
Abstract
α-Glucosidase, Agl2, from Xanthomonas campestris was successfully overexpressed in Escherichia coli BL21(DE3) cells and purified with Ni columns. The enzyme exhibits glycosylation abilities towards a wide range of phenolic substrates, including phenol, vanillin, and ethyl vanillin, with maltose as the glycosyl donor. The catalytic properties of the purified enzyme were further investigated. It was observed that the synthesized glycosides started to degrade with prolonged catalytic time, giving an “n”-shaped kinetic profile. To understand such catalytic behavior, the Agl2-catalyzed glycosylation process was investigated kinetically. Based on the obtained parameters, it was concluded that although the substrate conversions are thermodynamically restricted in a batch system, the glycosylation efficiency can be kinetically controlled by the glycosylation/hydrolysis selectivity. Glucose was produced by both glycosylation and hydrolysis, significantly impacting the glycosylation efficiency. This study provides a mechanistic understanding of the α-glucosidase-catalyzed glycosylation process in a water-based system. The developed kinetic model was successful in explaining and analyzing the catalytic process. It is suggested that when α-glucosidase is employed for glycosylation in a water-enriched environment, the catalytic efficiency is mainly impacted by the enzyme’s glycosylation/hydrolysis selectivity and glucose content in the catalytic environment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycosylation is the process by which the hydroxyl group(s) of a chemical is (are) etherified with one or more carbohydrate units through glycosidic linkages. The modified structure, named a glycoside, usually possesses advantageous physiochemical properties that differ from those of the original chemical. For example, water-immiscible compounds such as menthol and vanillin have been reported to have improved water solubility after glycosylation (Sato et al. 2012; Nakagawa et al. 2000), which favors their applications as food additives. On the other hand, the carbohydrate moiety has a positive influence on bioavailability due to its high affinity towards the hydrophilic phospholipid heads of the cell membrane (Moradi et al. 2016). Such favorable properties are observed for a number of glycosides, such as arbutin, glyceryl glycoside, and ascorbic acid 2-glycoside, in terms of their antioxidative, moisturizing, and skin-brightening effects (Li et al. 2017; Han et al. 2013; Schrader et al. 2012; Zhu et al. 2019a, b; Kurosu et al. 2002), which are substantially desired for cosmetic uses. Glycosylation also improves the stability of chemicals with enolic or polyphenolic structures, such as kojic acid, ascorbic acid, and flavonoids, by masking the hydroxyl groups, while their antioxidative effect is maintained after the chemicals are adsorbed and hydrolyzed intracellularly (Buchner et al. 2006; Kwon et al. 2007; Kitao and Serine 1994). In addition, after glycosylation, therapeutic proteins exhibit increased molecular stability in vivo, modulated pharmacodynamic responses, and improved pharmacokinetic profiles, which has aroused great pharmaceutical research interest (Sinclair and Elliott 2005; Solá and Griebenow 2010).
The natural presence of glycosides suggests a sustainable, enzyme-catalyzed method of glycosylation. Glycosyltransferase catalyzes the transfer of carbohydrate moieties from nucleotide diphosphate sugars (NDP-sugars) to aglycons, which are responsible for the natural establishment of glycosidic linkages (Weijers et al. 2008; Breton et al. 2006). However, the process is not suitable for in vitro glycosylation due to the expensiveness of the activated glycosyl donor (Wu et al. 2013). On the other hand, glucosidase, which mainly catalyzes the cleavage of glycosidic bonds in a water-enriched environment (Demir et al. 2018a), is capable of transferring or even forming glycosidic bonds under specific conditions, such as low water activity and high aglycon concentration (Hansson and Adlercreutz 2001; Rivas et al. 2013; Rather and Mishra 2013). Compared to glycosyltransferase, glucosidase employs naturally abundant, inactivated carbohydrates (glucose, xylose, mannose, galactose, maltose, etc.) directly as glycosyl donors (Gao et al. 2000; Vic et al. 1996; Drouet et al. 1994), making it a prominent catalyst for scaled production of glycosides.
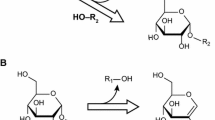
The catalysis of glucosidase follows either the inverting or retaining mechanism determined by the nature of the enzyme (Davies and Henrissat 1995). Both mechanisms require a carboxylic acid/base pair located at the catalytic center that is able to form a skew-boat enzyme-carbohydrate intermediate with the anomeric carbon, as shown in Fig. 1 (Rye and Withers 2000). As the glycosylation and hydrolysis processes catalyzed by glucosidase follow the same mechanism with only a difference in the glycosyl acceptor (either aglycon or H2O), the synthesis of glycoside is kinetically controlled when performed in a water-based, aglycon-enriched environment (Thuan and Sohng 2013; Lundemo et al. 2017). On the other hand, the kinetics of the process from a mechanistic perspective is still obscure because of the complexity of the process resulting from selective reaction routes as well as inhibition effects from multiple resultant products. Ojima et al. (2012) evaluated the kinetics of the glycerol glycosylation process under the catalysis of α-glucosidase (EC 3.2.1.20) (Scheme 1). Due to the presence of metal ions in their study, two states of the enzymes with different glycosylation activities were characterized. On the other hand, the impacts of the glycosyl donor, maltose in this case, as well as its hydrolysis, were overlooked, which lead to significant deviations between their simulation and experimental results. Bohlin et al. (2013) developed a more integrated model (Scheme 2) with considerations of both glycosylation and hydrolysis to explain the oligosaccharide synthesis process catalyzed by β-glucosidase (EC 3.2.1.21). However, the reversible interaction between glucose and glucosidase, as reported in many studies, was overlooked in the mechanism, which leads to incomplete characterization of the glucose inhibition effect (Andrić et al. 2010; Öhgren et al. 2007), as well as the reverse hydrolysis process (Ljunger et al. 1994; Kosary et al. 1998). In addition, Scheme 2 lacks the characterization of glycoside hydrolysis (irreversible interactions between glucosidase and the synthesized oligosaccharides), which makes the developed model more suitable to explain the initial catalytic state (with negligible glucose and glycoside concentrations) instead of the overall process.

The inverting (a) and retaining (b) catalytic schemes of α-glucosidase. ROH is either water or aglycon for the hydrolysis and glycosylation processes, respectively (Rye and Withers 2000)


The glycosylation scheme proposed by Bohlin et al. (2013), where cellobiose (G2) was employed in the synthesis of cellotriose (G3). Side reactions with glucose or H2O as glycosyl acceptors were also considered. However, the interactions between the enzyme and the resultant products (G, G2, and G3) were neglected, so the model can be applied only at the initial reaction state (Bohlin et al. 2013)
In this study, the glycosylation processes of multiple substances, including phenol, vanillin, and ethyl vanillin, catalyzed by α-glucosidase isolated from Xanthomonas campestris were evaluated. Maltose was employed as the glycosyl donor. An “n”-shaped kinetic profile of the obtained glycosides was observed, which cannot be properly explained by the existing schemes. Herein, for a profound understanding of the glucosidase-catalyzed glycosylation process, Scheme 3 was proposed with two major considerations: (1) selective glycosylation and hydrolysis processes with multiple glycosyl acceptors and (2) reversible interactions of both glucose and glycoside with glucosidase. The developed scheme was employed to characterize the kinetic profiles of the whole transglycosylation process with different glycosyl acceptors. The constants in the proposed scheme were quantified, which help reveal the restrictive factors in the glycosylation process.

The selective glycosylation/hydrolysis catalytic scheme of α-glucosidase in a water-enriched environment with maltose (M) as a glycosyl donor. The enzyme first interacts with maltose to generate a unit of glucose (G) and a unit of enzyme-glucose intermediate (EG*). EG* competitively reacts with either aglycon (S) or water, which is a kinetically controlled step. The generated glycoside (P) and glucose are considered to be reversibly interacting with the enzyme with product inhibition effects
Materials and methods
Plasmid and strain construction
The construction of the Escherichia coli BL21(DE3) (pET28a-Agl2) strain with target enzyme expression was performed by following similar previously reported protocols (Demir and Beydemir 2015). X. campestris sp. campestris 8004 is a spontaneous rifampicin-resistant strain derived from X. campestris sp. campestris NCPPB No. 1145 (He et al. 2007). The genomic DNA of X. campestris sp. campestris 8004 was extracted with a Bacterial DNA Extraction Kit (Omega Bio-Tek, Guangzhou, China). Based on the reported genome sequencing of the strain (Qian et al. 2005), the nucleotide sequences of the α-glucosidase genes (Agl1, NCBI Protein ID: AAY48076; Agl2, NCBI Protein ID: AAY48708; Agl3, NCBI Protein ID: AAY48711) were amplified by PCR with a Phanta® Max Super-Fidelity DNA Polymerase Kit (Vazyme Biotech, Nanjing, China) according to the manufacturer’s protocols. The resultant product was purified using a gel extraction kit (Omega Bio-Tek, Guangzhou, China). The plasmid pET28a was linearized by NdeI and HindIII (TaKaRa Bio, Dalian, China) and purified with a gel extraction kit (Omega Bio-Tek, Guangzhou, China). The purified PCR product and pET28a were ligated with a One Step Cloning Kit (Vazyme Biotech, Nanjing, China) and transformed into competent E. coli BL21(DE3) cells to obtain the recombinant E. coli strains. Agl2 was later identified as the target enzyme. The corresponding recombinant E. coli BL21(DE3) (pET28a-Agl2) strain was employed in the following studies.
Expression and purification of α-glucosidase
A seed culture of E. coli BL21(DE3) (pET28a-Agl2) strain was prepared in 50 mL LB medium with 50 μg/mL kanamycin at 37 °C. Batch culture was conducted in a 2.5 L fermenter (BioFlo 110; New Brunswick Scientific Co., Shanghai, China) with 1.0 L fermentation medium composed of 15 g/L yeast extract, 20 g/L tryptone, 10 g/L glycerol, 0.17 M KH2PO4, 0.72 M K2HPO4, and 0.5 g/L MgSO4·7H2O. The pH was maintained at 6.8 by adding 28% (v/v) ammonia hydroxide. Dissolved oxygen was maintained above 25% of saturation by coupling the agitation with an air feed at 2.5 L/min. Induction was started when the OD600 reached ~ 10. The culturing temperature was reduced to 24 °C prior to the feeding of glycerol (300 g/L) and lactose (200 g/L) at 12 mL/h and 36 mL/h, respectively. The cells were harvested when the OD600 reached ~ 42. The harvested cells were then centrifuged and resuspended in 50 mM phosphate buffer, followed by homogenization. After homogenization, the supernatant was collected and tested for the glycosylation abilities of Agl2 with phenol, vanillin, and ethyl vanillin in 0.5 g/L phosphate buffer, and the enzyme glycosylation activity (GA) was calculated via Eq. (1)
where GAU stands for the unit of the enzyme’s glycosylation activity, cP, t (g/L) is the glycoside concentration after 2 min of catalysis, and MP (g/mol) is the molecular weight of the corresponding glycoside.
The target enzyme was further purified with His60 Ni Superflow Resin & Gravity Columns (TaKaRa Bio, Dalian, China) according to the protocol instructions. The eluted purified Agl2 was desalted in 5 mM phosphate buffer through dialysis (14 kDa cutoff value) and then concentrated with solid PEG 20000. The obtained Agl2 was confirmed by SDS-PAGE analysis by following protocols similar to those previously reported (Demir et al. 2017, 2018b) and stored at − 20 °C for further experimental use. The hydrolytic activity of Agl2 was assayed by hydrolyzing 1 mmol/L p-NPG in 0.1 mol/L sodium phosphate buffer (pH = 8.0). One unit of enzyme activity is defined as the amount of enzyme that catalyzes the release of 1 μmol of p-NP released per minute (U/mL).
Quantitative analysis by HPLC
HPLC (Agilent Technologies 1260 Infinity II, USA) with a DAD detector (Agilent Technologies G7115A 1260 DAD WR, USA) and an InertSustain C18 (GL Sciences, Japan) column was employed to quantify the concentrations of glycosides and glycosyl acceptors in the samples. The HPLC conditions were as follows: mobile phase, methanol-water (20:80, v/v); flow rate, 1 mL/min; temperature, 40 °C; injection volume, 10 μL; and wavelength, 272 nm.
The glucose concentration was analyzed by HPLC with an ELSD detector (Agilent Technologies G4260B 1260 Infinity II ELSD, USA) and an Xtimate Sugar-Ca column (Welch Materials, Inc., Shanghai, China). The HPLC conditions were as follows: mobile phase, water; flow rate, 1 mL/min; temperature, 40 °C; and injection volume, 10 μL.
Enzyme deactivation
The denaturing effect of Agl2 in the presence of glycosyl acceptors was investigated. Twenty microliters of the purified enzyme was incubated in a 500 μL solution containing 1 g/L phenol, vanillin, or ethyl vanillin. After incubation for different durations, 500 μL maltose solution (700 g/L) was added, and samples were taken after 2 min of glycosylation at 40 °C. The glycoside concentrations were quantified based on HPLC protocols as described in the previous session. The resultant Agl2 glycosylation activity was calculated via Eq. (1)
Kinetic investigation of the glycosylation process
The glycosylation kinetics of phenol, vanillin, and ethyl vanillin was investigated with purified Agl2. Glycosylation was initiated by adding 20 μL enzyme into 1 mL phosphate buffer solutions (pH = 8.0) containing 350 g/L maltose and 0.5 g/L glycosyl acceptors, phenol, vanillin, and ethyl vanillin. Catalysis was performed at 40 °C, and samples were taken continuously on a time basis. The samples were added to 1 M HCl solutions to quench the reaction. Two more groups of ethyl vanillin glycosylation were studied with initial ethyl vanillin concentrations of 1 g/L and 2 g/L to ensure the accuracy of data interpretation.
The rates of glycosylation (rg) and hydrolysis (rh) in the catalytic system based on Scheme 3 are given as Eqs. (2) and (3), respectively.
The enzyme balance leads to
where kd represents the enzyme deactivation constant in the catalytic environment, which was characterized by the enzyme deactivation study.
The formation rates of glycosides and glucose can be obtained based on the reaction scheme by Eqs. (5) and (6), respectively, and these rates are later employed to characterize the kinetic parameters of the proposed scheme. The regressions were performed according to our previously reported method (Liu 2017).
Effect of glucose inhibition
The inhibitory effect of glucose on glycosylation activity was investigated. Five groups of experiments were carried out with 2 mL phosphate buffer solutions containing 350 g/L maltose and 0.5 g/L ethyl vanillin. Then, 0 mg, 20 mg, 40 mg, 80 mg, and 160 mg glucose were added to each group, followed by the addition of 20 μL purified to start the catalysis. The enzyme’s glycosylation activity was calculated based on Eq. (1). The relative activity (RA) of each group was defined by Eq. (7)
where GAG and GA0 are the glycosylation activities with and without the addition of glucose, respectively.
Results
Heterologous expression of Agl2 and its glycosylation abilities
Previous studies have reported that the Xanthomonas campestris strain displays high glycosylation activity with maltose as a glycosyl donor, which suggests the potential responsibility of α-glucosidase (EC 3.2.1.20) in the glycosylation process (Nakagawa et al. 2000; Kurosu et al. 2002). To identify the enzyme, the genetic sequences of α-glucosidases in X. campestris were cloned and expressed in E. coli BL21(DE3) cells. Glycosylation of multiple glycosyl acceptors, including phenol, vanillin, and ethyl vanillin, was performed using the cytoplasmic supernatant. High glycosylation activity and substrate adaptability were observed in Agl2. An extra signal was observed by HPLC prior to the aglycon in all investigated groups, which was further proven to be the corresponding glycoside by LC-MS (Figure S1).
For further investigations of the catalytic properties of Agl2, a two-stage fermentation strategy including a batch and a fed-batch was employed for the heterologous expression of Agl2 in the recombinant E. coli strain, followed by His-tag purification. The protein content of the purified Agl2 was measured to be 3.76 mg/mL with bovine serum albumin as a standard, and the hydrolytic activity was assayed to be 5983 U/mL. The molecular size of the purified enzyme was characterized to be approximately 60 kDa based on SDS-PAGE (Figure S2), which agrees with the cloned DNA length (1.662 kb, protein sequence + His-tag + T7-tag).
Enzyme deactivation during glycosylation
The stability of the glycosylation activity of Agl2 was investigated by incubating the purified enzyme in solutions containing 1 g/L glycosyl acceptors at the catalytic temperature. The glycosylation activity after incubation was quantified via Eq. (1), and it was observed that the impact of phenol, vanillin, and ethyl vanillin on the glycosylation activity of Agl2 at 40 °C is negligible after 2 h of cultivation (Figure S3). In this case, the deactivation coefficient in Eq. (4) is kd ≈ 0, which leads to the simplification of the enzyme balance equation as
Equation (8) is employed in the following kinetic characterization.
Glycosylation kinetics of Agl2 in a water-based environment
In the kinetic analysis, the corresponding glycoside concentrations of the three species were monitored during the glycosylation process, as shown in Figs. 2 a, 3 a, and 4 a. Agl2 displayed fast glycosylation in the first 10 min of catalysis with GAs of 6.11 GAU/mL, 0.55 GAU/mL, and 0.24 GAU/mL for phenol, vanillin, and ethyl vanillin, respectively, in the groups with an initial substrate concentration of 0.5 g/L. Interestingly, instead of reaching equilibrium, degradation of the glycosides was observed with prolonged reaction time in all three species. The maximum conversions were measured to be 74.5%, 12.3%, and 4.6%. An increase in the substrate concentration from 0.5 to 1.5 g/L in the ethyl vanillin group seemed to have little impact on the conversions, which were measured to be 4.6%, 4.4%, and 4.5%. This further suggested the negligible impact of the substrates on the enzyme’s glycosylation activity. By extending the reaction time, over 40% of the glycosides was degraded in the vanillin and ethyl vanillin groups. Phenol glycoside is relatively stable, experiencing only 16% degradation. The concentration of glucose, as the major by-product, was also monitored. A consistent increase in glucose in the catalytic environment was observed until the end of the experiment.



The selective glycosylation/hydrolysis process catalyzed by glucosidase was described kinetically as shown in Scheme 3. The parameters from Scheme 3 are given in Table 1 and were determined through the regression of kinetic data with Eqs. (2), (3), and (8). The values of k1,+, k1,−, and K1 vary among species, as the interactions between enzyme and aglycons are substrate specific. On the other hand, due to the same interaction between the enzyme and carbohydrates, KM, k2,+, k2,−, and K2 remain the same among the groups. E0 is also a constant due to the same amount of enzyme added. The equilibrium constants of glycosylation given as KG, calculated by Eq. (9), represent the thermodynamic control of glycosylation. The low glycosylation equilibrium constant (KG) compared to the hydrolysis equilibrium constant (KH) in the vanillin and ethyl vanillin groups suggests a significant thermodynamic limitation in glycosylating the two species.
The glycosylation/hydrolysis selectivity of Agl2 was determined by Eq. (11), which is based on the enzyme’s activity in glycosylating or hydrolyzing the enzyme-carbohydrate intermediate. The enzyme selectivity was 316.47 L/mol, 17.06 L/mol, and 10.59 L/mol for phenol, vanillin, and ethyl vanillin, respectively.
Impact of glucose on glycosylation activity
Glucose, as the by-product of the glycosylation process, is also generated through the hydrolysis of maltose. By observing the accumulation of glucose in the catalytic environment, the impact of glucose on the GA of Agl2 was evaluated by changing the initial glucose concentration. The relative glycosylation activity of Agl2 with ethyl vanillin as the glycosyl acceptor was calculated by Eq. (7). Agl2 encountered a 50% loss of GA when the glucose concentration was increased by 20 g/L. Over 80% of the glycosylation activity was lost when 80 g/L glucose was added, but the activity loss was less significant at higher glucose concentrations (Figure S4). As the GA was measured as the initial rate, the loss of activity at the initial catalytic stage suggests a fast and inhibitory interaction between Agl2 and glucose, as shown in Scheme 3.
Discussion
The glycosylation kinetic profiles of PhOH, vanillin, and ethyl vanillin shown in Figs. 2 a, 3 a, and 4 a suggest the high glycosylation activity and substrate adaptability possessed by Agl2. As seen by comparing the k1,+ values, the enzyme displays glycosylation activity in the order of phenol > vanillin > ethyl vanillin. The differences in glycosylation activity among the three aglycons can potentially result from the ether group in the ortho-position of the phenolic hydroxyl group, which causes steric hindrance for aglycons in attacking the enzyme-glucose intermediate.
It was observed in all three groups that after 10 min of fast glycosylation, the synthesized glycosides started to degrade with prolonged reaction time. To explain this phenomenon, the catalytic scheme of α-glucosidase, as shown in Scheme 3, needs to be followed. In the first step, α-glucosidase as a hydrolytic enzyme cleaves the α-1,4-glycosidic bond in maltose, which releases one molecule of glucose and leaves the other glucose unit attached to the enzyme in its activated form (EG*), ready to be attacked by a nucleophile. This step is considered reversible, as EG* can be attacked by the hydroxyl group of glucose, leading to oligomerization (Bohlin et al. 2013). More importantly, EG* can be attacked by aglycons and water, leading to glycosylation and hydrolysis, respectively. The reversed glycosylation is considered the only way for glycosides to degrade, following the attack by water that leads to glycoside degradation. Studies reporting product inhibition during hydrolysis (Andrić et al. 2010; Öhgren et al. 2007) and the use of glucose in synthesizing glycosides (Ljunger et al. 1994; Kosary et al. 1998) suggest the reversibility of the interaction between α-glucosidase and glucose, as well as the activation of glucose by α-glucosidase. In this case, the overall glycosylation (Eq. (12)) and hydrolysis (Eq. (13)) reactions catalyzed by α-glucosidase are interconnected by EG* and can be treated as two reversible reactions with general equations written as
By understanding this reaction scheme, it can be concluded that Agl2 glycosylation is dominant at the beginning of catalysis, which results in the fast synthesis of glucosides. On the other hand, glucose resulting from both glycosylation and hydrolysis slows down the glycosylation process to its first equilibrium point (point A in Fig. 5), where Eq. (12) has reached equilibrium but Eq. (13) has not. With prolonged catalysis, glucose is continuously obtained from the hydrolysis of maltose, as shown in Figs. 2 b, 3 b, and 4 b–d, shifting the glycosylation equilibrium to the substrate side and leading to the degradation of glycosides until the second equilibrium point (point B in Fig. 5), where both Eqs. (12) and (13) have reached equilibrium. In this case, the glycosylation catalyzed by Agl2 can be characterized as kinetically controlled at the beginning and thermodynamically controlled at the end.

The glycosylation of ethyl vanillin catalyzed by Agl2 with different initial maltose concentrations. The lines were obtained based on Eqs. (2)–(6) with the parameters given in Table 1. Initial ethyl vanillin concentration, CS,0 = 0.5 g/L. A high glycosyl donor concentration is desired for the synthesis of glycosides by α-glucosidase in a water-based environment
The low KG values given in Table 1 indicate that the glycosylation process is thermodynamically restrained, especially for vanillin and ethyl vanillin. Considering the poor water solubility for most phenolic compounds, the high glycosyl donor concentration in the catalytic environment (350 g/L maltose in this study) becomes the major driving force for glycosylation to occur (Fig. 5).
Selectivity is a major property in determining whether a substrate is suitable for glycosylation by a given glucosidase. Although a high selectivity does not necessarily result in high glycosylation activity, it impacts the synthetic yield at point A as well as the ease of reaction control. The high values of selectivity suggest that Agl2 is a proper tool in catalyzing glycosylation reactions.
Recent studies on glucosidase employed enzyme engineering technologies to create a new type of enzyme named glycosynthase, which is characterized by a complete elimination of its hydrolytic activity but preservation of its glycosylation activity (Min et al. 2007). Through such modification, the competitive attack of the enzyme-glucose intermediate by water is prevented, which improves the glycosylation efficiency. A comparison of the ethyl vanillin glycosylation process with Agl2 and glycosynthase is given in Fig. 6. It can be seen that with the same glycosylation properties (k1,+, k1,−, and K1 values), selectivity has a strong impact on the glycosylation efficiency.

Synthesis of ethyl vanillin glycoside with Agl2, glycosynthase, and Agl2 with constant glucose and maltose concentrations. The initial maltose, glucose, and ethyl vanillin concentrations are 350 g/L, 1 g/L, and 0.5 g/L, respectively
Another advantage of glycosynthase lies in its low glucose production. According to the catalytic scheme, the inhibition effect of glucose can be characterized by three aspects: (1) The generation of an activated enzyme-glucose intermediate (EG*) from a glycosyl donor is hindered by the accumulation of glucose; (2) glucose in the catalytic environment can bind to and inactivate glucosidase (EG), leading to product inhibition; and (3) the accumulation of glucose shifts the glycosylation equilibrium to the substrate side, resulting in degradation of glycosides. By understanding this mechanism, the timely removal of glucose from the catalytic environment with integrated catalysis-separation technologies can be used to promote glycosylation efficiency while preventing the synthesized glycosides from degradation (Fig. 6). Such technology has already been applied in the cellulose hydrolysis process, where glucose is removed in a timely manner by membrane reactors to avoid its inhibition of hydrolytic enzymes (Andrić et al. 2010). On the other hand, corresponding studies on glycoside synthesis have yet to be reported and are required for further investigations.
In summary, Agl2 exhibited significantly higher glycosylation efficiency of phenol than of vanillin and ethyl vanillin, and the synthesized glycosides display an n-shaped kinetic profile. Such kinetic properties can be properly explained based on Scheme 3 when reversible and selective glycosylation/hydrolysis processes in a water-enriched environment are considered. According to the obtained parameters, the glycosylation efficiency of vanillin and ethyl vanillin is restrained both thermodynamically and kinetically. Glycosylation/hydrolysis selectivity and glucose inhibition are the two key restrictive factors impacting glycosylation efficiency. Selectivity affects the kinetic control of the effective attack of the enzyme-glucose intermediate (EG*) by aglycons. Glucose, on the other hand, has both a kinetic impact on glycosylation activity and a thermodynamic impact on glycosylation equilibrium.
References
Andrić P, Meyer AS, Jensen PA, Dam-Johansen K (2010) Effect and modeling of glucose inhibition and in situ glucose removal during enzymatic hydrolysis of pretreated wheat straw. Appl Biochem Biotechnol 160:280–297
Bohlin C, Praestgaard E, Baumann MJ, Borch K, Praestgaard J, Monrad RN, Westh P (2013) A comparative study of hydrolysis and transglycosylation activities of fungal β-glucosidases. Appl Microbiol Biotechnol 97:159–169
Breton C, Šnajdrová L, Jeanneau C, Koča J, Imberty A (2006) Structures and mechanisms of glycosyltransferases. Glycobiology 16:29R–37R
Buchner N, Krumbein A, Rohn S, Kroh LW (2006) Effect of thermal processing on the flavonols rutin and quercetin. Rapid Commun Mass Spectrom 20:3229–3235
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859
Demir Y, Beydemir Ş (2015) Purification, refolding, and characterization of recombinant human paraoxonase-1. Turk. J. Chem. 39:764–776
Demir Y, Taslimi P, Ozaslan MS, Oztaskin N, Çetinkaya Y, Gulçin I, Beydemir Ş, Goksu S (2018a) Antidiabetic potential: in vitro inhibition effects of bromophenol and diarylmethanones derivatives on metabolic enzymes. Arch. Pharm. 351:1800263
Demir Y, Kotan MS, Dikbaş N, Beydemir Ş (2017) Phytase from Weissella halotolerans: purification, partial characterisation and the effect of some metals. Int. J. Food Prop. 20:S2127–S2137
Demir Y, Dikbaş N, Beydemir Ş (2018b) Purification and biochemical characterization of phytase enzyme from Lactobacillus coryniformis (MH121153). Mol. Biotechnol. 60:783–790
Drouet P, Zhang M, Legoy MD (1994) Enzymatic synthesis of alkyl β-D-xylosides by transxylosylation and reverse hydrolysis. Biotechnol Bioeng 43:1075–1080
Gao C, Mayon P, MacManus DA, Vulfson EN (2000) Novel enzymatic approach to the synthesis of flavonoid glycosides and their esters. Biotechnol Bioeng 71:235–243
Han R, Liu L, Shin HD, Chen RR, Du G, Chen J (2013) Site-saturation engineering of lysine 47 in cyclodextrin glycosyltransferase from Paenibacillus macerans to enhance substrate specificity towards maltodextrin for enzymatic synthesis of 2-O-D-glucopyranosyl-L-ascorbic acid (AA-2G). Appl Microbiol Biotechnol 97:5851–5860
Hansson T, Adlercreutz P (2001) Enhanced transglucosylation/hydrolysis ratio of mutants of Pyrococcus furiosus β-glucosidase: effects of donor concentration, water content, and temperature on activity and selectivity in hexanol. Biotechnol Bioeng. 75:656–665
He Y, Zhang L, Jiang B, Zhang Z, Xu R, Tang D, Qin J, Jiang W, Zhang X, Liao J, Cao J, Zhang S, Wei M, Liang X, Lu G, Feng J, Chen B, Cheng J, Tang J (2007) Comparative and functional genomics reveals genetic diversity and determinants of host specificity among reference strains and a large collection of Chinese isolates of the phytopathogen Xanthomonas campestris pv. campestris. Genome Biol. 8:1–26
Kitao S, Serine H (1994) Syntheses of two kojic acid glycosides with sucrose phosphorylase from Leuconostoc mesenteroides. Biosci Biotechnol Biochem 58:419–420
Kosary J, Stefanovits-Banyai E, Boross L (1998) Reverse hydrolytic process for O-alkylation of glucose catalysed by immobilized α-and β-glucosidases. J Biotechnol 66:83–86
Kurosu J, Sato T, Yoshida K, Tsugane T, Shimura S, Kirimura K, Kino K, Usami S (2002) Enzymatic synthesis of α-arbutin by α-anomer-selective glucosylation of hydroquinone using lyophilized cells of Xanthomonas campestris WU-9701. J Biosci Bioeng 93:328–330
Kwon T, Kim CT, Lee JH (2007) Transglucosylation of ascorbic acid to ascorbic acid 2-glycoside by a recombinant sucrose phosphorylase from Bifidobacterium longum. Biotechnol Lett 29:611
Li Z, Han H, Wang B, Gao J, Zhu B, Peng R, Yao Q (2017) Transglucosylation of ascorbic acid to ascorbic acid 2-glycoside by a truncated version of α-glucosidase from Aspergillus niger. J Food Biochem 41:e12432
Liu S (2017) Kinetic theory and reaction kinetics. In: Liu S (ed) Bioprocess engineering: kinetics, sustainability, and reactor design, 2nd edn. Elsevier, pp 259-295
Ljunger G, Adlercreutz P, Mattiasson B (1994) Enzymatic synthesis of octyl-β-glycoside in octanol at controlled water activity. Enzyme Microb Technol 16:751–755
Lundemo P, Karlsson EN, Adlercreutz P (2017) Eliminating hydrolytic activity without affecting the transglycosylation of a GH1 β-glucosidase. Appl Microbiol Biotechnol 101:1121–1131
Min Y, Gideon JD, Benjamin GD (2007) A glycosynthase catalyst for the synthesis of flavonoid glycosides. Angew Chem Int Ed 46:3885–3888
Moradi SV, Hussein WM, Varamini P, Simerska P, Toth I (2016) Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem Sci 7:2492–2500
Nakagawa H, Dobashi Y, Sato T, Yoshida K, Tsugane T, Shimura S, Kirimura K, Kino K, Usami S (2000) α-Anomer-selective glucosylation of menthol with high yield through a crystal accumulation reaction using lyophilized cells of Xanthomonas campestris WU-9701. J Biosci Bioeng 89:138–144
Öhgren K, Bura R, Lesnicki G, Saddler J, Zacchi G (2007) A comparison between simultaneous saccharification and fermentation and separate hydrolysis and fermentation using steam-pretreated corn stover. Process Biochem 42:834–839
Ojima T, Saburi W, Yamamoto T, Kudo T (2012) Characterization of Halomonas sp. Strain H11 α-glucosidase activated by monovalent cations and its application for efficient synthesis of α-D-glucosylglycerol. Appl Environ Microbiol 78:1836–1845
Qian W, Jia Y, Ren S, He Y, Feng J, Lu L, Sun Q, Ying G, Tang H, Wu W, Hao P, Wang L, Jiang B, Zeng S, Gu W, Lu G, Rong L, Tian Y, Yao Z, Fu G, Chen B, Fang R, Qiang B, Chen Z, Zhao G, Tang J, He C (2005) Comparative and functional genomic analyses of the pathogenicity of phytopathogen Xanthomonas campestris pv. campestris. Genome Res. 15:757–767
Rather MY, Mishra S (2013) β-Glucosidases: an alternative enzyme based method for synthesis of alkyl-glycosides. Sustainable Chem Processes 1(1):7
Rivas F, Parra A, Martinez A, Garcia-Granados A (2013) Enzymatic glycosylation of terpenoids. Phytochem Rev 12:327–339
Rye CS, Withers SG (2000) Glucosidase mechanisms. Curr Opin Chem Biol 4:573–580
Sato D, Eshita Y, Katsuragi H, Hamada H, Shimoda K, Kubota N (2012) Glycosylation of vanillin and 8-nordihydrocapsaicin by cultured Eucalyptus perriniana cells. Molecules 17:5013–5020
Schrader A, Siefken W, Kueper T, Breitenbach U, Gatermann C, Sperling G, Biernoth T, Scherner C, Stäb F, Wenck H, Wittern KP (2012) Effects of glyceryl glycoside on AQP3 expression, barrier function and hydration of human skin. Skin Pharmacol Physiol 25:192–199
Sinclair AM, Elliott S (2005) Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci 94:1626–1635
Solá RJ, Griebenow K (2010) Glycosylation of therapeutic proteins. BioDrugs 24:9–21
Thuan NH, Sohng JK (2013) Recent biotechnological progress in enzymatic synthesis of glycosides. J Ind Microbiol Biotechnol 40:1329–1356
Vic G, Hastings JJ, Crout DH (1996) Glucosidase-catalysed synthesis of glycosides by an improved procedure for reverse hydrolysis: application to the chemoenzymatic synthesis of galactopyranosyl-(1-4)-O-α-galactopyranoside derivatives. Tetrahedron: Asymmetry 7:1973–1984
Weijers CAGM, Franssen MCR, Visser GM (2008) Glycosyltransferase-catalyzed synthesis of bioactive oligosaccharides. Biotechnol Adv 26:436–456
Wu X, Chu J, Wu B, Zhang S, He B (2013) An efficient novel glycosylation of flavonoid by β-fructosidase resistant to hydrophilic organic solvents. Bioresour Technol 129:659–662
Zhu L, Jiang D, Zhou Y, Lu Y, Fan Y, Chen X (2019a) Batch-feeding whole-cell catalytic synthesis of α-arbutin by amylosucrase from Xanthomonas campestris. J Ind Microbiol Biotechnol 46:759–767
Zhu L, Xu M, Lu C, Chen L, Xu A, Fang J, Chen H, Lu Y, Fan Y, Chen X (2019b) Optimization of whole-cell biotransformation for scale-up production of α-arbutin from hydroquinone by the use of recombinant Escherichia coli. AMB Express 9:94
Funding
This study was funded by the National Natural Science Foundation of China (No. 31601390, No. 21908196, and No. 21572206).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 461 kb)
Rights and permissions
About this article

Cite this article
Chen, H., Yang, S., Xu, A. et al. Insight into the glycosylation and hydrolysis kinetics of alpha-glucosidase in the synthesis of glycosides. Appl Microbiol Biotechnol 103, 9423–9432 (2019). https://doi.org/10.1007/s00253-019-10205-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-10205-6