Relative expression levels are the means of three independent experiments, and the error bars indicate the standard deviations. Letters indicate significant differences between groups (p < 0.05, one-way ANOVA)
SrpkF participates in cellulose-responsive expression of cellulolytic and xylanolytic enzyme genes in A. aculeatus
To assess the effect of overexpression of srpkF on the expression of genes encoding cellulosic biomass-degrading enzymes in A. aculeatus, we constructed an srpkF-overexpressing strain that constitutively expresses srpkF under the control of the translation elongation factor 1α gene promoter (Ptef), a high-level constitutive promoter in Aspergillus (Kunitake et al. 2015). The srpkF-overexpressing cassette was introduced into the pyrG locus in ΔsrpkF as a single copy by homologous recombination, as confirmed by Southern blotting (Supplementary Fig. S3), yielding the srpkF-overexpressing strain (OEsrpkF). Expression of srpkF increased four to seven-fold in the presence of cellobiose with 1-deoxynojirimycin (DNJ) and approximately nine-fold in the presence of d-xylose (Supplementary Fig. S5). Since A. aculeatus produces β-glucosidase, which effectively hydrolyzes cellobiose to glucose, DNJ was added as a β-glucosidase inhibitor (Tani et al. 2012). The physiological phenotype of the OEsrpkF strain was no different from MR12, ΔCsrpkF, and ΔsrpkF (data not shown).
We assessed the effect of srpkF overexpression on the expression of cellulosic biomass-degrading enzyme genes in response to physiological inducers. cbhI, cmc2, and cel7b were upregulated in response to cellobiose via ManR-dependent signaling. Expression of cbhI, cmc2, and cel7b generally increased in OEsrpkF compared with MR12 under inducing conditions (Fig. 5a, b); however, expression profiles varied. Expression of chbI was induced at 2, 3, and 4 h postinduction in OEsrpkF and resulted in a significant fold increase (Fig. 5a, b). By contrast, overexpression of srpkF did not significantly increase either expression or fold induction of cmc2 (Fig. 5a, b). Expression of cel7b was significantly stimulated under both inducing and noninducing conditions and showed no significant difference in the fold induction. Thus, SprkF stimulated expression of cel7b at a basal level (Fig. 5a, b).

Effect of srpkF overexpression on expression of cellulase and hemicellulase genes. qRT-PCR results for each gene in MR12 (M, black bars) and the srpkF-overexpressing strain OEsrpkF (O, striped bars). a, b RNA was prepared from strains grown for 2–4 h in the presence of 0.1% cellobiose with 50 μg/L DNJ. Relative expression corresponds to the ratio of each gene divided by mean expression levels of gpdA (a). Fold induction of each test gene reflects the gene expression level under inducing conditions divided by expression under noninducing condition (b). c, d RNA was prepared from strains grown for 1.5–3.0 h in the presence of 1% (w/v) D-xylose. Relative expression corresponds to the ratio of each gene divided by mean expression levels of gpdA (c). d Fold induction of each test gene reflects gene expression under inducing conditions divided by expression under noninducing conditions (d). For all panels, the results shown are the means of three independent experiments, and the error bars indicate the standard deviations. An asterisk indicates a significant difference between the expression of test genes in MR12 and OEsrpkF (p < 0.05, Student’s t test)
Expression of xynIb and cmc1 is induced in response to d-xylose via XlnR-dependent signaling. Expression of xynIb and cmc1 was enhanced markedly at 1.5 and 3.0 h postinduction with d-xylose in OEsrpkF (Fig. 5c, d). These data confirm that SrpkF has a potential to participate in cellulose- and d-xylose-responsive signaling pathways.
Discussion
We identified SrpkF as a positive regulator that induces cellulolytic and xylanolytic enzyme gene expression in response to cellulose. An srpkF-overexpressing strain demonstrated the potential for this gene to stimulate the d-xylose-responsive induction via the XlnR-dependent signaling pathway. However, SrpkF functions as a positive regulator to stimulate cellulose-responsive induction via both ManR- and XlnR-dependent signaling under physiological conditions. srpkF expression was stimulated under cellulose and carbon starvation condition but not by the presence of d-xylose (Fig. 6).

Schematic illustration of possible mechanisms for SrpkF induction of cellulolytic and xylanolytic enzyme genes in A. aculeatus. S SrpkF protein
A comparison of amino acid sequences using the FASTA algorithm indicated that srpkF orthologs are highly conserved in the Aspergillus section Nigri. Orthologs are also present in some strains from other sections and other genera, such as A. nidulans (AN10082, E value = 2e-91; Identities = 42%), Penicillium rubens (Pc12g16110, E value = 1e-104; Identities = 47%), and Coccidioides immitis (CIMG_04484, E value = 6e-103; Identities = 44%) (Fig. 7). AN10082 in A. nidulans encodes a serine–arginine protein kinase F (SrpkF) for which a deletion mutant did not show a distinguishing phenotype (de Souza et al. 2013). SRPK was first identified as a cell cycle-regulated kinase specific for SR proteins, which are a family of pre-mRNA splicing factors containing SR domains that consist largely of serine/arginine repeats (Gui et al. 1994a, b). Members of this kinase family are known to phosphorylate serines within SR domains and are widely conserved in eukaryotes. S. cerevisiae encodes a single SRPK family member, Sky1, but A. nidulans and A. aculeatus encode seven SRPKs from SrpkA to G (de Souza et al. 2013) and four SRPKs, respectively. SrpkA proteins in Aspergillus are highly conserved along with S. cerevisiae Sky1. Thus, these proteins may modulate subcellular localization and function of Ser-Arg rich splicing-factor proteins (Dagher and Fu 2001; Gui et al. 1994a). However, SRPKs possess various domains, suggesting functional diversification. Few non-splicing functions of SRPKs are reported (Gou et al. 2020; Hong et al. 2012; Wang et al. 2017), and it remains unclear whether SRPKs have evolved further regulatory roles (Bustos et al. 2020). Functions of SRPKs might be illuminated by close examination of gene expression since some genes exhibited highly tissue-specific profiles (Nakagawa et al. 2005; Wang et al. 1998).

Phylogenetic analysis of serine–arginine protein kinase-like genes. Phylogenetic relationships among srpk orthologs are shown as a consensus neighbor-joining tree based on sequences orthologous to srpkF in A. aculeatus. Alignment used ClustalW. Individual nodes were examined with 1000 bootstrap replicates; only values below 1000 are shown
The expression of srpkF increased under carbon-limited conditions, such as the no carbon and Avicel® conditions (Fig. 3), which showed strong correlations between SrpkF functions in cellulose-responsive induction and expression profiles. We focused on two signaling pathways involved in cellulose-responsive induction in A. aculeatus to narrow possible pathways. One is ManR-dependent signaling that induces expression of cbhI, cmc2, and cel7b. We expected that deletion and overexpression of srpkF would show a consistent effect on these three genes. However, effects varied, suggesting that transcription factors other than ManR could be involved. Overexpression of clbR, a putative transcription factor involved in cellobiose-responsive induction in A. aculeatus, did not affect the expression of cbhI and cmc2 but did reduce the expression of cel7b. Still, deletion of clbR reduced their expression levels in response to Avicel® (Kunitake et al. 2015). These data suggest that various factors participate in ManR-dependent signaling in response to cellulosic carbon sources in A. aculeatus.
Interestingly, overexpression of srpkF promoted xylanase gene expression in response to d-xylose, but no effect of srpkF deletion was seen for xylose-responsive expression of the xylanase gene. SrpkF is thus critically regulated at the transcription level. How SrpkF participates in the two different signaling pathways in response to cellulose and d-xylose is not clear. XlnR is constitutively expressed in A. oryzae and is phosphorylated in the presence of d-xylose then rapidly dephosphorylated by removing d-xylose from the medium (Noguchi et al. 2011). ManR phosphorylation status is unknown and SrpkF is a candidate kinase that could be involved in ManR- and XlnR-dependent signaling (Fig. 6).
A recent study addressed levels of conservation and diversity in the regulatory mechanisms of cellulolytic enzyme genes in Ascomycete fungi (Kunitake and Kobayashi 2017). XlnR controls the transcription of 20–30 genes encoding cellulolytic and xylanolytic enzymes in the presence of cellulose in A. niger (Stricker et al. 2008). XlnR mainly regulates the expression of xylanolytic enzyme genes for A. oryzae and A. aculeatus and is only marginally involved in the expression of cellulolytic enzyme genes in response to cellulose (Marui et al. 2002; Tani et al. 2012). In contrast, Xyr1, an XlnR ortholog in T. reesei, is a master regulator that modulates the expression of xylanolytic and cellulolytic enzyme genes in response to various carbon sources, such as d-xylose, sophorose, galactose, and lactose (Stricker et al. 2006, 2007). XLR-1 in N. crassa participates in the induction of xylanolytic but not significantly involved in the induction of cellulolytic enzyme genes (Sun et al. 2012). The expression of cellulolytic enzyme genes is mainly regulated by ManR in A. oryzae and A. aculeatus (Ogawa et al. 2013; Tsumura et al. 2021) and its orthologs ClrB in A. nidulans and CLR-2 in N. crassa (Coradetti et al. 2012, 2013). Complex regulation mechanisms can be conferred by the acquisition of paralogous genes that establish new signal transduction pathways (Baker et al. 2013). Orthologous genes of srpkF are absent from the genomes of several cellulase-producing fungi, such as Trichoderma and Neurospora species. The acquisition of srpkF in Aspergillus might lead to differential regulation of cellulolytic enzyme genes. A logical next step is to identify target proteins of SrpkF, which will help understand the complex regulatory mechanisms of cellulolytic enzyme genes in Aspergillus.
References
Adachi H, Tani S, Kanamasa S, Sumitani J, Kawaguchi T (2009) Development of a homologous transformation system for Aspergillus aculeatus based on the sC gene encoding ATP-sulfurylase. Biosci Biotechnol Biochem 73:1197–1199. https://doi.org/10.1271/bbb.80772
Baba Y, Sumitani J, Tani S, Kawaguchi T (2015) Characterization of Aspergillus aculeatus β-glucosidase 1 accelerating cellulose hydrolysis with Trichoderma cellulase system. AMB Express 5:3. https://doi.org/10.1186/s13568-014-0090-3
Baker CR, Hanson-Smith V, Johnson AD (2013) Following gene duplication, paralog interference constrains transcriptional circuit evolution. Science 342:104–108. https://doi.org/10.1126/science.1240810
Bischof RH, Ramoni J, Seiboth B (2016) Cellulases and beyond: the first 70 years of the enzyme producer Trichoderma reesei. Microb Cell Fact 15:106. https://doi.org/10.1186/s12934-016-0507-6
Bustos F, Segarra-Fas A, Nardocci G, Cassidy A, Antico O, Davidson L, Brandenburg L, Macartney TJ, Toth R, Hastie CJ, Moran J, Gourlay R, Varghese J, Soares RF, Montecino M, Findlay GM (2020) Functional diversification of SRSF protein kinase to control ubiquitin-dependent neurodevelopmental signaling. Dev Cell 55(629–647):e627. https://doi.org/10.1016/j.devcel.2020.09.025
Coradetti S, Craig J, Xiong Y, Shock T, Tian C, Glass N (2012) Conserved and essential transcription factors for cellulase gene expression in Ascomycete fungi. Proc Natl Acad Sci USA 109:7397–7402. https://doi.org/10.1073/pnas.1200785109
Coradetti ST, Xiong Y, Glass NL (2013) Analysis of a conserved cellulase transcriptional regulator reveals inducer-independent production of cellulolytic enzymes in Neurospora crassa. MicrobiologyOpen 2:595–609. https://doi.org/10.1002/mbo3.94
Dagher SF, Fu XD (2001) Evidence for a role of Sky1p-mediated phosphorylation in 3’ splice site recognition involving both Prp8 and Prp17/Slu4. RNA 7:1284–1297. https://doi.org/10.1017/s1355838201016077
de Souza CP, Hashmi SB, Osmani AH, Andrews P, Ringelberg CS, Dunlap JC, Osmani SA (2013) Functional analysis of the Aspergillus nidulans kinome. PLoS One 8:e58008. https://doi.org/10.1371/journal.pone.0058008
Gui JF, Chandler SD, Fu XD (1994a) Purification and characterization of a kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc Natl Acad Sci U S A 91:10824–10828. https://doi.org/10.1073/pnas.91.23.10824
Gui JF, Lane WS, Fu XD (1994b) A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 1994:678–682. https://doi.org/10.1038/369678a0
Gou LT, Lim DH, Ma W, Aubol BE, Hao Y, Wang X, Zhao J, Liang Z, Shao C, Zhang X et al (2020) Initiation of parental genome reprogramming in fertilized oocyte by splicing kinase SRPK1-catalyzed protamine phosphorylation. Cell 180:1212–1227, e1214. https://doi.org/10.1016/j.cell.2020.02.020
Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807. https://doi.org/10.1126/science.1137016
Hong Y, Chan CB, Kwon IS, Li X, Song M, Lee HP, Liu X, Sompol P, Jin P, Lee HG, Yu SP, Ye K (2012) SRPK2 phosphorylates tau and mediates the cognitive defects in Alzheimer's disease. J Neurosci 32:17262–17272. https://doi.org/10.1523/JNEUROSCI.3300-12.2012
Kunitake E, Kobayashi T (2017) Conservation and diversity of the regulators of cellulolytic enzyme genes in ascomycete fungi. Curr Genet 63:951–958. https://doi.org/10.1007/s00294-017-0695-6
Kunitake E, Tani S, Sumitani J, Kawaguchi T (2011) Agrobacterium tumefaciens-mediated transformation of Aspergillus aculeatus for insertional mutagenesis. AMB Express 1:46. https://doi.org/10.1186/2191-0855-1-46
Kunitake E, Tani S, Sumitani J, Kawaguchi T (2013) A novel transcriptional regulator, ClbR, controls the cellobiose- and cellulose-responsive induction of cellulase and xylanase genes regulated by two distinct signaling pathways in Aspergillus aculeatus. Appl Microbiol Biotechnol 97:2017–2028. https://doi.org/10.1007/s00253-012-4305-8
Kunitake E, Kawamura A, Tani S, Takenaka S, Ogasawara W, Sumitani J, Kawaguchi T (2015) Effects of clbR overexpression on enzyme production in Aspergillus aculeatus vary depending on the cellulosic biomass-degrading enzyme species. Biosci Biotechnol Biochem 79:488–495. https://doi.org/10.1080/09168451.2014.982501
Kunitake E, Li Y, Uchida R, Nohara T, Asano K, Hattori A, Kimura T, Kanamaru K, Kimura M, Kobayashi T (2019) CreA-independent carbon catabolite repression of cellulase genes by trimeric G-protein and protein kinase A in Aspergillus nidulans. Curr Genet 65:941–952. https://doi.org/10.1007/s00294-019-00944-4
Marui J, Kitamoto N, Kato M, Kobayashi T, Tsukagoshi N (2002) Transcriptional activator, AoXlnR, mediates cellulose-inductive expression of the xylanolytic and cellulolytic genes in Aspergillus oryzae. FEBS Lett 528:279–282. https://doi.org/10.1016/s0014-5793(02)03328-8
Murao S, Kanamoto J, Arai M (1979) Isolation and identification of a cellulolytic enzyme producing microorganism. J Ferment Technol 57:151–156
Nakagawa O, Arnold M, Nakagawa M, Hamada H, Shelton JM, Kusano H, Harris TM, Childs G, Campbell KP, Richardson JA, Nishino I, Olson EN (2005) Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev 19:2066–2077. https://doi.org/10.1101/gad.1338705
Nakazawa H, Kawai T, Ida N, Shida Y, Kobayashi Y, Okada H, Tani S, Sumitani J, Kawaguchi T, Morikawa Y, Ogasawara W (2012) Construction of a recombinant Trichoderma reesei strain expressing Aspergillus aculeatus β-glucosidase 1 for efficient biomass conversion. Biotechnol Bioeng 109:92–99. https://doi.org/10.1002/bit.23296
Noguchi Y, Tanaka H, Kanamaru K, Kato M, Kobayashi T (2011) Xylose triggers reversible phosphorylation of XlnR, the fungal transcriptional activator of xylanolytic and cellulolytic genes in Aspergillus oryzae. Biosci Biotechnol Biochem 75:953–959. https://doi.org/10.1271/bbb.100923
Ogawa M, Kobayashi T, Koyama Y (2013) ManR, a transcriptional regulator of the β-mannan utilization system, controls the cellulose utilization system in Aspergillus oryzae. Biosci Biotechnol Biochem 77:426–429. https://doi.org/10.1271/bbb.120795
Payne CM, Knott BC, Mayes HB, Hansson H, Himmel ME, Sandgren M, Ståhlberg J, Beckham GT (2015) Fungal cellulases. Chem Rev 115:1308–1448. https://doi.org/10.1021/cr500351c
Schuster A, Tisch D, Seidl-Seiboth V, Kubicek CP, Schmoll M (2012) Roles of protein kinase A and adenylate cyclase in light-modulated cellulase regulation in Trichoderma reesei. Appl Environ Microbiol 78:2168–2178. https://doi.org/10.1128/AEM.06959-11
Stricker AR, Grosstessner-Hain K, Würleitner E, Mach RL (2006) Xyr1 (xylanase regulator 1) regulates both the hydrolytic enzyme system and D-xylose metabolism in Hypocrea jecorina. Eukaryot Cell 5:2128–2137. https://doi.org/10.1128/ec.00211-06
Stricker AR, Steiger MG, Mach RL (2007) Xyr1 receives the lactose induction signal and regulates lactose metabolism in Hypocrea jecorina. FEBS Lett 581:3915–3920. https://doi.org/10.1016/j.febslet.2007.07.025
Stricker AR, Mach RL, de Graaff LH (2008) Regulation of transcription of cellulases- and hemicellulases-encoding genes in Aspergillus niger and Hypocrea jecorina (Trichoderma reesei). Appl Microbiol Biotechnol 78:211–220. https://doi.org/10.1007/s00253-007-1322-0
Sun J, Tian C, Diamond S, Glass NL (2012) Deciphering transcriptional regulatory mechanisms associated with hemicellulose degradation in Neurospora crassa. Eukaryot Cell 11:482–493. https://doi.org/10.1128/EC.05327-11
Tani S, Kanamasa S, Sumitani J, Arai M, Kawaguchi T (2012) XlnR-independent signaling pathway regulates both cellulase and xylanase genes in response to cellobiose in Aspergillus aculeatus. Curr Genet 58:93–104. https://doi.org/10.1007/s00294-012-0367-5
Tani S, Tsuji A, Kunitake E, Sumitani J, Kawaguchi T (2013) Reversible impairment of the ku80 gene by a recyclable marker in Aspergillus aculeatus. AMB Express 3:4. https://doi.org/10.1186/2191-0855-3-4
Tani S, Kawaguchi T, Kobayashi T (2014) Complex regulation of hydrolytic enzyme genes for cellulosic biomass degradation in filamentous fungi. Appl Microbiol Biotechnol 98:4829–4837. https://doi.org/10.1007/s00253-014-5707-6
Tani S, Yuki S, Kunitake E, Sumitani J, Kawaguchi T (2017) Dipeptidyl peptidase IV is involved in the cellulose-responsive induction of cellulose biomass-degrading enzyme genes in Aspergillus aculeatus. Biosci Biotechnol Biochem 81:1227–1234. https://doi.org/10.1080/09168451.2017.1295800
Tsumura R, Sawada K, Kunitake E, Sumitani J, Kawaguchi T, Tani S (2021) A component of the septation initiation network complex, AaSepM, is involved in multiple cellulose-responsive signaling pathways in Aspergillus aculeatus. Appl Microbiol Biotechnol 105:1535–1546. https://doi.org/10.1007/s00253-021-11110-7
van Peij NN, Visser J, de Graaff LH (1998) Isolation and analysis of XlnR, encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger. Mol Microbiol 27:131–142. https://doi.org/10.1046/j.1365-2958.1998.00666.x
Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu XD (1998) SRPK2: A differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol 140:737–750. https://doi.org/10.1083/jcb.140.4.737
Wang M, Dong Y, Zhao Q, Wang F, Liu K, Jiang B, Fang X (2014) Identification of the role of a MAP kinase Tmk2 in Hypocrea jecorina (Trichoderma reesei). Sci Rep 4:6732. https://doi.org/10.1038/srep06732
Wang ZH, Liu P, Liu X, Manfredsson FP, Sandoval IM, Yu SP, Wang JZ, Ye K (2017)Delta-secretase phosphorylation by SRPK2 enhances its enzymatic activity, provoking pathogenesis in Alzheimer's disease. Mol cell 67:812–825, e815. https://doi.org/10.1016/j.molcel.2017.07.018
Znameroski EA, Li X, Tsai JC, Galazka JM, Glass NL, Cate JH (2014) Evidence for transceptor function of cellodextrin transporters in Neurospora crassa. J Biol Chem 289:2610–2619. https://doi.org/10.1074/jbc.M113.533273
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science under Grant 19K05777. Part of this work was also supported by the New Energy and Industrial Technology Development Organization Project under Grant P07015.
Funding
This work was supported by JSPS KAKENHI under Grant 19K05777. Part of this work was also supported by the New Energy and Industrial Technology Development Organization (NEDO) Project under Grant P07015.
Author information
Authors and Affiliations
Contributions
S.T. and T.K. conceived and designed the project. R.K. and N.K. conducted the experiments. S.T wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethical approval
This article does not contain any studies that involve human participants or animals.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files. DNA sequences will be available at the DDBJ database when this article is released for publication.
Code availability
Not applicable.
Additional information
Communicated by Michael Polymenis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
294_2021_1207_MOESM1_ESM.pdf
Supplementary file1 Supplementary Fig. S1. Construction of ΔsrpkF and srpkF+ strains. The upper portion of the figure shows restriction enzyme maps of srpkF loci in MR12, Δ srpkF, and srpkF+ strains. Underlined DNA regions were used as DNA probes for Southern blotting to detect DNA fragments after digestion with BamHI (lower portion of figure). Supplementary Fig. S2. Construction of ΔCsrpkF and complement strains (CsrpkF+). The upper portion of the figure shows restriction enzyme maps of srpkF loci in MR12, ΔCsrpkF, and complement strains. Asterisks indicate positions of stop codons. Underlined DNA regions were used as DNA probes for Southern blotting to detect DNA fragments after digestion with DraI (lower portion of figure). Supplementary Fig. S3. Construction of the OEsrpkF strain. The upper portion of the figure shows restriction enzyme maps of srpkF loci in MR12 and OEsrpkF strains. Underlined DNA regions were used as DNA probes for Southern blotting to detect DNA fragments after digestion with PstI (lower portion of figure). Supplementary Fig. S4. Effect of srpkF deletion on expression of cbhI and xynIb. qRT-PCR results for each gene in MR12 (M), ΔsrpkF (Δ), srpkF+ (+), ΔCsprkF (ΔC), and the complement strain of ΔCsprkF (C+) incubated for 9 h under noninducing and the 1% Avicel®-inducing conditions. Expression of genes was normalized to gpdA expression. Fold induction of test genes (gene expression under inducing conditions divided by that under the noninducing condition) is presented as means of three independent experiments, and error bars indicate standard deviations. Letters indicate significant differences between groups (p < 0.05, one-way ANOVA). Supplementary Fig. S5. Expression of srpkF in the overexpression strain. qRT-PCR analysis of srpkF in MR12 (M) and the srpkF-overexpressing strain (OEsrpkF, OE). RNA was prepared from strains grown for 24 h in the presence of 0.1% cellobiose with 50 μg/L DNJ (a) or 1% d-xylose (b). Relative expression is the ratio of mean expression in srpkF divided by gpdA expression. Relative expression is presented as means of three independent experiments, and error bars indicate standard deviations. M: MR12, filled bars; OE: OEsrpkF, striped bars. (PDF 1593 kb)
Rights and permissions
About this article

Cite this article
Katayama, R., Kobayashi, N., Kawaguchi, T. et al. Serine–arginine protein kinase-like protein, SrpkF, stimulates both cellobiose-responsive and d-xylose-responsive signaling pathways in Aspergillus aculeatus. Curr Genet 68, 143–152 (2022). https://doi.org/10.1007/s00294-021-01207-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-021-01207-x