
Abstract
The fungus Fusarium fujikuroi causes bakanae disease of rice due to its ability to produce the plant hormones, the gibberellins. The fungus is also known for producing harmful mycotoxins (e.g., fusaric acid and fusarins) and pigments (e.g., bikaverin and fusarubins). However, for a long time, most of these well-known products could not be linked to biosynthetic gene clusters. Recent genome sequencing has revealed altogether 47 putative gene clusters. Most of them were orphan clusters for which the encoded natural product(s) were unknown. In this review, we describe the current status of our research on identification and functional characterizations of novel secondary metabolite gene clusters. We present several examples where linking known metabolites to the respective biosynthetic genes has been achieved and describe recent strategies and methods to access new natural products, e.g., by genetic manipulation of pathway-specific or global transcritption factors. In addition, we demonstrate that deletion and over-expression of histone-modifying genes is a powerful tool to activate silent gene clusters and to discover their products.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rice-pathogenic fungus Fusarium fujikuroi (formerly Gibberella fujikuroi), a member of the monophyletic but diverse Fusarium fujikuroi species complex (FFC), is one of the first described plant pathogens (Kvas et al. 2009). The fungus causes bakanae (foolish seedling) disease which led and still leads to serious crop losses in all rice-growing countries: typical symptoms are excessively elongated rice seedlings with chlorotic stems and leaves. The infected plants are infertile and therefore produce no/few edible grains (Ou 1985). The unusual early elongation of seedlings is attributed to the production of gibberellins (GAs), a family of plant hormones that are secreted by the fungus (Tudzynski et al. 2016; Bömke and Tudzynski 2009). Sometimes, the fungus causes stunting of rice shoots, probably due to the production of fumonisins and fusaric acid. The type of symptoms (stunting or hyper-elongation) and the severity of the disease depend not only on the ability of the strains to produce high levels of fumonisins and fusaric acid on the one hand, and GAs on the other hand, but also on the resistance levels of the host (Fiyaz et al. 2016; Niehaus et al. 2017a).
Today, the fungus is used worldwide for the industrial-scale production of GAs which are used to manage fruit size and quality (e.g., grapes, apples and oranges), to malt barley for beer production, to increase the size of ornamental flowers (e.g., gardenia or geranium flowers), and to increase the sugar yield in sugarcane. GAs are also extensively used to induce parthenocarpic seedless fruits (e.g., grapes and melons) which are at the same time larger and sweeter (Sponsel and Hedden 2010).
Besides GAs, the fungus is also known to produce additional secondary metabolites (SMs), such as harmful mycotoxins as well as pigments (Fig. 1a–j). For example, it produces the mycotoxins fusaric acid, moniliformin, fusarins, fumonisins, and beauvericin (Moretti et al. 1996; Desjardins et al. 1997; Desjardins and Proctor 2007; Kvas et al. 2009; Barrero et al. 1991; Bacon et al. 1996) as well as the pigments neurosporaxanthin (carotenoid), bikaverin, and fusarubins (Avalos et al. 2012; Balan et al. 1970; Linnemannstöns et al. 2002b; Studt et al. 2012). However, the biosynthetic genes for the production of these SMs and the genetic capacity of the fungus to produce even more yet unknown products remained unknown for most of them. Only in 1998, the genes for the biosynthesis of the best known SMs of F. fujikuroi, the GAs, were cloned by differencial cDNA screenings. Subsequent chromosome walking revealed that all seven pathway genes are located adjacent to each other in a gene cluster (Tudzynski and Hölter 1998). At that time, an increasing number of identified SM biosynthetic genes provided evidence that clustering of those genes is a common feature not only for prokaryotic metabolic pathways but also for genes involved in fungal secondary metabolism (Keller and Hohn 1997).

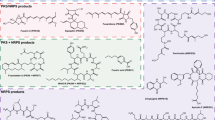
Chemical structures of F. fujikuroi secondary metabolites. Gibberellin GA3 is produced by a terpene cyclase (a), metabolites highlighted in blue (b, c, f and h) are polyketide synthase (PKS) products, those highlighted in yellow (g, j) are non-ribosomal peptide synthetase (NRPS)-derived, and metabolites shown in green (d, e and i) are PKS-NRPS products
Some years later, the key enzyme-encoding genes for bikaverin and carotenoid biosynthesis were cloned by PCR and heterologous hybridization of a genomic library, respectively, and were also shown to be organized in gene clusters (Linnemannstöns et al. 2002b; Linnemannstöns et al. 2002a; Wiemann et al. 2009). However, only the availability of the genome sequences of F. fujikuroi (Wiemann et al. 2013) and highly related Fusarium species, such as F. verticillioides (Ma et al. 2010), F. circinatum (Wingfield et al. 2012), F. mangiferae and F. proliferatum (Niehaus et al. 2016b), revealed a complete overview of the genetic capacity of the members of the FFC to produce SMs. The in silico analyses revealed not only common gene clusters but also surprising differences between the related species. These FFC genome sequences allowed the researchers to create a preliminary catalog of genes encoding polyketide synthases (PKSs), non-ribosomal peptide synthetases (NRPSs), dimethylallyltryptophan synthases (DMATSs), and terpene cyclases (TCs), the key enzymes for SM biosynthesis (Wiemann et al. 2013; Niehaus et al. 2016b; Hansen et al. 2015).
The purpose of this review is to briefly summarize our current knowledge on secondary metabolism in F. fujikuroi. As most of the SM gene clusters are not expressed under standard laboratory conditions, different approaches have been developed for the activation of those silent gene clusters, e.g., optimization of culture conditions (pH, nutrient availability, light), genetic manipulation of pathway-specific transcription factors (TFs), global regulators, and histone modifications affecting epigenetic control. We describe the approaches by which cryptic and silent gene clusters have been deciphered and new products identified.
In silico analysis of the genome of F. fujikuroi
To estimate the genetic potential of F. fujikuroi to produce SMs other than GAs, the genome of F. fujikuroi strain IMI58289 was searched for genes predicted to encode the four classes of core enzymes: PKSs, NRPSs, TCs, and DMATSs. The genes were identified in silico by the presence of characteristic protein domain content and by BLAST analyses (Wiemann et al. 2013). This analysis revealed 47 potential SM key genes: 17 genes that encode putative type I PKSs, one type III PKS, 15 NRPSs, 2 DMATSs, and 12 TCs. The latter enzymes include nine sesquiterpene cyclases (STC1–STC9), one diterpene cyclase (ent-kaurene synthase, DTC1), one triterpene cyclase (TrTC1), and one tetraterpene cyclase (phytoene synthase, TeTC1) (Niehaus et al. 2016b). Four of the PKSs contain typical domains of both PKS and NRPS and are also referred to as PKS-NRPS hybrids. Based on domain architecture, 14 of the predicted PKSs belong to the class of reducing-type PKSs (R-PKSs): they contain the characteristic ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) domains. The remaining three PKSs are non-reducing-type PKSs (NR-PKSs) because they lack the KR, DH, and ER domains (Chiang et al. 2010). NR-PKSs synthesize aromatic polyketides, e.g., the red pigment bikaverin (Wiemann et al. 2009).
At the time of genome sequencing, only some of the gene clusters and their products were known: the GA gene cluster with the bifunctional ent-kaurene/ent-copalyl diphosphate synthase (DTC1; Cps/ks; Fig. 2a), and the pigment gene clusters for bikaverin (PKS4; Bik1; Fig. 2b) and carotenoid (TeTC1) biosynthesis (Tudzynski and Hölter 1998; Wiemann et al. 2009; Studt et al. 2012; Linnemannstöns et al. 2002a) (Table 1). To unravel cryptic gene clusters, it was important to find the correct conditions for their expression. Therefore, genome-wide expression studies under high and low nitrogen, as well as acidic and alkaline pH conditions, were performed. These experiments revealed that about half of the key genes are not expressed under these standard conditions. For the others, there were clear indications for a nitrogen and/or pH-dependent gene expression (Wiemann et al. 2013). Due to the high level of genome conservation between the Fusarium species, especially between members of the FFC, the products of some of the newly identified putative gene clusters could be predicted by comparative genomics, e.g., the products for four NRPS genes required for synthesis of the siderophores ferricrocin, fusarinine, and ferrichrome and the insecticidal mycotoxin beauvericin (BEA), as well as the PKS genes required for synthesis of fusaric acid, equisetin, and fumonisins (Niehaus et al. 2016b) (Table 1).

Gene clusters responsible for the biosynthesis of F. fujikuroi secondary metabolites. The designations (a-j) correspond to the panels in Fig. 1. The GA gene cluster Key enzyme- and transcription factor-encoding genes are highlighted in blue and violet, respectively. Genes that do not belong to the clusters are gray. The direction of transcription is indicated with arrows and white bars represent introns (color figure online)
Linking biosynthetic gene clusters to their metabolites via comparative genomics
Due to the genome sequencing of some members of the FFC by our and other groups, comparative genomics helped to identify several gene clusters and link them to their products (Ma et al. 2010; Niehaus et al. 2016b; Wiemann et al. 2013; Chiara et al. 2015). In F. verticillioides, the gene clusters for fumonisins, fusaric acid, and fusarins were already well characterized (Brown et al. 2007; Brown et al. 2012). Based on these data, highly conserved orthologous gene clusters have now been identified and functionally studied in the genome of F. fujikuroi (Fig. 2c–e).
After the identification of the PKS Fum1, the key enzyme of fumonisin (FUM) biosynthesis in the maize pathogen F. verticillioides, subsequent deletion of the adjacent genes resulted in the identification of the FUM biosynthetic cluster consisting of 17 co-expressed genes (Proctor et al. 1999; Brown et al. 2012; Brown et al. 2007). The FUM gene cluster of F. verticillioides, the main FUM producer among the genus Fusarium, shows a high homology to the predicted PKS11 gene cluster in F. fujikuroi (FFUJ_09240 to FFUJ_09254). Exceptions are FUM20 which is absent and FUM17 which is non-functional due to a premature truncation (Fig. 2c) (Rösler et al. 2016a). Both genes are probably not involved in FUM biosynthesis in F. verticillioides (Proctor et al. 2003). However, despite the strong similarities of the FUM clusters that occur in both fungi, the gene expression and FUM production levels are much lower in F. fujikuroi. Only the over-expression of the cluster-specific Zn(II)2Cys6-type TF gene FUM21 in F. fujikuroi led to the activation of FUM gene expression and an about 1000-fold elevation of toxin levels compared to the wild type (WT) (Rösler et al. 2016a). The data provide evidence that even the low-level producer F. fujikuroi can become a potent FUM producer, leading to potentially high contamination of rice products.
Only recently, genome sequencing of eight additional F. fujikuroi isolates from different rice-growing countries revealed strain-specific differences: while most of the isolates produce GAs but not FUM, one Korean isolate (B14) is able to produce FUM but not GAs and, therefore, causes stunting and early withering instead of the typical hyper-elongation of rice seedlings (Niehaus et al. 2017a).
One of the oldest known SMs of F. fujikuroi and some other Fusarium species is fusaric acid (FSA), a mycotoxin with high phytotoxic properties but low toxicity to animals and humans (Fig. 1d). The key gene for fusaric acid biosynthesis, the PKS-encoding gene FUB1, was first identified in F. verticillioides. Its deletion resulted in total loss of FSA production. Four adjacent genes were shown to be co-regulated with FUB1 suggesting that five genes belong to the putative FSA gene cluster (Brown et al. 2012). An orthologous gene cluster exists in F. fujikuroi, and its function was studied by targeted gene replacement of all five postulated biosynthetic genes (FUB1-FUB5) (Fig. 2d) and subsequent product analysis. Only two of the five cluster genes were shown to be essential for the biosynthesis of FSA (Niehaus et al. 2014b). However, the biosynthetic pathway leading to FSA and especially the origin of the nitrogen atom, which is incorporated into the FSA backbone, remained unknown. Then, in 2016, an extended FUB gene cluster encoding a second key enzyme (NRPS34) and two Zn(II)2Cys6-type TFs, Fub10 and Fub12, was identified independently in both fungi (Studt et al. 2016b; Brown et al. 2015) (Fig. 2d).
Besides FSA, various Fusarium species also produce the potent mutagen fusarin C and some drivatives (Fig. 1e). The first fusarin (FUS) biosynthetic gene, fusS, encoding a hybrid of a type I iterative PKS fused to an NRPS module, was found in F. moniliforme and F. venenatum (Song et al. 2004). Later, functional fusS homologs have been discovered also in F. graminearum (GzFUS1), F. verticillioides (FUS1), and F. fujikuroi (fusA), respectively (Brown et al. 2012; Diaz-Sanchez et al. 2012; Gaffoor et al. 2005). Although many Fusarium species inside and outside the FFC contain fusS orthologs and predicted FUS gene clusters, there are surprising differences in gene cluster organization and gene expression between closely related members of the FFC. For instance, F. mangiferae, belonging to the Asian clade of the FFC together with F. fujikuroi, has only remnants of the FUS cluster (Wiemann et al. 2013).
Besides the members of the genus Fusarium, a similar FUS gene cluster has been identified in the distantly related fungus Metarhizium anisopliae where it is responsible for the biosynthesis of 7-desmethyl analogs of fusarin C and (8Z)-fusarin C, named NG-391 and NG-393, respectively (Krasnoff et al. 2006).
After genome sequencing, the entire FUS gene cluster containing nine co-regulated genes (FUS1-FUS9) (Fig. 2e) has been functionally characterized in F. fujikuroi. Beside FUS1, encoding the PKS-NRPS key enzyme, the other genes encode a putative α/β hydrolase with a predicted peptidase domain (FUS2), a glutathione S-transferase (FUS3), a peptidase A1 (FUS4), a serine hydrolase (FUS5), a major facilitator superfamily (MFS) transporter (FUS6), an aldehyde dehydrogenase (FUS7), a cytochrome P450 monooxygenase (FUS8), and a methyltransferase (FUS9) (Kleigrewe et al. 2012; Niehaus et al. 2013). Deletion of each single gene and subsequent product analyses revealed that only four of them (FUS1, FUS2, FUS8, and FUS9) are required for FUS biosynthesis, while WT-like FUS levels were found in the culture filtrates of ∆fus3, ∆fus4, ∆fus5, ∆fus6, and ∆fus7 (Niehaus et al. 2013). Previously, it has been shown for F. moniliforme (and probably for other Fusarium species, too) that the PKS-NRPS forms the amide linkage between the heptaketide part of the PKS and the activated l-homoserine of the NRPS part (Rees et al. 2007). Analysis of the biosynthetic pathway in F. fujikuroi revealed that the product of Fus1, called pre-fusarin, is released as an alcohol with an open ring structure. Pre-fusarin is then hydroxylated by the P450 monooxygenase Fus8 at carbon C-20 to 20-hydroxy-fusarin (Niehaus et al. 2013). The subsequent ring closure is most likely catalyzed by Fus2 what enables the P450 monooxygenase Fus8 to catalyze an additional hydroxylation at the C-20 atom. In a last reaction, the methyltransferase Fus9 methylates the hydroxy group at C-21 and forms fusarin C (Kleigrewe et al. 2012; Niehaus et al. 2013).
F. fujikuroi produces a second PKS-derived red pigment—fusarubin
At the time of genome sequencing, the only characterized PKS gene cluster in F. fujikuroi was the one for bikaverin (BIK) biosynthesis (Wiemann et al. 2009). The BIK genes were shown to be strongly repressed by conditions of high nitrogen and an alkaline pH (Wiemann et al. 2009). Surprisingly, the culture fluid was still deeply red pigmented in cultures with low levels of sodium nitrate, which makes the medium alkaline (pH 6.5–7.5) within the first 24 h of cultivation. High-performance liquid chromatography coupled to UV light and Fourier transformation mass spectrometry (HPLC-UV-FTMS) detection clearly demonstrated that the red pigment was indeed not BIK, but an unknown compound (Studt et al. 2012). We assumed the involvement of a second NR-PKS because all green and red naphthoquinone piments so far studied were shown to be synthesized by this group of enzymes. A BLASTp analysis with the Bik1 sequence against the just sequenced genome of F. fujikuroi IMI58289 led to the identification of a second NR-PKS, designated Fsr1, with 40% identity to Bik1. Phylogenetic analysis of the KS domains of known fungal NR-PKSs revealed the highest similarity of Fsr1 to the Pgl1 proteins of F. verticillioides and F. graminearum, which are both responsible for perithecial pigmentation (Gaffoor et al. 2005; Proctor et al. 2007). However, no chemical structure had been proposed for these perithecial pigments.
Deletion of FSR1 and the five co-regulated genes (Fig. 2f) downstream revealed that FSR4 and FSR5 are not involved in biosynthesis of the unknown pigment, whereas deletion of FSR1 and FSR6, the latter encoding a putative Zn(II)2Cys6 TF, resulted in the total loss of pigmentation (Studt et al. 2012). Expression studies confirmed that Fsr6 acts as a pathway-specific TF. The structure of the main compounds found in the cultures of the WT and deletion mutants was extensively elucidated by MS, characteristic UV spectra, and nuclear magnetic resonance (NMR) data. By these methods, a whole set of chemically related naphthoquinones, the fusarubins (FSR), was identified (Fig. 1f). Based on these data, a model for the biosynthetic pathway of FSR was suggested (Studt et al. 2012). It is noteworthy that naphthoquinones from Fusarium javanicum with a similar structure have been described seven decades ago and were designated javanicin and oxyjavanicin (Arnstein and Cook 1947). Oxyjavanicin was later renamed FSR.
Genome mining by over-expression of cluster-specific transcription factor genes
Our genome-wide expression studies showed that most of the in silico identified gene clusters in F. fujikuroi are orphan clusters for which the products are still unknown. In most cases, the genes are not expressed or expressed at very low levels under laboratory conditions making the chemical analysis of the products difficult (Wiemann et al. 2013). To identify as many new SMs as possible, we searched for those putative gene clusters which contain typical TF-encoding genes (Table 2). In F. fujikuroi, 17 out of the 47 gene clusters (36%) contain one or two TFs genes. There are several successful examples demonstrating that over-expression of pathway-specific TF genes by using strong or inducible promoters is a sufficient approach to activate normally silent gene clusters and to identify new SMs (Brakhage 2013).
Two of the predicted clusters with PKS19 and NRPS31 (Fig. 2g, h) as key enzymes were of special interest because they were present only in the genome of F. fujikuroi, but absent from any other sequenced genomes of highly related FFC species (Wiemann et al. 2013). A gene cluster similar to the NRPS31 (FFUJ_00003) cluster has been characterized in the distantly related species Fusarium semitectum and shown to be responsible for the synthesis of the cyclic tetrapeptide apicidin, an inhibitor of histone deacetylases (HDACs) (Jin et al. 2010). In both species, the cluster encodes an atypical “bank” TF, which contains a basic DNA-binding domain and four ankyrin repeats. However, in contrast to typical bZIP TFs, bank TFs do not contain leucine zipper or helix-loop-helix motifs. Over-expression of this TF gene in F. fujikuroi led to elevated expression of all cluster genes and a 10-fold enhanced product formation under inducing high nitrogen conditions. De-regulated gene expression was observed even under repressing conditions of low nitrogen (Wiemann et al. 2013). However, instead of the expected apicidin, NMR and MS analyses of the over-expression mutant revealed the presence of a similar but distinct product designated apicidinF (APF) (Fig. 1g).
In contrast to apicidin, APF contains l-phenylalanine instead of l-isoleucine, and l-2-aminooctanedioic acid instead of l-2-amino-8-oxodecanoic acid. The other two amino acids, N-methoxy-l-tryptophan and d-pipecolic acid, are common in both compounds. To study the biosynthesis of APF, knock-out mutants of several cluster genes were generated and analyzed for their ability to produce APF or intermediates by HPLC coupled to high-resolution MS (Niehaus et al. 2014a). These studies showed that l-phenylalanine can be directly activated, while the three non-proteinogenic amino acids are synthesized by pathway-specific enzymes encoded by cluster genes. Besides APF, new derivatives were identified in the cultures of deletion mutants, which were designated apicidin J and K. Based on chemical analysis of the deletion mutants, a model for the APF biosynthetic pathway was suggested (Niehaus et al. 2014a).
The predicted PKS19 cluster consists of five genes encoding PKS19 (FFUJ_12239), a protein with unknown function (FFUJ_12240), a ToxD-like protein (FFUJ_12241), a putative MFS transporter (FFUJ_12242), a Zn(II)2Cys6 TF (FFUJ_12243), and a P450 monooxygenase (FFUJ_12244) (Fig. 2h) (Wiemann et al. 2013). Microarray and Northern blot analyses indicated that the predicted cluster genes are only expressed at very low levels. To activate the expression of the PKS19 cluster genes and potentially induce production of the yet unknown product(s), the cluster-specific TF-encoding gene (FFUJ_12243) was over-expressed resulting in enhanced expression of PKS19 (FFUJ_12239) and three additional predicted cluster genes (FFUJ_12240, FFUJ_12242, and FFUJ_12243). However, only simultaneous double over-expression of the TF- and PKS19-encoding genes (OE::PKS19/OE::TF) led to identification of four metabolites that were not produced by the WT. These compounds have similar molecular formula and UV spectra (Wiemann et al. 2013). The structures of the four newly discovered products, named fujikurins A-D (Fig. 1h), were elucidated using a combined approach of NMR and MS. They are all cyclic lactones with 1,3-diketo elements (von Bargen et al. 2015). Recently, orthologous PKS19 clusters have been found in the genomes of two newly sequenced F. proliferatum strains (Niehaus et al. 2016b).
Beside the FUS key gene FUS1, three additional PKS-NRPS genes have been identified in the genome of F. fujikuroi. One of them is PKS-NRPS1 (FFUJ_02219), the key gene of a predicted gene cluster with high similarity to the equisetin gene cluster from the distantly related fungus Fusarium heterosporum (Kakule et al. 2013). However, the F. fujikuroi cluster lacks the equisetin N-methyltransferase gene eqxD and, consequently, equisetin cannot be the final product. Both the F. fujikuroi and the F. heterosporum clusters harbor two genes encoding Zn(II)2Cys6 TFs (Table 2; Fig. 2i). One of them was shown to act as positive regulator of the cluster genes in F. heterosporum (Kakule et al. 2013).
Microarray analyses under different conditions revealed that the PKS-NRPS1 cluster is silent in F. fujikuroi (Janevska et al. 2017a). Over-expression of TF22 (FFUJ_02222), one of the two cluster-specific TF genes, led to an activation of all five cluster genes, including the PKS-NRPS key gene and the second TF-encoding gene (Fig. 2i). The mutant strains showed severe growth defects demonstrating that the produced compound is toxic to the fungus. The metabolic profile of the OE::TF22 mutant revealed a new peak which was not present in the WT. Structure elucidation confirmed that this product is trichosetin (Fig. 1i). Furthermore, over-expression of eqxD from F. heterosporum into the OE::TF22 mutant of F. fujikuroi led to the accumulation of the N-methylated equisetin, the final product of the F. heterosporum cluster (Janevska et al. 2017a). In contrast to TF22, over-expression of TF23 (FFUJ_02223) did not result in an activation of the cluster genes. Instead, TF23 was essential for inducing the cluster-encoded MFS transporter gene (FFUJ_02224) by the final product trichosetin. Therefore, TF23 plays an important role in detoxification of trichosetin and self-protection of the producing fungus (Janevska et al. 2017a).
However, not all TF genes which are located adjacent to key enzyme-encoding genes act as positive regulators of those clusters. Attempts to identify the products of PKS13 (gibepyrones), PKS16 (unknown product), STC1 (germacrene D), or NRPS22 (BEA) by over-expressing the TF genes in near proximity to the key genes have failed (Table 2). The strong upregulation of the TF-encoding genes did not result in elevated expression of the remaining cluster genes. Similarly, the TF adjacent to the dimethylallyltryptophan synthase-encoding gene DMATS1 (FFUJ_09179) is not functional or at least does not regulate DMATS1 expression. However, over-expression of the key gene DMATS1 resulted in production of a new metabolite, a reversely N-prenylated tryptophan with a rare form of prenylation (Arndt et al. 2017).
Global regulators affect growth, differentiation, and secondary metabolism
Besides cluster-specific TFs, the expression of SM biosynthetic genes is controlled by a hierarchical network of global regulators that respond to multiple environmental signals (Lind et al. 2015). The best studied regulatory complex is the fungal-specific velvet complex. The founding member of this group is the Aspergillus nidulans velvet protein VeA. VeA1 mutant strains obtained by classic mutagenesis were described more than 40 years ago, but only in 2002, the veA gene was cloned by complementation of the veA1 mutant strain with a multicopy plasmid library (Calvo 2008). Deletion of veA resulted in loss of the ability to form cleistothecia and the production of SMs, e.g., sterigmatocystin or penicillin, demonstrating a regulatory connection between secondary metabolism and development (Bayram and Braus 2012). In 2004, another global regulator of secondary metabolism was identified: LaeA (loss of aflR expression) (Bok and Keller 2004). Besides sterigmatocystin and penicillin, the A. nidulans laeA mutant was unable to produce lovastatin and some other SMs (Bok and Keller 2004). In 2008, it was found that LaeA and two proteins of the velvet family, VeA and VelB, form a trimeric complex that is essential for coordination of secondary metabolism and development (Bayram et al. 2008). Since then, the velvet complex and its impact on secondary metabolism has been studied in F. fujikuroi (Wiemann et al. 2010) and several other fungi, e.g., Penicillium chrysogenum, Trichoderma reesei, and Aspergillus fumigatus (Hoff et al. 2010; Karimi-Aghcheh et al. 2013; Perrin et al. 2007; Dhingra et al. 2012).
Deletion of vel1, vel2, and lae1 (the orthologs of veA, velB and laeA) in F. fujikuroi revealed common and different functions of the complex partners: while deletion of the three components of the F. fujikuroi velvet complex almost totally abolished biosynthesis of GAs, FUM, FUS, and FSA, Vel1 could simultaneously act as repressor of BIK biosynthesis (Wiemann et al. 2010; Niehaus et al. 2014b) (Fig. 3a). After genome sequencing, the role of Lae1 on secondary metabolism was analyzed again on a genome-wide basis (Niehaus et al. 2017c). The expression profile of the WT was compared with those of the ∆lae1 and a LAE1 over-expressing mutant (OE:LAE1). Both deletion and over-expression of LAE1 resulted in upregulation of some of the known, and five yet unknown, gene clusters. Thus, the expression of all GA, FUS, FUM, FSR, and the recently identified silent BEA genes was significantly increased in the OE:LAE1 mutant under inducing and in some cases also under repressing conditions. For instance, the strongest upregulation of FUS genes was observed under repressing low nitrogen conditions indicating that the nitrogen regulation is overcome by over-expression of LAE1. Similarly, the FSR genes, which are only expressed at alkaline pH in the WT (Studt et al. 2012), were upregulated in the OE:LAE1 mutant under acidic conditions suggesting that the pH regulation is circumvented by over-expressing LAE1. The most prominent result was the elevated expression of the GA biosynthetic genes and increased product levels under otherwise repressing nitrogen sufficent conditions (Mihlan et al. 2003; Pfannmüller et al. 2017).

Global regulators that affect the biosynthesis of the major F. fujikuroi secondary metabolites (SMs). Schematic representation of the clusters responsible for the biosynthesis of the nitrogen-repressed SMs gibberellins (GA), bikaverin (BIK), and fumonisins (FUM), as well as of the nitrogen-induced SMs fusarins (FUS), fusaric acid (FSA), and apicidin F (APF). The regulatory network is shown for the velvet complex, Sge1 and Csm1 (a) and for the nitrogen regulators AreA and AreB (b). Black arrows: direct or indirect gene cluster activation; red bars: direct or indirect repression (color figure online)
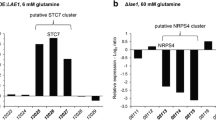
In contrast, the expression of the BIK genes (Wiemann et al. 2009) was increased in the ∆lae1 mutant under normally repressing high nitrogen indicating that Lae1 acts as repressor of BIK biosynthesis, similar to what was shown before for Vel1 (Wiemann et al. 2010). Lae1 and Vel1 also act as repressors for PKS13 which was recently shown to be responsible for gibepyrone biosynthesis (Janevska et al. 2016). Besides, some cryptic putative gene clusters, e.g., the yet uncharacterized STC7 and NRPS4 genes, were upregulated in the ∆lae1 and OE::LAE1 mutants. Based on the co-expression of adjacent genes, the borders of the putative STC7 and NRPS4 gene clusters could be predicted (Niehaus et al. 2017c).
Another global regulator affecting secondary metabolism in some Fusarium species is Sge1, a homolog of the morphological switch regulators Wor1 and Ryp1 in Candida albicans and Histoplasma capsulatum, respectively (Michielse and Rep 2009; Michielse et al. 2015; Brown et al. 2014). In contrast to other fungi, this TF is not required for conidiogenesis and pathogenicity in F. fujikuroi (Michielse et al. 2015). Microarray analysis of the Δsge1 mutant in comparison to the WT revealed that Sge1 functions as a global activator of secondary metabolism in F. fujikuroi. Under inducing low nitrogen conditions, the transcript and product levels of GA and FUM biosynthesis were significantly reduced, while under high nitrogen, the optimal condition for FSA, FUS, and APF (Niehaus et al. 2013; Niehaus et al. 2014a; Niehaus et al. 2014b), the gene expression and production levels for these three SMs were significantly reduced in the Δsge1 mutant (Michielse et al. 2015) (Fig. 3a). Interestingly, the almost total loss of APF biosynthesis in the Δsge1 mutant could not be overruled by over-expression of the cluster-specific TF gene APF2, suggesting that Sge1 affects the accessibility of this SM gene cluster for its activating TF, possibly through modification of the histone landscape (Michielse et al. 2015).
Over-expression of SGE1 in the WT background led to elevated FUM, FSA, and APF productions under favorable and FUS and FSA productions even under non-favorable conditions, indicating that nitrogen regulation can be overruled in this mutant. Most significantly, Sge1 was also required for expression of the yet uncharacterized non-canonical NRPS34 gene and six adjacent genes. All these genes were downregulated in the Δsge1 mutant and upregulated in the OE:SGE1 strain (Michielse et al. 2015). The product of the new cluster was identified by comparing the metabolite profiles between the OE:SGE1 mutant and the NRPS34 deletion strain. Surprisingly, instead of a new compound, a peak with the corresponding mass for FSA was identified in the OE:SGE1 mutant. As the new gene cluster is located in close proximity to the already known FSA biosynthetic gene cluster (Niehaus et al. 2014b), we suggested that both cluster segments are functionally linked and involved in FSA biosynthesis (Studt et al. 2016b) (Fig. 2d). During the course of this study, a second segment of the FUB gene cluster was also reported for two related fungi, F. verticillioides and Fusarium oxysporum (Brown et al. 2015). In all three species, the five FUB genes of the first cluster segment and the seven genes of the second segment are separated by three to four genes which are not involved in FSA biosynthesis (Fig. 2d). The extended FSA gene cluster encodes two TFs with specific functions, similar to the trichosetin (PKS-NRPS1) gene cluster (Janevska et al. 2017a). One of them, i.e., Fub10, regulates the expression of all cluster genes while the over-expression of the second TF, FUB12, did not result in a significant upregulation of FUB gene expression and FSA product formation. Instead, Fub12 is involved in the regulation of the derivatization of FSA into the less toxic derivatives dehydrofusaric acid and fusarinolic acid (Studt et al. 2016b).
Recently, another TF was found to be a major regulator of secondary metabolism and conidiation in F. fujikuoi, the GATA-type TF Csm1 (Niehaus et al. 2017b). This conserved GATA TF is an ortholog of A. nidulans NsdD which was shown to be an activator of sexual development and key repressor of conidiation in A. nidulans (Lee et al. 2014). In Botrytis cinerea, this TF was described as light-regulated repressor of macroconidia formation (Ltf1) and regulator of secondary metabolism (Schumacher et al. 2014). Deletion of CSM1 in F. fujikuroi resulted in strong elevation of microconidia formation compared to the WT indicating that Csm1 also acts as repressor of conidiogenesis in this fungus. Furthermore, expression patterns were compared between the F. fujikuroi WT and the ∆csm1 mutant under different nitrogen conditions. This microarray analysis revealed a strong impact of Csm1 on the expression of 19 out of the 47 gene clusters present in the genome of F. fujikuroi. Among them were the genes involved in biosynthesis of known products, e.g., APF, BEA, BIK, FSR, FSA, fujikurins, gibepyrones, and FUM (Fig. 3a). For some of them, we observed a de-regulated gene expression under non-favorable nitrogen or pH conditions (Niehaus et al. 2017b). For instance, the expression of APF, BEA, FSA, and FUS biosynthetic genes was elevated under usually repressing low nitrogen conditions, and the expression of the BIK and FSR biosynthetic genes was de-regulated under otherwise repressing pH conditions. Besides, some putative silent gene clusters, e.g., those with STC1, NRPS4, NRPS11 , and NRPS20 as key enzyme-encoding genes, were strongly upregulated in the ∆csm1 mutant. The STC1 cluster with the sesquiterpene cyclase gene STC1 and two additional co-regulated genes was analyzed in more detail. Heterologous expression of STC1 in Escherichia coli led to the identification of the product as the volatile bioactive compound (−)-germacrene D (Niehaus et al. 2017b). The function of the remaining genes is currently under investigation.
It is currently not known whether the global TFs can directly bind the promoters of cluster genes, or whether they affect the expression of gene clusters rather indirectly, e.g., by regulating the expression of other TFs and chromatin modifiers.
The role of the global nitrogen regulators AreA and AreB on secondary metabolism in F. fujikuroi
It has been known for a long time that the commercial production of GAs delivers highest yields under low nitrogen conditions (Borrow et al. 1964; Bu'Lock et al. 1974; Jefferys 1970). However, the molecular mechanism of nitrogen regulation of GA biosynthesis remained elucive. The GAs were the first fungal SMs for which an essential role of the major nitrogen regulator, the GATA-type TF AreA, was shown: deletion of areA resulted in almost total loss of GA biosynthesis (Tudzynski et al. 1999) and GA gene expression (Mihlan et al. 2003). These findings were unexpected because AreA was only known as global regulator required for the activation of alternative nitrogen assimilation pathways which are not expressed when preferred nitrogen sources, such as glutamine and ammonium, are available (Caddick et al. 1994; Arst and Cove 1973; Marzluf 1997).
Later, genome-wide microarray analyses under low and high nitrogen conditions revealed that not only the GA biosynthetic genes, but half of the SM gene clusters in F. fujikuroi depend on nitrogen availability (Wiemann et al. 2013). However, not all nitrogen-repressed SM gene clusters are regulated in the same way. For instance, the expression of the BIK cluster genes, though co-regulated with the GA genes, do not depend on the presence of AreA (Wiemann et al. 2009).
Recently, a second GATA-type TF, termed AreB, was shown to be involved in nitrogen-dependent regulation of SM genes in F. fujikuroi (Michielse et al. 2014; Pfannmüller et al. 2017). Whereas the AreB orthologs in A. nidulans and P. chrysogenum were generally regarded as the negative counterparts to AreA, acting as major repressors of AreA-activated nitrogen catabolism genes (Haas et al. 1997; Wong et al. 2008), its function appears to be more complex in F. fujikuroi. In this fungus, AreB can positively or negatively regulate SM gene clusters under both nitogen-limiting and nitrogen-sufficient conditions (Michielse et al. 2014). For instance, AreB is essential for the expression of APF, FUS, and FSA genes under high nitrogen and for GA and FUM genes under low nitrogen (Pfannmüller et al. 2017) (Fig. 3b). In addition to their activity as TFs, both AreA and AreB are likely required for chromatin remodeling processes at the GA gene cluster (Pfannmüller et al. 2017) (Fig. 4a). The most surprising function of AreB in regulating secondary metabolism is the one as repressor of BIK biosynthesis (Fig. 3b). In accordance with elevated BIK transcription, the cultures of the ΔareB mutant were deeply red colored under both low and repressing high nitrogen conditions, and significantly elevated amounts of BIK were produced (Pfannmüller et al. 2017).

Interplay of pathway-specific transcription factors, global regulators, and histone modifying enzymes that regulate gibberellin (a), beauvericin (b), and trichosetin (c) biosynthesis in F. fujikuroi. Black arrows indicate (direct or indirect) transcriptional activation, while red bars show (direct or indirect) gene cluster repression. Dotted arrows represent unclear interactions. The histone landscape around the secondary metabolite gene clusters is shown schematically: the trimethylation of lysine 27 at histone H3 (K27me3) is a repressive mark (facultative heterochromatin), while the acetylation of H3K9 (K9ac) and H3K27 (K27ac) is likely responsible for active transcription at these gene clusters
In addition, AreB acts as repressor of some orphan SM clusters, e.g., the PKS9, PKS20, NRPS4, and NRPS21 clusters, which are never expressed in the WT. Therefore, the ∆areB mutant can be used to activate these unknown gene clusters and to identify their potential products in near future.
Activation of silent gene clusters by genetic manipulation of histone modifying genes
Chromatin-based regulation through histone modifications, such as acetylation and methylation, has now been shown to precisely affect the expression of physically linked SM genes belonging to one gene cluster in several fungi (Brakhage 2013).
Due to the high number of silent gene clusters in F. fujikuroi, several efforts were made to activate them either by inactivation of HDACs or by deletion or silencing of histone methyltransferase genes. Actively trancribed genes typically carry high levels of histone acetylation catalyzed by histone acetyltransferases, while histone deacetylation often results in gene silencing (Bannister and Kouzarides 2011). Deletion of three HDAC-encoding genes, HDA1, HDA2, and HDA4, indicated that Hda1 and Hda2 are involved in regulation of secondary metabolism, while Hda4 regulates growth and development but is dispensable for SM production in F. fujikuroi (Studt et al. 2013). Single deletions of both HDA1 and HDA2 led to up- and downregulation of several SM gene clusters, indicating that HDACs not only are involved in gene silencing but also participate in the activation of gene expression (Table 2). For instance, the expression of GA biosynthetic genes and GA production levels were reduced in both single deletion mutants, resulting in a loss of bakanae symptom development. Furthermore, Hda1 and Hda2 were essential for WT-like expression of BIK, FSR, and FSA biosynthetic genes (Studt et al. 2013). The most striking result was the activation of a silent and orphan gene cluster in the ∆hda1 mutant (Niehaus et al. 2016a). Comparison between the SM profiles of the WT and the ∆hda1 mutant revealed a novel peak in the mutant, which was identified as the cyclooligomer depsipeptide BEA (Figs. 1j and 4b). This mycotoxin is well known from the insect pathogen Beauveria bassiana (Xu et al. 2008) but was also detected in some Fusarium species (Zhang et al. 2013; Moretti et al. 1996). Phylogenetic analysis revealed that NRPS22 from F. fujikuroi grouped together with BeaS, the key enzyme for BEA biosynthesis in B. bassiana. Deletion of the respective NRPS22 (BEA1) gene in the ∆hda1 mutant abolished BEA biosynthesis, confirming that it is the ortholog of beaS. Together with the key enzyme-encoding gene, three additional genes are involved in BEA biosynthesis and regulation, encoding a ketoisovalerate reductase (KivR; Bea2), an ATP-binding cassette (ABC) transporter (Bea3), and a Zn(II)2Cys6 TF (Bea4) (Fig. 2j) (Niehaus et al. 2016a). BEA2 and BEA3 were strongly upregulated in the ∆hda1 mutant together with BEA1, while BEA4 expression was unaffected by the deletion of HDA1.
Recent chromatin immunoprecipitation combined with high-throughput sequencing (ChIP-Seq) showed that many of the silent SM gene clusters in F. fujikuroi, mainly located at subtelomeric regions, are enriched for the silencing methylation mark at lysine 27 of histone 3 (H3K27me3) (Studt et al. 2016a). The gene for the putative H3K27 methyltransferase, KMT6, was recently identified in two fungi, F. graminearum and Epichloë festucae (Connolly et al. 2013; Chujo and Scott 2014). Deletion of the KMT6 homologs in these two ascomycetes resulted in upregulation of several yet uncharacterized SM-related genes demonstrating that H3K27me3 is a promising target for the induction of otherwise silent SM gene clusters. It is worth to mention that several other model fungi, e.g., A. nidulans and the human pathogen A. fumigatus, lack KMT6 and all other components of the known polycomb repressive complex 2 (PRC2) (Connolly et al. 2013).
Surprisingly, the KMT6 gene is an essential gene in F. fujikuroi and could not be deleted. Therefore, a knockdown approach was performed to reduce the H3K27me3 level at a genome-wide level. Several yet unknown and silent SM key enzyme-encoding genes were upregulated in the KMT6 kd mutant (Table 2). Among them was again the BEA (NRPS22) cluster (Studt et al. 2016a) which was shown to be repressed by a whole set of negatively acting regulators: Hda1, Kmt6, the cluster-specific TF Bea4 and the ABC transporter Bea3 (Niehaus et al. 2016a) (Fig. 4b). The latter seems to act as a regulator of gene expression rather than as transporter.
Besides the BEA cluster, several additional silent gene clusters were activated by downregulating KMT6 (Table 2). Among them were the PKS-NRPS1 (Fig. 4c), NRPS4, STC4, STC8, and STC5 clusters. All of them are located in regions of high H3K27me3 levels, either at subtelomeric regions (NRPS22, PKS-NRPS1, NRPS4) or in central regions of the chromosomes (STC4, STC5, STC8) (Studt et al. 2016a). The sesquiterpene cyclase STC5 was analyzed in more detail and shown to be responsible for the production of the sesquiterpene hydrocarbon (1R,4R,5S)-guaia-6,10(14)-diene (Studt et al. 2016a; Burkhardt et al. 2016). The product of a second silent gene cluster, PKS-NRPS1, was identified as trichosetin by an alternative approach as described above: the over-expression of the cluster-specific TF gene (Janevska et al. 2017a) (Fig. 4c).
An important role for the regulation of secondary metabolism was shown for the histone acetyltransferase (HAT) Gcn5, a member of the SAGA complex. This HAT is responsible for acetylation of several histone 3 lysines in F. fujikuroi, i.e., H3K4, H3K9, H3K18, and H3K27 (Rösler et al. 2016b) (Fig. 4a, b). In total, the transcription of 28 out of the 47 bioinformatically identified key genes of putative SM gene clusters was affected by GCN5 deletion (Table 2). The expression of the majority (18 out of 28) of these SM clusters, e.g., those for GA and FSR biosynthesis, was downregulated or abolished in ∆gcn5 compared to the WT, suggesting a mainly activating role of Gcn5 in SM gene regulation. However, some gene clusters, e.g., those for BIK and FSA biosynthesis, as well as some apparently silent gene clusters (NRPS4, PKS-NRPS1, STC5, STC9, DMATS1) were upregulated under at least one tested condition in the mutant (Rösler et al. 2016b).
The methylation marks of H3K4 and H3K36 have been described as hallmarks of euchromatin in budding and fission yeasts as well as higher eukaryotes (Rando and Chang 2009; Wagner and Carpenter 2012). The euchromatic localization of H3K4me2 could be verified by ChIP-Seq (Wiemann et al. 2013), while H3K36me3 was found to cover whole chromosomes in F. fujikuroi (Janevska et al. 2017b). Set2 is the major H3K36-specific methyltransferase, and it has already been studied in Neurospora crassa and F. verticillioides (Adhvaryu et al. 2005; Gu et al. 2017). In addition, only filamentous fungi, but not yeasts, contain a second H3K36-specific methyltransferase gene, ASH1, which was functionally analyzed in F. fujikuroi for the first time (Janevska et al. 2017b). Thus, F. fujikuroi Set2 and Ash1 were shown to deposit their methylation at euchromatic and subtelomeric regions, respectively. Intriguingly, deletion of ASH1 resulted in the loss of subtelomeric regions and of the accessory chromosome XII, likely counteracting H3K27me3 at these regions and mediating DNA repair mechanisms. Although secondary metabolism was de-regulated in Δset2 and Δash1 mutants, the effects are probably indirect because the expression of SM biosynthetic genes did not correlate with H3K36me3 and/or H3K27me3 levels at the analyzed gene clusters (Janevska et al. 2017b).
Furthermore, the loss of H3K4me3 upon CCL1 deletion had an impact on secondary metabolism in F. fujikuroi (Studt et al. 2017). Ccl1 is a critical component for the trimethylation of H3K4 by the complex of proteins associated with Set1 (COMPASS). Also in this case, many of the effects are likely to be indirect, although enhanced levels of H3K4me2 at the BIK cluster correlated with an elevated BIK biosynthesis in Δccl1 (Studt et al. 2017). However, further analyses are required to fully understand the impact of histone modifications on fungal secondary metabolism.
Conclusion
Sequencing of large numbers of fungal genomes revealed the enormous genetic capacity of fungi to produce a battery of low molecular weight natural products. However, most of the SM gene clusters remain silent under standard laboratory conditions and, therefore, their products are still unknown. The increasing interest in identification of novel fungal SMs with potential applications in medicine, agriculture, and the food industry resulted in efforts to activate them by the use of different molecular techniques. This review summarizes the recent work on secondary metabolism of the rice pathogenic fungus F. fujikuroi. Whereas only three gene clusters for GA, BIK, and carotenoid biosynthesis were identified and experimentally characterized before the genome was sequenced in 2013, the products of 20 out of the 47 potential SM gene clusters are now known due to genetic manipulations of key enzyme-encoding genes, pathway-specific and global regulators, and histone-modifying enzymes. By these approaches, we were able to shed more light on the multi-level regulation of secondary metabolism. For example, we gained much more knowledge on the complex regulation of GA biosynthesis, the most prominent SM of F. fujikuroi studied over a long period. At the beginning, only the repression of GA formation by high levels of nitrogen was described. Now, a number of transcription factors and histone modifiers were shown to affect the expression of GA genes (Fig. 4a). Besides the major nitrogen regulators, AreA and AreB, all three components of the F. fujikuroi velvet complex are essential for high gene expression. In addition, global regulators such as Sge1 and the acetylation status of histones were shown significantly to affect GA gene expression (Fig. 4a). One of the most surprising results of these studies was that each single gene cluster is regulated by a specific and unique network of regulators, probably due to their specific biological function.
References
Adhvaryu KK, Morris SA, Strahl BD, Selker EU (2005) Methylation of histone H3 lysine 36 is required for normal development in Neurospora crassa. Eukaryot Cell 4(8):1455–1464. https://doi.org/10.1128/EC.4.8.1455-1464.2005
Arndt B, Janevska S, Schmid R, Hübner F, Tudzynski B, Humpf HU (2017) A fungal N-dimethylallyltryptophan metabolite from Fusarium fujikuroi. Chembiochem 18(10):899–904. https://doi.org/10.1002/cbic.201600691
Arnstein H, Cook A (1947) Production of antibiotics by fungi. Part III. Javanicin. An antibacterial pigment from Fusarium javanicum. J Chem Soc (Resumed):1021–1028
Arst HN, Cove DJ (1973) Nitrogen metabolite repression in Aspergillus nidulans. Mol Gen Genet 126(2):111–141. https://doi.org/10.1007/BF00330988
Avalos J, Prado-Cabrero A, Estrada AF (2012) Neurosporaxanthin production by Neurospora and Fusarium. Microb Carotenoids Fungi: Methods Protoc:263–274, DOI: https://doi.org/10.1007/978-1-61779-918-1_18
Bacon CW, Porter JK, Norred WP, Leslie JF (1996) Production of fusaric acid by Fusarium species. Appl Environ Microbiol 62(11):4039–4043
Balan J, Fuska J, Kuhr I, Kuhrova V (1970) Bikaverin, an antibiotic from Gibberella fujikuroi, effective against Leishmania brasiliensis. Folia Microbiol (Praha) 15(6):479–484. https://doi.org/10.1007/BF02880192
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395. https://doi.org/10.1038/cr.2011.22
Barrero AF, Sánchez JF, Oltra JE, Tamayo N, Cerdá-Olmedo E, Candau R, Avalos J (1991) Fusarin C and 8Z-fusarin C from Gibberella fujikuroi. Phytochemistry 30(7):2259–2226. https://doi.org/10.1016/0031-9422(91)83625-U
Bayram O, Braus GH (2012) Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol Rev 36(1):1–24. https://doi.org/10.1111/j.1574-6976.2011.00285.x
Bayram O, Krappmann S, Ni M, Bok JW, Helmstaedt K, Valerius O, Braus-Stromeyer S, Kwon NJ, Keller NP, JH Y, Braus GH (2008) VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320(5882):1504–1506. https://doi.org/10.1126/science.1155888
Bok JW, Keller NP (2004) LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot Cell 3(2):527–535. https://doi.org/10.1128/EC.3.2.527-535.2004
Bömke C, Tudzynski B (2009) Diversity, regulation, and evolution of the gibberellin biosynthetic pathway in fungi compared to plants and bacteria. Phytochemistry 70(15-16):1876–1893. https://doi.org/10.1016/j.phytochem.2009.05.020
Borrow A, Brown S, Jefferys E, Kessell R, Lloyd EC, Lloyd P, Rothwell A, Rothwell B, Swait J (1964) The kinetics of metabolism of Gibberella fujikuroi in stirred culture. Can J Microbiol 10(3):407–444. https://doi.org/10.1139/m64-054
Brakhage AA (2013) Regulation of fungal secondary metabolism. Nat Rev Microbiol 11(1):21–32. https://doi.org/10.1038/nrmicro2916
Brown DW, Busman M, Proctor RH (2014) Fusarium verticillioides SGE1 is required for full virulence and regulates expression of protein effector and secondary metabolite biosynthetic genes. Mol Plant-Microbe Interact 27(8):809–823. https://doi.org/10.1094/MPMI-09-13-0281-R
Brown DW, Butchko RAE, Busman M, Proctor RH (2012) Identification of gene clusters associated with fusaric acid, fusarin, and perithecial pigment production in Fusarium verticillioides. Fungal Genet Biol 49(7):521–532. https://doi.org/10.1016/j.fgb.2012.05.010
Brown DW, Butchko RAE, Busman M, Proctor RH (2007) The Fusarium verticillioides FUM gene cluster encodes a Zn(II)2Cys6 protein that affects FUM gene expression and fumonisin production. Eukaryot Cell 6(7):1210–1218. https://doi.org/10.1128/EC.00400-06
Brown DW, Lee SH, Kim LH, Ryu JG, Lee S, Seo Y, Kim YH, Busman M, Yun SH, Proctor RH, Lee T (2015) Identification of a 12-gene fusaric acid biosynthetic gene cluster in Fusarium species through comparative and functional genomics. Mol Plant-Microbe Interact 28(3):319–332. https://doi.org/10.1094/MPMI-09-14-0264-R
Bu'Lock J, Detroy R, Hošťálek Z, Munim-Al-Shakarchi A (1974) Regulation of secondary biosynthesis in Gibberella fujikuroi. Trans Br Mycol Soc 62(2):377–389. https://doi.org/10.1016/S0007-1536(74)80046-X
Burkhardt I, Siemon T, Henrot M, Studt L, Rösler S, Christmann M, Tudzynski B, Dickschat JS (2016) Mechanistic characterization of two sesquiterpene cyclases from the plant pathogen Fusarium fujikuroi. Angew Chem 55(30):8748–8751. https://doi.org/10.1002/anie.201603782
Caddick MX, Peters D, Platt A (1994) Nitrogen regulation in fungi. Antonie Van Leeuwenhoek 65(3):169–177. https://doi.org/10.1007/BF00871943
Calvo AM (2008) The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet Biol 45(7):1053–1061. https://doi.org/10.1016/j.fgb.2008.03.014
Chiang Y, Oakley BR, Keller NP, Wang CC (2010) Unraveling polyketide synthesis in members of the genus Aspergillus. Appl Microbiol Biotechnol 86(6):1719–1736. https://doi.org/10.1007/s00253-010-2525-3
Chiara M, Fanelli F, Mule G, Logrieco AF, Pesole G, Leslie JF, Horner DS, Toomajian C (2015) Genome sequencing of multiple isolates highlights subtelomeric genomic diversity within Fusarium fujikuroi. Genome Biol Evol 7(11):3062–3069. https://doi.org/10.1093/gbe/evv198
Chujo T, Scott B (2014) Histone H3K9 and H3K27 methylation regulates fungal alkaloid biosynthesis in a fungal endophyte–plant symbiosis. Mol Microbiol 92(2):413–434. https://doi.org/10.1111/mmi.12567
Connolly LR, Smith KM, Freitag M (2013) The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet 9(10):e1003916. https://doi.org/10.1371/journal.pgen.1003916
Desjardins AE, Plattner RD, Nelson PE (1997) Production of fumonisin B(inf1) and moniliformin by Gibberella fujikuroi from rice from various geographic areas. Appl Environ Microbiol 63(5):1838–1842
Desjardins AE, Proctor RH (2007) Molecular biology of Fusarium mycotoxins. Int J Food Microbiol 119(1-2):47–50. https://doi.org/10.1016/j.ijfoodmicro.2007.07.024
Dhingra S, Andes D, Calvo AM (2012) VeA regulates conidiation, gliotoxin production, and protease activity in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot Cell 11(12):1531–1543. https://doi.org/10.1128/EC.00222-12
Diaz-Sanchez V, Avalos J, Limon MC (2012) Identification and regulation of fusA, the polyketide synthase gene responsible for fusarin production in Fusarium fujikuroi. Appl Environ Microbiol 78(20):7258–7266. https://doi.org/10.1128/AEM.01552-12
Fiyaz RA, Yadav AK, Krishnan SG, Ellur RK, Bashyal BM, Grover N, Bhowmick PK, Nagarajan M, Vinod K, Singh NK (2016) Mapping quantitative trait loci responsible for resistance to bakanae disease in rice. Rice 9(1):45. https://doi.org/10.1186/s12284-016-0117-2
Gaffoor I, Brown DW, Plattner R, Proctor RH, Qi W, Trail F (2005) Functional analysis of the polyketide synthase genes in the filamentous fungus Gibberella zeae (anamorph Fusarium graminearum). Eukaryot Cell 4(11):1926–1933. https://doi.org/10.1128/EC.4.11.1926-1933.2005
Gu Q, Wang Z, Sun X, Ji T, Huang H, Yang Y, Zhang H, Tahir HAS, Wu L, Wu H, Gao X (2017) FvSet2 regulates fungal growth, pathogenicity, and secondary metabolism in Fusarium verticillioides. Fungal Genet Biol 107:24–30. https://doi.org/10.1016/j.fgb.2017.07.007
Haas H, Angermayr K, Zadra I, Stöffler G (1997) Overexpression of nreB, a new GATA factor-encoding gene of Penicillium chrysogenum, leads to repression of the nitrate assimilatory gene cluster. J Biol Chem 272(36):22576–22582. https://doi.org/10.1074/jbc.272.36.22576
Hansen FT, Gardiner DM, Lysoe E, Fuertes PR, Tudzynski B, Wiemann P, Sondergaard TE, Giese H, Brodersen DE, Sorensen JL (2015) An update to polyketide synthase and non-ribosomal synthetase genes and nomenclature in Fusarium. Fungal Genet Biol 75:20–29. https://doi.org/10.1016/j.fgb.2014.12.004
Hoff B, Kamerewerd J, Sigl C, Mitterbauer R, Zadra I, Kurnsteiner H, Kuck U (2010) Two components of a velvet-like complex control hyphal morphogenesis, conidiophore development, and penicillin biosynthesis in Penicillium chrysogenum. Eukaryot Cell 9(8):1236–1250. https://doi.org/10.1128/EC.00077-10
Janevska S, Arndt B, Niehaus EM, Burkhardt I, Rösler SM, Brock NL, Humpf HU, Dickschat JS, Tudzynski (2016) Gibepyrone biosynthesis in the rice pathogen Fusarium fujikuroi is facilitated by a small polyketide synthase gene cluster. J Biol Chem 291(53):27403–27420. https://doi.org/10.1074/jbc.M116.753053
Janevska S, Arndt B, Baumann L, Apken LH, Mauriz Marques LM, Humpf HU, Tudzynski B (2017a) Establishment of the inducible Tet-on system for the activation of the silent trichosetin gene cluster in Fusarium fujikuroi. Toxins (Basel) 9:https://doi.org/10.3390/toxins9040126
Janevska S, Baumann L, Sieber CMK, Münsterkötter M, Ulrich J, Kämper J, Güldener U, Tudzynski B (2017b) Elucidation of the two H3K36me3 histone methyltransferases Set2 and Ash1 in Fusarium fujikuroi unravels their different chromosomal targets and a major impact of Ash1 on genome stability. Genetics. https://doi.org/10.1534/genetics.117.1119
Jefferys E (1970) The gibberellin fermentation. Adv Appl Microbiol 13:283–316. https://doi.org/10.1016/S0065-2164(08)70407-6
Jin JM, Lee S, Lee J, Baek SR, Kim JC, Yun SH, Park SY, Kang S, Lee YW (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol Microbiol 76(2):456–466. https://doi.org/10.1111/j.1365-2958.2010.07109.x
Kakule TB, Sardar D, Lin Z, Schmidt EW (2013) Two related pyrrolidinedione synthetase loci in Fusarium heterosporum ATCC 74349 produce divergent metabolites. ACS Chem Biol 8(7):1549–1557. https://doi.org/10.1021/cb400159f
Karimi-Aghcheh R, Bok JW, Phatale PA, Smith KM, Baker SE, Lichius A, Omann M, Zeilinger S, Seiboth B, Rhee C, Keller NP, Freitag M, Kubicek CP (2013) Functional analyses of Trichoderma reesei LAE1 reveal conserved and contrasting roles of this regulator. G3 (Bethesda) 3(2):369–378. https://doi.org/10.1534/g3.112.005140
Keller NP, Hohn TM (1997) Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol 21(1):17–29. https://doi.org/10.1006/fgbi.1997.0970
Kleigrewe K, Niehaus EM, Wiemann P, Tudzynski B, Humpf HU (2012) New approach via gene knockout and single-step chemical reaction for the synthesis of isotopically labeled fusarin C as an internal standard for the analysis of this Fusarium mycotoxin in food and feed samples. J Agric Food Chem 60(34):8350–8355. https://doi.org/10.1021/jf302534x
Krasnoff SB, Sommers CH, Moon Y, Donzelli BGG, Vandenberg JD, Churchill ACL, Gibson DM (2006) Production of mutagenic metabolites by Metarhizium anisopliae. J Agric Food Chem 54(19):7083–7088. https://doi.org/10.1021/jf061405r
Kvas M, Marasas WFO, Wingfield BD, Wingfield MJ, Steenkamp ET (2009) Diversity and evolution of Fusarium species in the Gibberella fujikuroi complex. Fungal Divers 34:1–21
Lee MK, Kwon NJ, Choi JM, Lee IS, Jung S, JH Y (2014) NsdD is a key repressor of asexual development in Aspergillus nidulans. Genetics 197(1):159–173. https://doi.org/10.1534/genetics.114.161430
Lind AL, Wisecaver JH, Smith TD, Feng X, Calvo AM, Rokas A (2015) Examining the evolution of the regulatory circuit controlling secondary metabolism and development in the fungal genus Aspergillus. PLoS Genet 11(3):e1005096. https://doi.org/10.1371/journal.pgen.1005096
Linnemannstöns P, Prado MM, Fernandez-Martin R, Tudzynski B, Avalos J (2002a) A carotenoid biosynthesis gene cluster in Fusarium fujikuroi: the genes carB and carRA. Mol Gen Genomics 267:593–602
Linnemannstöns P, Schulte J, Del Mar Prado M, Proctor RH, Avalos J, Tudzynski B (2002b) The polyketide synthase gene pks4 from Gibberella fujikuroi encodes a key enzyme in the biosynthesis of the red pigment bikaverin. Fungal Genet Biol 37(2):134–148. https://doi.org/10.1016/S1087-1845(02)00501-7
Ma LJ, van der Does HC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M, Henrissat B, Houterman PM, Kang S, Shim WB, Woloshuk C, Xie X, Xu JR, Antoniw J, Baker SE, Bluhm BH, Breakspear A, Brown DW, Butchko RA, Chapman S, Coulson R, Coutinho PM, Danchin EG, Diener A, Gale LR, Gardiner DM, Goff S, Hammond-Kosack KE, Hilburn K, Hua-Van A, Jonkers W, Kazan K, Kodira CD, Koehrsen M, Kumar L, Lee YH, Li L, Manners JM, Miranda-Saavedra D, Mukherjee M, Park G, Park J, Park SY, Proctor RH, Regev A, Ruiz-Roldan MC, Sain D, Sakthikumar S, Sykes S, Schwartz DC, Turgeon BG, Wapinski I, Yoder O, Young S, Zeng Q, Zhou S, Galagan J, Cuomo CA, Kistler HC, Rep M (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464(7287):367–373. https://doi.org/10.1038/nature08850
Marzluf GA (1997) Genetic regulation of nitrogen metabolism in the fungi. Microbiol Mol Biol Rev 61(1):17–32
Michielse C, Pfannmüller A, Macios M, Rengers P, Dzikowska A, Tudzynski B (2014) The interplay between the GATA transcription factors AreA, the global nitrogen regulator and AreB in Fusarium fujikuroi. Mol Microbiol 91(3):472–493. https://doi.org/10.1111/mmi.12472
Michielse CB, Rep M (2009) Pathogen profile update: Fusarium oxysporum. Mol Plant Pathol 10(3):311–324. https://doi.org/10.1111/j.1364-3703.2009.00538.x
Michielse CB, Studt L, Janevska S, Sieber CM, Arndt B, Espino JJ, Humpf HU, Güldener U, Tudzynski B (2015) The global regulator FfSge1 is required for expression of secondary metabolite gene clusters but not for pathogenicity in Fusarium fujikuroi. Environ Microbiol 17(8):2690–2708. https://doi.org/10.1111/1462-2920.12592
Mihlan M, Homann V, Liu TD, Tudzynski B (2003) AREA directly mediates nitrogen regulation of gibberellin biosynthesis in Gibberella fujikuroi, but its activity is not affected by NMR. Mol Microbiol 47(4):975–991. https://doi.org/10.1046/j.1365-2958.2003.03326.x
Moretti A, Logrieco A, Bottalico A, Ritieni A, Fogliano V, Randazzo G (1996) Diversity in beauvericin and fusaproliferin production by different populations of Gibberella fujikuroi (Fusarium section Liseola). Sydowia 48:44–56
Niehaus EM, Studt L, von Bargen KW, Kummer W, Humpf HU, Reuter G, Tudzynski B (2016a) Sound of silence: the beauvericin cluster in Fusarium fujikuroi is controlled by cluster-specific and global regulators mediated by H3K27 modification. Environ Microbiol 18(11):4282–4302. https://doi.org/10.1111/1462-2920.13576
Niehaus EM, Janevska S, von Bargen KW, Sieber CMK, Harrer H, Humpf HU, Tudzynski B (2014a) Apicidin F: characterization and genetic manipulation of a new secondary metabolite gene cluster in the rice pathogen Fusarium fujikuroi. PLoS One 9(7):e103336. https://doi.org/10.1371/journal.pone.0103336
Niehaus EM, Kleigrewe K, Wiemann P, Studt L, Sieber CMK, Connolly LR, Freitag M, Güldener U, Tudzynski B, Humpf HU (2013) Genetic manipulation of the Fusarium fujikuroi fusarin gene cluster yields insight into the complex regulation and fusarin biosynthetic pathway. Chem Biol 20(8):1055–1066. https://doi.org/10.1016/j.chembiol.2013.07.004
Niehaus EM, Munsterkotter M, Proctor RH, Brown DW, Sharon A, Idan Y, Oren-Young L, Sieber CM, Novak O, Pencik A, Tarkowská D, Hromadová K, Freeman S, Maymon M, Elazar M, Youssef SA, El-Shabrawy ESM, Shalaby AA, Houterman P, Brock NL, Burkhardt I, Tsavkelova EM, Dickschat JS, Galuszka P, Güldener U, Tudzynski B (2016b) Comparative “omics” of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biol Evol 8(11):3574–3599. https://doi.org/10.1093/gbe/evw259
Niehaus EM, Kim HK, Münsterkötter M, Janevska S, Arndt B, Kalinina SA, Houterman PM, Ahn IP, Alberti I, Tonti S, Kim DW, Sieber CMK, Humpf HU, Yun SH, Güldener U, Tudzynski B (2017a) Comparative genomics of geographically distant Fusarium fujikuroi isolates revealed two distinct pathotypes correlating with secondary metabolite profiles. PLoS Pathog 13(10):e1006670. https://doi.org/10.1371/journal.ppat.1006670
Niehaus EM, Schumacher J, Burkhardt I, Rabe P, Münsterkötter M, Güldener U, Sieber CMK, Dickschat JS, Tudzynski B (2017b) The GATA-type transcription factor Csm1 regulates conidiation and secondary metabolism in Fusarium fujikuroi. Front Microbiol 8:1175. https://doi.org/10.3389/fmicb.2017.01175
Niehaus EM, Rindermann L, Janevska S, Münsterkötter M, Güldener U, Tudzynski B (2017c) Analysis of the global regulator Lae1 uncovers a connection between Lae1 and the histone acetyltransferase HAT1 in Fusarium fujikuroi. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-017-8590-0
Niehaus EM, von Bargen KW, Espino JJ, Pfannmüller A, Humpf HU, Tudzynski B (2014b) Characterization of the fusaric acid gene cluster in Fusarium fujikuroi. Appl Microbiol Biotechnol 98(4):1749–1762. https://doi.org/10.1007/s00253-013-5453-1
Oide S, Berthiller F, Wiesenberger G, Adam G, Turgeon BG (2015) Individual and combined roles of malonichrome, ferricrocin, and TAFC siderophores in Fusarium graminearum pathogenic and sexual development. Frontiers in Microbiology. 5:759
Ou SH (1985) Rice diseases. Published by Slough, Commonwealth Mycological Institute
Perrin RM, Fedorova ND, Bok JW, Cramer RA Jr, Wortman JR, Kim HS, Nierman WC, Keller NP (2007) Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog 3(4):e50. https://doi.org/10.1371/journal.ppat.0030050
Pfannmüller A, Leufken J, Studt L, Michielse CB, Sieber CMK, Güldener U, Hawat S, Hippler M, Fufezan C, Tudzynski B (2017) Comparative transcriptome and proteome analysis reveals a global impact of the nitrogen regulators AreA and AreB on secondary metabolism in Fusarium fujikuroi. PLoS One 12(4):e0176194. https://doi.org/10.1371/journal.pone.0176194
Proctor RH, Brown DW, Plattner RD, Desjardins AE (2003) Co-expression of 15 contiguous genes delineates a fumonisin biosynthetic gene cluster in Gibberella moniliformis. Fungal Genet Biol 38(2):237–249. https://doi.org/10.1016/S1087-1845(02)00525-X
Proctor RH, Butchko RAE, Brown DW, Moretti A (2007) Functional characterization, sequence comparisons and distribution of a polyketide synthase gene required for perithecial pigmentation in some Fusarium species. Food Addit Contam 24(10):1076–1087. https://doi.org/10.1080/02652030701546495
Proctor RH, Desjardins AE, Plattner RD, Hohn TM (1999) A polyketide synthase gene required for biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi mating population A. Fungal Genet Biol 27(1):100–112. https://doi.org/10.1006/fgbi.1999.1141
Rando OJ, Chang HY (2009) Genome-wide views of chromatin structure. Annu Rev Biochem 78(1):245–271. https://doi.org/10.1146/annurev.biochem.78.071107.134639
Rees DO, Bushby N, Cox RJ, Harding JR, Simpson TJ, Willis CL (2007) Synthesis of [1, 2-13C2, 15N]-L-homoserine and its incorporation by the PKS-NRPS system of Fusarium moniliforme into the mycotoxin fusarin C. Chembiochem 8(1):46–50. https://doi.org/10.1002/cbic.200600404
Rösler SM, Sieber CMK, Humpf HU, Tudzynski B (2016a) Interplay between pathway-specific and global regulation of the fumonisin gene cluster in the rice pathogen Fusarium fujikuroi. Appl Microbiol Biotechnol 100(13):5869–5882. https://doi.org/10.1007/s00253-016-7426-7
Rösler SM, Kramer K, Finkemeier I, Humpf HU, Tudzynski B (2016b) The SAGA complex in the rice pathogen Fusarium fujikuroi: structure and functional characterization. Mol Microbiol 102(6):951–974. https://doi.org/10.1111/mmi.13528
Schumacher J, Simon A, Cohrs KC, Viaud M, Tudzynski P (2014) The transcription factor BcLTF1 regulates virulence and light responses in the necrotrophic plant pathogen Botrytis cinerea. PLoS Genet 10(1):e1004040. https://doi.org/10.1371/journal.pgen.1004040
Song Z, Cox RJ, Lazarus CM, Simpson TJTJ (2004) Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum. Chembiochem 5(9):1196–1203. https://doi.org/10.1002/cbic.200400138
Sponsel VM, Hedden P (2010) Gibberellin biosynthesis and inactivation. In: Plant hormones. (Anonymous ), pp. 63–94. Springer Netherlands, DOI: https://doi.org/10.1007/978-1-4020-2686-7_4
Studt L, Janevska S, Arndt B, Boedi S, Sulyok M, Humpf HU, Tudzynski B, Strauss J (2017) Lack of the COMPASS component Ccl1 reduces H3K4 trimethylation levels and affects transcription of secondary metabolite genes in two plant-pathogenic Fusarium species. Front Microbiol 7:2144. https://doi.org/10.3389/fmicb.2016.02144
Studt L, Rösler SM, Burkhardt I, Arndt B, Freitag M, Humpf HU, Dickschat JS, Tudzynski B (2016a) Knock-down of the methyltransferase Kmt6 relieves H3K27me3 and results in induction of cryptic and otherwise silent secondary metabolite gene clusters in Fusarium fujikuroi. Environ Microbiol 18(11):4037–4054. https://doi.org/10.1111/1462-2920.13427
Studt L, Janevska S, Niehaus EM, Burkhardt I, Arndt B, Sieber CMK, Humpf HU, Dickschat JS, Tudzynski B (2016b) Two separate key enzymes and two pathway-specific transcription factors are involved in fusaric acid biosynthesis in Fusarium fujikuroi. Environ Microbiol 18(3):936–956. https://doi.org/10.1111/1462-2920.13150
Studt L, Schmidt FJ, Jahn L, Sieber CM, Connolly LR, Niehaus EM, Freitag M, Humpf HU, Tudzynski B (2013) Two histone deacetylases, FfHda1 and FfHda2, are important for Fusarium fujikuroi secondary metabolism and virulence. Appl Environ Microbiol 79(24):7719–7734. https://doi.org/10.1128/AEM.01557-13
Studt L, Wiemann P, Kleigrewe K, Humpf HU, Tudzynski B (2012) Biosynthesis of fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl Environ Microbiol 78(12):4468–4480. https://doi.org/10.1128/AEM.00823-12
Tobiasen C, Aahman J, Ravnholt KS, Bjerrum MJ, Grell MN, Giese H (2007). Nonribosomal peptide synthetase (NPS) genes in Fusarium graminearum, F. culmorum and F. pseudograminearium and identification of NPS2 as the producer of ferricrocin. Curr Genet 51:43–58
Tudzynski B, Homann V, Feng B, Marzluf G (1999) Isolation, characterization and disruption of the areA nitrogen regulatory gene of Gibberella fujikuroi. Mol Gen Genet 261(1):106–114
Tudzynski B, Hölter K (1998) Gibberellin biosynthetic pathway in Gibberella fujikuroi: evidence for a gene cluster. Fung Genet Biol 25:157–170
Tudzynski B, Studt L, Cecilia M (2016) Gibberellins in fungi, bacteria and lower plants: biosynthesis, function and evolution. Annual Plant Reviews 49:121–152
Varga J, Kocsubé S, Tóth B, Mesterházy A (2005) Nonribosomal peptide synthetase genes in the genome of Fusarium graminearum, causative agent of wheat head blight. Acta Biologica Hungarica 56:375–388
von Bargen KW, Niehaus EM, Krug I, Bergander K, Wurthwein EU, Tudzynski B, Humpf HU (2015) Isolation and structure elucidation of fujikurins A-D: products of the PKS19 gene cluster in Fusarium fujikuroi. J Nat Prod 78(8):1809–1815. https://doi.org/10.1021/np5008137
Wagner EJ, Carpenter PB (2012) Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13(2):115–126. https://doi.org/10.1038/nrm3274
Wiemann P, Brown DW, Kleigrewe K, Bok JW, Keller NP, Humpf HU, Tudzynski B (2010) FfVel1 and FfLae1, components of a velvet-like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol Microbiol 77(4):972–994. https://doi.org/10.1111/j.1365-2958.2010.07263.x
Wiemann P, Sieber CMK, von Bargen KW, Studt L, Niehaus EM, Espino JJ, Huss K, Michielse CB, Albermann S, Wagner D, Bergner SV, Connolly LR, Fischer A, Reuter G, Kleigrewe K, Bald T, Wingfield BD, Ophir R, Freeman S, Hippler M, Smith KM, Brown DW, Proctor RH, Münsterkötter M, Freitag M, Humpf HU, Güldener U, Tudzynski B (2013) Deciphering the cryptic genome: genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog 9(6):e1003475. https://doi.org/10.1371/journal.ppat.1003475
Wiemann P, Willmann A, Straeten M, Kleigrewe K, Beyer M, Humpf HU, Tudzynski B (2009) Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Mol Microbiol 72(4):931–946. https://doi.org/10.1111/j.1365-2958.2009.06695.x
Wingfield BD, Steenkamp ET, Santana QC, Coetzee M, Bam S, Barnes I, Beukes CW, Yin Chan W, De Vos L, Fourie G (2012) First fungal genome sequence from Africa: a preliminary analysis. S Afr J Sci 108:01–09
Wong KH, Hynes MJ, Davis MA (2008) Recent advances in nitrogen regulation: a comparison between Saccharomyces cerevisiae and filamentous fungi. Eukaryot Cell 7(6):917–925. https://doi.org/10.1128/EC.00076-08
Xu Y, Orozco R, Wijeratne EK, Gunatilaka AL, Stock SP, Molnár I (2008) Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem Biol 15(9):898–907. https://doi.org/10.1016/j.chembiol.2008.07.011
Zhang T, Zhuo Y, Jia X, Liu J, Gao H, Song F, Liu M, Zhang L (2013) Cloning and characterization of the gene cluster required for beauvericin biosynthesis in Fusarium proliferatum. Sci China Life Sci 56(7):628–637. https://doi.org/10.1007/s11427-013-4505-1
Acknowledgements
We thank Brian Williamson for the critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Janevska, S., Tudzynski, B. Secondary metabolism in Fusarium fujikuroi: strategies to unravel the function of biosynthetic pathways. Appl Microbiol Biotechnol 102, 615–630 (2018). https://doi.org/10.1007/s00253-017-8679-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-017-8679-5