
Abstract
Streptococcus pneumoniae is the main cause of pneumonia, meningitis, and other conditions that kill thousands of children every year worldwide. The replacement of pneumococcal serotypes among the vaccinated population has evidenced the need for new vaccines with broader coverage and driven the research for protein-based vaccines. Pneumococcal surface protein A (PspA) protects S. pneumoniae from the bactericidal effect of human apolactoferrin and prevents complement deposition. Several studies indicate that PspA is a very promising target for novel vaccine formulations. Here we describe a production and purification process for an untagged recombinant fragment of PspA from clade 4 (PspA4Pro), which has been shown to be cross-reactive with several PspA variants. PspA4Pro was obtained using lactose as inducer in Phytone auto-induction batch or glycerol limited fed-batch in 5-L bioreactor. The purification process includes two novel steps: (i) clarification using a cationic detergent to precipitate contaminant proteins, nucleic acids, and other negatively charged molecules as the lipopolysaccharide, which is the major endotoxin; and (ii) cryoprecipitation that eliminates aggregates and contaminants, which precipitate at −20 °C and pH 4.0, leaving PspA4Pro in the supernatant. The final process consisted of cell rupture in a continuous high-pressure homogenizer, clarification, anion exchange chromatography, cryoprecipitation, and cation exchange chromatography. This process avoided costly tag removal steps and recovered 35.3 ± 2.5% of PspA4Pro with 97.8 ± 0.36% purity and reduced endotoxin concentration by >99.9%. Circular dichroism and lactoferrin binding assay showed that PspA4Pro secondary structure and biological activity were preserved after purification and remained stable in a wide range of temperatures and pH values.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Streptococcus pneumoniae causes diseases as pneumonia, meningitis, and sepsis, representing a major cause of mortality worldwide, mainly among young children and elderly people (Walker et al. 2013). Conjugated pneumococcal vaccines based on capsular polysaccharides (PS) effectively prevent invasive diseases, but only of the serotypes included in formulations. As a consequence, the vaccine serotypes are being replaced by non-vaccine serotypes, which have emerged as a new challenge (Pichon et al. 2013), because the need for multiple PS in formulations increases the costs associated with the development of wide-spectrum vaccines and hinders their use in low- and middle-income countries.
The use of protein antigens shared by different S. pneumoniae serotypes should allow for the elaboration of serotype-independent pneumococcal vaccines. Among candidates, pneumococcal surface protein A (PspA) represents one of the most promising. Every pneumococcal strain evaluated to date expresses this protein attached to the cell wall and exposed outside the polysaccharide layer. PspA inhibits complement deposition on the pneumococcal surface (Ren et al. 2004) and binds to apolactoferrin, preventing its bactericide action in mucosal areas (Mirza et al. 2004). Immunization with PspA protected mice in challenge experiments (Briles et al. 2000; Briles et al. 2003; Darrieux et al. 2008) and sera from human volunteers immunized with PspA protected mice in passive immunization assays (Briles et al. 2000). Moreover, pre-existing serum antibodies against PspA, but not PS, protected humans against carriage after intranasal inoculation with S. pneumoniae (McCool et al. 2002).
The PspA structure comprises four distinct domains: a coiled-coil α-helix-rich N-terminal domain that is exposed outside the polysaccharide layer, a proline-rich domain, a choline-binding domain, and a short hydrophobic tail at the C-terminal domain (Jedrzejas et al. 2001). PspA variants are divided into three families and six clades depending on the identity of amino acid sequences of the α-helix region (Hollingshead et al. 2000). Several studies have shown that more than 90% of pneumococcal strains express PspA belonging to families 1 or 2 (Coral et al. 2001; Croney et al. 2012; Qian et al. 2012). Some N-terminal fragments of PspA elicit a broadly protective cross-response against several clades (Darrieux et al. 2008; Oliveira et al. 2010; Moreno et al. 2010; Goulart et al. 2011).
In large-scale recombinant protein production, downstream processes account for up to 80% of total production costs (Banki et al. 2005). These costs, while potentially acceptable for high-end products, pose an obstacle to the large-scale production of vaccines that should benefit entire populations in countries with different investment potentials. Therefore, the development of simple and reliable purification processes for recombinant proteins represents an important goal in vaccine production.
Tags are often added to recombinant proteins to facilitate the purification process, improve yield, increase solubility, and aid refolding. However, tags may also have detrimental effects such as changing protein conformation, inhibiting enzyme activity, altering biological activity, and toxicity (Arnau et al. 2006). Even commonly used small tags, like histidine tags, may interfere with protein properties (Wu and Filutowicz 1999). Because of these negative effects, ideally, tags should be cleaved off and an additional purification step added to eliminate leftover-tagged proteins, cleaving enzymes, and cleaved tags. Tag removal poses some technical difficulties: the cleavage may not be complete or the proteases may cleave the target protein at secondary sites; the separation often has low yield and depends on expensive chromatography resins due to the similarity between hydrolyzed and tagged molecules (Arnau et al. 2006). Furthermore, the affinity step alone does not always suffice, and further purification steps are required for a product with the desired purity (Hou et al. 2004). An alternative and novel strategy was developed using self-cleaving tags, thus eliminating the need for proteases (Lu et al. 2011; Shi et al. 2014). However, cleavage may be incomplete and further purification steps must be included to separate the target protein from the cleavage products.
At industrial scale, on the other hand, a tag is adopted only if it presents clear advantages in production or to the therapeutic, because of extra time and cost associated with tag removal and the subsequent purification steps required when a tag system is used (Bell et al. 2013). Furthermore, few studies describe the production of untagged recombinant proteins (Hou et al. 2004; Wear et al. 2005; Ludwig et al. 2010).
In the present work, we describe a fully scalable process for the production and purification of a recombinant fragment of PspA from clade 4 (PspA4Pro), which contains the N-terminal α-helix domain and the first block of the proline-rich domain and cross-reacts with PspA variants from clades 1 to 5 (Moreno et al. 2010). This fragment, cloned without tags, was produced in the intracellular soluble fraction of Escherichia coli. The purification strategy was designed to reach at least the purity recommended by the World Health Organization (WHO 2009) for carrier proteins used in pneumococcal conjugated vaccines with low endotoxin content.
Materials and methods
Materials
The shovel mixer X-520 was supplied by Ingenieurbüro CAT M. Zipperer GmbH (Staufen im Breisgau, Germany). The cell disruption system was composed of a high-pressure continuous homogenizer from APV GAULIN (Crawley, UK), a tube-and-shell heat exchanger from Exergy (NY, USA), a jacketed reservoir (ArtePeças, São Paulo, Brazil), and a chiller from PolyScience (Nilles, USA). The centrifuge Avanti J-251 was from Beckman Coulter (Brea, USA). All preparative chromatographic media and preparative chromatographic system (Äkta Avant 150) were from GE Lifescience (USA). The densitometer GS-800 was from BioRad (Hercules, USA). A size exclusion high-performance chromatography (HPSEC) column TSK-GEL G2000SWXL (TOSOH BIOSCIENCE, Minato, Japan) connected to a high-performance liquid chromatography (HPLC) system SCL-10AVP (SHIMADZU, Kyoto, Japan) was used for lactoferrin binding and molecular mass assays. Circular dichroism spectra were obtained in a spectropolarimeter J-810 (Japan Spectroscopic, Tokyo, Japan) equipped with a Peltier unit for temperature control. Human milk lactoferrin, phenylmethilsulfonyl fluoride (PMSF), Triton X-100, cationic detergent cetyltrimethylammonium bromide (CTAB), globular protein molecular mass standards, and dextran linear polymer standards were purchased from Sigma-Aldrich (St. Louis, USA). Phytone peptone and yeast extract were provided by BD/Difco (Sparks, USA). All other reagents were of analytical grade, and water for injection was used in the process.
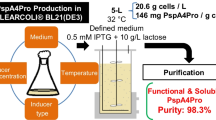
PspA4Pro production
The N-terminal domain plus the first proline block of PspA4Pro gene (GenBank EF649969.1) was inserted in the plasmid pET37b+ (Novagen) and expressed in E. coli BL21(DE3) (Invitrogen, Carlsbad, CA, USA). A 5-L bioreactor described by Horta et al. (2011) was monitored and controlled by the software SuperSys_HCDC (Horta et al. 2014). The pH was controlled at 6.7 by addition of NH4OH (30%). Temperature was set at 31 °C, and dissolved oxygen concentration was kept no lower than 30% by a controller, which automatically changed the stirrer speed between 200 and 900 rpm and added pure oxygen to the air stream supplied to the bioreactor. The total gas flow rate was maintained at 4 L/min. Two strategies were applied for cultivation: a simple batch with an auto-induction medium (see medium composition in Campani et al. 2016) and a fed-batch with chemically defined medium using glycerol as sole carbon source and lactose as inducer, according to Horta et al. (2012).
Cell concentration was measured by culture broth optical density (λ = 600 nm) and dry cell weight (g/L). Acetate and carbon source concentrations were analyzed by HPLC using an Aminex HPX-87H column (Bio-Rad) and 5 mM sulfuric acid as the mobile phase (0.6 mL/min) at 60 °C, with a refraction index detector (Waters 410) for sugars and glycerol, and UV-detector (Waters 486) at 210 nm for organic acids. For plasmid stability measurement, diluted samples of culture broth were plated on LB agar and incubated for 24 h at 37 °C. Then, at least 50 colonies from each sample were replicated in LB agar with and without kanamycin. After incubation at the same conditions, plasmid stability was calculated as the percentage of antibiotic-resistant colonies in relation to the number of colonies formed in plates without antibiotic.
PspA4Pro purification
The protein was purified with the following steps: cell separation, cell rupture, clarification, anion exchange chromatography, cryoprecipitation at pH 4.0, and cation exchange chromatography. Three clarification procedures were evaluated: CTAB or acidic pH precipitation and centrifugation. Mix mode chromatography was evaluated as an alternative to the two ion exchange chromatographies. After the purification process was established, it was repeated once with the biomass produced with auto-induction medium, and a second time with the biomass from fed-batch cultivation with defined medium and induction with lactose.
Cell separation
Biomass was harvested from culture broth by centrifugation, at 4 °C and 6693×g, for 30 min. The cell pellet was then transferred to plastic bags and stored at −80 °C until use.
Cell rupture
For cell lysis, 100 g of frozen wet cell pellet was suspended in 1 L of lysis buffer: 10 mM sodium phosphate buffer pH 6.5 with 2.5 mM EDTA, 0.1% (m/v) Triton X-100, and 1 mM PMSF, using the shovel mixer at 4 °C and 11,000 rpm until no more clumps were observed. The cell suspension was then recirculated through the high-pressure continuous homogenizer system. Chiller temperature was adjusted to 4 °C and cell rupture was performed circulating the suspension into the close loop for 8 min at 500 bar and 1 L/min. Samples were collected from the homogenizer reservoir every 2 min to analyze the evolution of cell rupture by optical density (OD) at 600 nm and protein release in the medium (absorbance at 280 nm) during the process. The recovered fraction was named Homogenate.
Establishing clarification conditions
Three different pH values and the detergent CTAB were evaluated to improve debris removal efficiency: pH 4.0, 5.5, and 6.5, and 0.1% (m/v) CTAB at pH 6.5. Samples (10 mL) from the Homogenate of the first purification process were adjusted to the desired pH or mixed with CTAB for 60 min at room temperature. These samples were centrifuged at 17,696×g and 4 °C for 90 min, and PspA4Pro purity and recovery were analyzed in the supernatant. The best condition for clarification was then applied to the entire Homogenate volume and to the two following purifications. The Homogenate was centrifuged at 17,696×g and 4 °C for 90 min. The supernatant containing PspA4Pro was called Clarified fraction.
Anion exchange chromatography
Anion exchange chromatography was carried out using 200 mL Q-Sepharose FF resin packed in a XK 50/30 column. The volumetric flow was 2.50 mL/(min cm2) (50 mL/min). The column was equilibrated with five column volumes (CV) of 10 mM sodium phosphate buffer, pH 6.5, and conductivity 1.2 mS/cm. The elution was performed with a discontinuous gradient of NaCl (150, 300, and 1000 mM) in 10 mM phosphate buffer pH 6.5, using 5CV for each step.
Cryoprecipitation
The pH of PspA4Pro fraction eluted from Q-Sepharose was decreased to 4.0 using glacial acetic acid. After pH adjustment, the sample was frozen (−20 °C) for 24 h. After this period, it was thawed and centrifuged (17,696×g, 60 min, 4 °C). The supernatant was recovered and called Cryo-pH 4.0.
Cation exchange chromatography
For cation exchange chromatography, 75 mL of SP-Sepharose FF resin was packed in an XK 26/20 column. The volumetric flow was 2.3 mL/(min cm2) (12 mL/min). The column was equilibrated with 5 CV of 25 mM sodium acetate buffer pH 4.0. The elution was performed by a discontinuous gradient of NaCl (500, 800, and 1000 mM) in 25 mM acetate buffer pH 4.0, using 5CV for each step.
Mix mode chromatography
For the mix mode chromatography, 25 mL of CAPTO-MMC resin was packed in a XK 26/20 column. The flow was 0.75 mL/(min cm2), i.e., 4 mL/min. This chromatography was used at two time points in the process: after clarification and after cryoprecipitation. In the first experiment, 150 mL of Clarified fraction was applied to the resin previously equilibrated with 5 CV of 10 mM sodium phosphate buffer, pH 6.5, and conductivity 1.5 mS/cm. A discontinuous gradient of l-arginine (100, 200, 300, 500, and 1000 mM) in 10 mM phosphate buffer pH 6.5 was performed using 5CV for each elution step. In the second experiment, 450 mL of Cryo-pH 4.0 fraction was applied to the resin previously equilibrated with 5 CV of 25 mM acetate buffer with 300 mM NaCl, pH 4.0, and conductivity 40 mS/cm. The elution was performed by a discontinuous gradient of l-arginine (500, 800, and 1000 mM) in 25 mM acetate buffer pH 4.0, using 5CV for each step.
Protein quantification and PspA4Pro purity and recovery determination
Protein concentration was determined by the Lowry methodology (Lowry et al. 1951). PspA4Pro purity was determined by densitometry of the bands in 12% SDS-PAGE with 2-mercaptoethanol (Laemmli 1970) and calculated as the percentage of PspA4Pro band in relation to the sum of all other bands in the lane. Lipopolysaccharide (LPS) was measured as endotoxin unities by the Limulus amebocyte lysate test (Bang 1956).
The purification factor (PF) and protein recovery (Rc) were calculated according to Eqs. (1) to (3) and the percentage of endotoxin reduction (Er) according to Eq. (4).
where PspA4Pro (%) is the purity determined by densitometry of PspA4Pro band in the Homogenate fraction (h) and in the step (n).
where PspA4Pro(g) is the PspA4Pro total amount and Prot(g) is the total protein amount measured according to Lowry.
where PspA4Pro(g)h is the total mass of PspA4Pro in the Homogenate fraction and PspA4Pro(g)n is the total mass of PspA4Pro in the step.
where E(EU/mL) is the endotoxin concentration and V(mL) is the volume in the Homogenate fraction (h) and in the step (n).
Binding of PspA4Pro to human lactoferrin
The binding ability of PspA4Pro to human lactoferrin was analyzed to verify if the protein preserved its biological function after purification. In this assay, lactoferrin was added to the purified PspA4Pro in three different proportions: 2 mg PspA4Pro plus 0.5 mg lactoferrin, 1 mg PspA4Pro plus 1 mg lactoferrin, and 1 mg PspA4Pro plus 2 mg lactoferrin. The mixtures were incubated at 37 °C for 1 h and applied to the HPSEC. The mobile phase was 300 mM sodium phosphate buffer pH 7.0 with 500 mM NaCl, and the flow rate was 0.6 mL/min.
Determination of PspA4Pro molecular mass
To evaluate the molecular mass of PspA4Pro, two standard curves were prepared in HPSEC. The first curve included the globular proteins β-amylase (220 kDa), albumin (66 kDa), carbonic anhydrase (30 kDa), cytochrome c (12 kDa), and aprotinin (6 kDa). The second curve included linear dextran polymers with molecular masses of 6, 10, 70, and 229 kDa. The molecular mass of PspA4Pro was calculated using the linear regression equation of the molecular mass logarithm versus the retention time of both standard curves.
Secondary structure and stability of purified PspA4Pro
Secondary structure and stability of PspA4Pro at different temperatures and pH values and in the presence of CTAB were verified by circular dichroism (CD). Measurements were obtained from 185 to 260 nm, and final CD spectra resulted from the mean of five measurements. PspA4Pro samples (0.1 mg/mL) were prepared in 10 mM sodium phosphate buffer pH 7.0 as follows: with buffer only, with 0.15% CTAB, and with buffer at pH 3.5 to pH 9.0. The sample in buffer only was heated (1 °C/min) from 15 to 95 °C and cooled back to 15 °C (1 °C/min). Deconvolution was calculated based on the Dichroweb online database (Whitmore and Wallace 2004) with Contin and CDSSTR algorithms (Sreerama and Woody 2000).
Results
PspA4Pro production
The auto-induction animal component-free medium employed in the simple batch cultivation has three carbon sources: glucose, consumed first; glycerol, the main carbon source during the induction phase; and lactose, the inducer of recombinant protein synthesis. Glucose exerts a catabolic repression effect on lac promoter and inhibits PspA4Pro synthesis. Glucose was exhausted in the medium within 8 h of cultivation, and the small amount of acetate produced was completely consumed after 6 h (Fig. 1). Glycerol consumption started after 4 h of cultivation, while lactose consumption started after 8 h, i.e., only after glucose exhaustion (Fig. 1). The combination of glycerol and lactose resulted in an almost unnoticeable diauxic growth. Lactose consumption increased after 9 h cultivation (Fig. 1), in parallel with the beginning of protein synthesis (Fig. 2a). In this process, the biomass concentration reached 39 g/L of dry cell weight in 16 h of cultivation (Fig. 2a). The plasmid remained quite stable, as the percentage of plasmid retention was higher than 88% within 14 h of cultivation and dropped to 70% at the end of the process (Fig. 2a). The volumetric production reached 7.6 g/L of PspA4Pro, the productivity 0.45 g/(L h) and the specific production 200 mg PspA4Pro/ g biomass (Fig. 2a).

Production of PspA4Pro in 5-L bioreactor batch cultivation. Time profiles of biomass (stars), acetate (solid triangles), glucose (open squares), glycerol (open circles), and lactose (solid squares) in Phytone auto-induction medium

PspA4Pro volumetric production (circles), PspA4Pro productivity (triangles), specific PspA4Pro production (diamonds), and plasmid retention (squares) in Phytone auto-induction simple batch (a) and in glycerol-limited fed-batch with lactose induction (b)
For the fed-batch strategy, the biomass increased up to 104 g/L within 36 h cultivation, leading to a PspA4Pro production reached 11.9 g/L after 11 h of induction. The plasmid retention was 95% at the end of cultivation (Fig. 2b). As expected, the fed-batch cultivation reached higher biomass concentration, but the cultivation was longer. Therefore, the PspA4Pro productivity was lower than in auto-induction batch, 0.33 g/(L h), as well as the specific production, 100 mg PspA4Pro/g biomass (Fig. 2b).
Cell rupture and clarification
Soluble PspA4Pro was produced in the cytoplasm of recombinant E. coli. Frozen cells were suspended in lysis buffer and ruptured in a high-pressure continuous homogenizer. During the circulation of cells inside the closed loop of the homogenizer, samples were taken to measure cell lysis and protein release, respectively, by OD at 600 nm and absorbance at 280 nm. There was a rapid decrease in OD, which indicates that most of the cells were ruptured in the first 2 min. After 4 min, both OD and absorbance at 280 nm reached an equilibrium plateau, indicating that cell rupture was completed after four residence times.
After cell lysis, four conditions were evaluated for the clarification step: pH 4.0, pH 5.5, pH 6.5, and 0.1% CTAB at pH 6.5. The purity and recovery increased with increasing pH (Table 1), but the pellet produced after centrifugation at pH 6.5 was soft and difficult to separate from the supernatant. The clarification with CTAB showed the greatest increase in purity and recovery of PspA4Pro (Table 1), and a hard and compact pellet was produced and easily removed, suggesting that the centrifugation time could be decreased.
CTAB also decreased endotoxin concentration (Table 2), because this cationic detergent interacts with negatively charged contaminants and parts of LPS, which is the main endotoxin molecule.
Comparison between ion exchange and mix mode chromatography
Both anion and cation exchange chromatographic steps allowed higher PspA4Pro recovery with similar purification factor than mix mode chromatography (data not shown). The low recovery in mix mode chromatography was associated with the combined interaction modes of the resin: ionic and hydrophobic, which led to a very strong binding with PspA4Pro that was not eluted from the resin even with 1 M l-arginine. Due to this low recovery, mix mode chromatography was removed from the purification process and the sequence of chromatographic steps was limited to an anion exchange followed by a cation exchange. It is important to emphasize that the opposite sequence is not possible, since pH reduction of the Homogenate led to a huge loss of PspA4Pro in the clarification step (Table 1).
Cryoprecipitation
A precipitate was observed when the pH of the fraction eluted from Q-Sepharose was adjusted to 4.0 in order to apply it to the cation exchange chromatography, but PspA4Pro remained in the supernatant and the purity increased from 65.2 ± 10.7 to 93.7 ± 4.9%. The freezing/thawing of the Q-fraction without pH adjustment also precipitated contaminants, while PspA4Pro was recovered in the supernatant. When the clarification step was performed at pH 4.0, it resulted in the loss of almost all PspA4Pro (Table 1). On the other hand, PspA4Pro did not precipitate at pH 4.0 after Q-Sepharose. Hence, pH adjustment and freezing were combined in a cryoprecipitation step, which resulted in a great increase of purity and high step recovery (Table 2).
Final purification process and endotoxin removal
The complete purification process was established based on the steps selected above: cell rupture in a continuous high-pressure homogenizer, clarification in the presence of the cationic detergent CTAB, anion exchange chromatography, cryoprecipitation at pH 4.0, and cation exchange chromatography. At the end of the three purification processes, 35.3 ± 2.5% of the initial PspA4Pro was recovered, with 97.8 ± 0.36% purity. In addition, the purification process successfully removed more than 99.9% of the endotoxin unities (EU) from the product (Table 2). The clarification step removed 96.5% of the EU, while centrifugation of Homogenate without CTAB removed 32.1% of the EU. Almost all EU that remained after clarification with CTAB was removed during anion exchange chromatography and cryoprecipitation at pH 4.0. The increase in EU/microgram observed in the cation exchange chromatography resulted from the loss of PspA4Pro during this step (Table 2). However, a slight EU reduction from 4.51 to 4.48 log was detected (data not shown). Figure 3 shows a representative SDS-PAGE of all purification fractions and the Western Blot of the purified protein. PspA4Pro has no enzymatic activity; thus, Western Blot and the lactoferrin binding assay were performed to verify, respectively, if PspA4Pro antigenicity and biological activity were preserved after the purification process.

a SDS-PAGE of untagged recombinant PspA4Pro in the purification process: 1 molecular marker, 2 Homogenate, 3 CTAB Clarified fraction, 4 anion exchange chromatography fraction, 5 Cryo-pH 4.0 fraction, 6 cation exchange chromatography fraction, 7 purified PspA4Pro (10 µg). b Western Blot of different amounts of purified PspA4Pro: 8 2 μg, 9 5 μg, and 10 10 μg
PspA4Pro binding to lactoferrin and molecular mass
After mixing PspA4Pro and human lactoferrin in different proportions, distinct peaks were observed in HPSEC: one corresponding to the PspA4Pro-lactoferrin complex (RT = 12.56 min) and the other to excess PspA4Pro (RT = 13.05 min) or excess lactoferrin (RT = 15.28 min). These results show that purified PspA4Pro maintained the capacity to bind to human lactoferrin, suggesting that biological activity was unchanged (Fig. 4). Based on the mass of human lactoferrin (80 kDa) and PspA4Pro (43 kDa), our results suggest that two PspA4Pro molecules can bind to each lactoferrin molecule (Fig. 4b), which is not in accordance with a previous calculated stoichiometry of approximately 1:1 for this binding (Senkovich et al. 2007). It is also important to notice that the PspA4Pro binding capacity was not affected by the high ionic force of the mobile phase, which contains 300 mM NaCl and 500 mM phosphate buffer, indicating high affinity between the two molecules.

Binding of PspA4Pro to human lactoferrin. a Two micrograms per microliter purified PspA4Pro (gray) and 1 mg/mL human lactoferrin (black). b Mixture of PspA4Pro and human lactoferrin. Two micrograms per microliter PspA4Pro and 0.5 mg/mL lactoferrin (black curve). One microgram per microliter PspA4Pro and 1 mg/mL lactoferrin (gray curve). One microgram per microliter PspA4Pro and 2 mg/mL lactoferrin (bold curve)
In order to calculate the PspA4Pro molecular mass, two different standard curves were prepared: one using globular proteins and the other using linear dextran polymers of different sizes (curves not shown). The theoretical mass of PspA4Pro is 43 kDa (PROTEIN CALCULATOR 2013). However, only the molecular mass calculated by the dextran standard curve was close to this value. PspA4Pro displayed a retention time of 12.85 min (Fig. 4a), which corresponds to a molecular mass of 111 kDa when calculated based on the globular protein standard curve, and 49 kDa when calculated based on the linear dextran standard curve. PspA4Pro is rich in α-helix and has a coiled-coil structure, which gives it a lengthened shape, like a rod. Thus, the Stokes radius is larger and retention time is lower for PspA4Pro than for globular proteins, which results in the overestimation of PspA4Pro mass when calculated with the globular protein standard curve. These results are in accordance with previous works that characterized recombinant N-terminal fragments of PspA and found that those fragments had linear coiled-coil structures (Jedrzejas et al. 2000; Jedrzejas et al. 2001).
Secondary structure and stability of purified PspA4Pro
CD spectra were obtained to determine the secondary structure of purified PspA4Pro in the presence or absence of CTAB, during thermal treatment and at different pH values. In all spectra, two valleys were observed, with minimum values at 208 and 222 nm (Fig. 5), which suggests α-helix-rich protein, as expected for the N-terminal fragment cloned, as demonstrated for N-terminal fragments of PspA from different pneumococcal strains (Jedrzejas et al. 2000; Lamani et al. 2000; Jedrzejas et al. 2001; Haughney et al. 2013). In the presence of CTAB, the molar ellipticity at 222 nm increased from −45,881 to −19,739, which indicates a loss of α-helix structure (Fig. 5). This loss, however, seems to be reversible, since the spectrum in the absence of CTAB was obtained with a PspA4Pro that was treated with CTAB at the beginning of the purification process.

Circular dichroism spectra of purified PspA4Pro in the presence (dotted line) or absence (solid line) of CTAB. Molar ellipticity from 185 to 260 nm
The overlap of heating and cooling curves obtained at 222 nm during the thermal treatment, in which PspA4Pro was heated from 15 to 95 °C and cooled back to the initial temperature, indicates that although PspA4Pro loses α-helix structures during heating, it regains its original structure when cooled (Table 3; Fig. 6). In addition, a region where PspA4Pro presented an intermediate structure that was more resistant to temperature was observed between 40 and 50 °C (Fig. 6). In order to calculate the Tm, the temperature at which the protein loses half of its original structure, the curve was divided in two parts: one between 15 and 50 °C, another between 40 and 95 °C. The similarity of Tm values obtained for heating and cooling confirms that PspA4Pro recovered its original structure after thermal treatment (Fig. 6). This property could be advantageous for downstream processing, as thermal treatment could be employed to remove contaminant proteins.

Changes in PspA4Pro molar ellipticity at 222 nm during heating (solid line) and cooling (dotted line) from 15 to 95 °C and from 95 to 15 °C, respectively, at 1 °C/min. The arrows indicate Tm during heating (h) and cooling (c) between 15 and 50 °C, Tmh1 = 36.781 °C, Tmc1 = 35.827 °C, and between 40 and 95 °C, Tmh2 = 67.501 °C, Tmc2 = 65.215 °C
PspA4Pro also maintained its secondary structure in a wide range of pH values according to Contin and CDSSTR algorithms (Table 3). The maintenance of secondary structure in face of pH changes suggests that the PspA4Pro loss during clarification at pH 4.0 (Table 1) was due to a dragging effect of other contaminants precipitating at acid pH. Therefore, precipitation steps based on pH variation are suited for PspA4Pro purification, as long as a certain level of purity has already been reached, as found here after anion exchange chromatography.
Discussion
In industrial bioprocesses, glucose-based mineral salt media are normally used and the cell metabolism is controlled by glucose limited fed-batch to reach high cell densities and high-level recombinant protein production (Krause et al. 2016). Due to the reduced acetate formation, our group has been using glycerol as carbon source in fed-batch cultivation, and lactose instead IPTG as inducer for recombinant protein production with important results (Carvalho et al. 2012; Horta et al. 2011, 2012, 2014; Vélez et al. 2014). Another strategy explored to improve protein production was the auto-induction system, which was originally developed for shaken flasks (Studier 2005) but was successfully adapted for bioreactor cultivation using an animal-free origin peptone to avoid the risks of bovine spongiform encephalopathy transmission (Vélez et al. 2014; Campani et al. 2016). Here, we show that the auto-induction system for bioreactor cultivation has the advantage of being simpler and faster than the glycerol limited fed-batch, with higher specific production and productivity (Fig. 2). Moreover, both cultivation strategies gave similar results in downstream processing (Table 2), which means that the purification process developed is robust enough to deal with differences in the raw material.
According to WHO (2009), diphtheria CRM197, a carrier protein commonly used in pneumococcal conjugate vaccines, should present more than 90% purity. Among the purification processes of recombinant protein candidates for vaccines, a purity ranging from 90 to 95% was described (Frace et al. 1999; Zhang and Pan 2005; Cheng et al. 2009). Thus, our process is among those with higher purity.
In the purification process developed, 35% of PspA4Pro was recovered and the purity increased from 33 to 97.8%, which translates into an overall purification factor of 2.96-fold. Hou et al. (2004) devised two protocols to purify His-tagged and untagged HIV-1 reverse transcriptase, obtaining similar activities and yields in both processes. Although we did not purify tagged PspA in this work, our group described purification processes for obtaining His-tagged PspA from families 1 and 2. The first one used anion exchange chromatography and IMAC Sepharose to increase the purity of PspA from 34.9 to 96.6%, while recovering 23% of the initial PspA (Barazzone et al. 2011). The second consisted of an anion exchange chromatography, metal affinity chromatography, and a cation exchange chromatography, increasing PspA purity from 22.1 to 96.5% with 22% recovery (Carvalho et al. 2012). Compared to the previous processes, the present one managed to obtain higher purity and better recovery of an untagged PspA employing two precipitation steps and two chromatographic steps. Moreover, this process is cheaper than the traditional purification using His-tag for metal affinity chromatography, since the elimination of the IMAC-Sepharose would reduce 4.5 times the process cost, without taking into account the costs of the steps to remove the tag.
Another important aspect of the purification process is the LPS removal in the clarification step. The detergent CTAB has been used for cell disruption and precipitation of polysaccharides and nucleic acids (Huang and Kim 2013; Tomanee et al. 2004; Lander et al. 2002). In addition, Panda and Chakraborty (1998) demonstrated that purified LPS can bind to cationic detergents forming mixed micelles. Yoshihisa (1999) employed CTAB for LPS removal in the purification process of RTX toxin family proteins in a process where these toxins were precipitated with detergent, leaving LPS in the supernatant. However, these studies did not evaluate the capacity of CTAB to precipitate LPS and other contaminants during protein purification.
Branston et al. (2015) described LPS removal from pre-purified Phage M13, which was precipitated three times with 2% (w/v) PEG 6000 and 500 mM NaCl with 97% yield and 90% endotoxin removal in the supernatant. After performing three new rounds of precipitation with 2% (w/v) PEG 6000, 500 mM NaCl, and 2% (v/v) Triton X-100, a 5.7 log endotoxin reduction was achieved. In contrast, we showed here that after only one precipitation step with CTAB, which was added to the crude raw material, the Homogenate, 100% of PspA4Pro was recovered in the supernatant and 96.5% of endotoxin was removed in the precipitate (Table 2). The purification process presented in this work reached a 5.3 log endotoxin reduction. In addition, Branston and colleagues do not state if the treatment with PEG and Triton X-100 removed contaminants other than endotoxins, while the clarification step described here increased PspA4Pro purity with high recovery, removed a large quantity of endotoxins, and helped centrifugation by forming a compact pellet. Characteristics of PspA, such as high solubility and a rod shape with well-defined electronegative and electropositive regions, might have contributed to the success of CTAB in the process. Removal of LPS in early steps of the purification process is especially important, as more free binding sites are available in the anion exchange resin to interact with PspA4Pro. It is noteworthy that due to CTAB positive charge, any remaining detergent is eliminated in the non-adsorbed fraction of Q-Sepharose.
Briles et al. (2000) immunized healthy human adults with two doses of 125 μg PspA in a phase 1 clinical trial. Thus, considering 42 weeks of work in 5-L auto-induction batches and 35% final recovery, the whole process described here would produce 4.46 million doses annually, which would be enough to immunize all Brazilian newborns in 2015 (IBGE 2016) with the same dose and immunization schedule. It is worth to note that the purification process was performed with 100 g of wet biomass, so in order to purify the whole biomass produced in 5-L (around 700 g wet biomass), the process have to be scaled seven times up, which means only 7 L of Homogenate. Therefore, this volume can be processed using fundamentally the same equipment as described here and no major problems are envisaged. In addition, scaling up ion exchange chromatography is extensively studied and easily done by increasing diameter or height of the column (Rathore and Velayudhan 2003; Al-Jibbouri 2006).
In conclusion, the auto-induction simple batch provides higher PspA4Pro specific production and productivity than the glycerol limited fed-batch. The purification process developed for untagged PspA4Pro is simple, easy to scale up, and adequate for vaccine production. The purification strategy adopted here allowed us not only to obtain a superior purity and yield in comparison to our previous works but also to eliminate >99.9% of the LPS. The protein displayed a CD spectrum that resembled previously reported spectra. The purified PspA4Pro was stable in a wide range of pH values and temperatures and maintained its capacity to bind human lactoferrin.
References
Al-Jibbouri S (2006) Scale-up of chromatographic ion-exchange processes in biotechnology. J Chromatogr A 1116:135–142. doi:10.1016/j.chroma.2006.03.033
Arnau J, Lauritzen C, Petersen GE, Pedersen J (2006) Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expression and Purif 48:1–13. doi:10.1016/j.pep.2005.12.002
Bang FBA (1956) Bacterial disease of Limulus polyphemus. Bull Johns Hopkins Hosp 98:325–351
Banki MR, Gerngross TU, Wood DW (2005) Novel end economical purification of recombinant proteins: intein-mediated protein purification using in vivo polyhydroxybutyrate (PHB) matrix association. Protein Sci 14:1387–1395. doi:10.1110/ps.041296305
Barazzone GC, Carvalho RJ, Kraschowetz S, Horta ACLH, Silva AJ, Zangirolami TC, Goulart C, Leite CCL, Tanizaki MM, Gonçalves VM, Cabrera-Crespo J (2011) Production and purification of recombinant fragment of pneumococcal surface protein A (PspA) in Escherichia coli. Procedia Vacinol 4:27–35. doi:10.1016/j.provac.2011.07.005
Bell MR, Engleka MJ, Malik A, Strickler JE (2013) To fuse or not to fuse: what is your purpose? Protein Sci 22:1466–1477. doi:10.1002/pro.2356
Branston SD, Wright J, Keshavarz-Moore E (2015) A non-chromatographic method for the removal of endotoxins from bacteriophages. Biotechnol and Biochem 112:1714–1719. doi:10.1002/bit.25571
Brazilian Institute of geography and Statistics (IBGE) available at <http://brasilemsintese.ibge.gov.br/populacao/taxas-brutas-de-natalidade.html> Accessed 01 July 2016.
Briles DE, Hollingshead SK, King J, Braun PA, Park MK, Ferguson LM, Nabors GS (2000) Immunization of humans with recombinant pneumococcal surface protein a (rPspA) elicits antibodies that passively protect mice from infection with Streptococcus pneumoniae bearing heterologous PspA. J Infect Dis 186:1694–1701. doi:10.1086/317602
Briles DE, Hollingshead SK, Paton JC, Ades EW, Novak L, van Ginkel FW, Benjamin WH Jr (2003) Immunizations with pneumococcal surface protein A and pneumolysin are protective against pneumonia in a murine model of pulmonary infection with Streptococcus pneumoniae. J Infect Dis 188:339–348. doi:10.1086/376571
Campani G, Santos MP, Silva GG, Horta ACL, Badino AC, Giordano RC, Gonçalves VM, Zangirolami TC (2016) Recombinant protein production by engineered Escherichia coli in a pressurized airlift bioreactor: a techno-economic analysis. Chem Eng Process Process Intensif. doi:10.1016/j.cep.2015.10.020
Carvalho RJ, Cabrera-Crespo J, Tanizaki MM, Gonçalves VM (2012) Development of production and purification processes of recombinant fragment of pneumococcal surface protein A in Escherichia coli using carbon sources and chromatography sequences. Appl Microbiol Biotechnol 94:683–694. doi:10.1007/s00253-011-3649-9
Cheng Y, Feng Y, Luo P, J G, Yu S, Zhang W, Liu Y, Wang Q, Zou Q, Mao X (2009) Fusion expression and immunogenicity of EHEC EspA-Stx2A1 protein: implications for vaccine development. J Microbiol (Seoul, Repub Korea) 47:498–505. doi:10.1007/s12275-009-0116-8
Coral MCV, Fonseca N, Castaneda E, Di Fabio JL, Hollingshead JK, Briles DE (2001) Pneumococcal surface protein A of invasive Streptococcus pneumoniae isolates from Colombian children. Emerg Infect Dis 7:832–836. doi:10.3201/eid0705.017510
Croney CM, Coats MT, Nahm MH, Briles DE, Crain MJ (2012) PspA family distribution, unlike capsular serotype, remains unaltered following introduction of the heptavalent pneumococcal conjugate vaccine. Clin Vaccine Immunol 19:891–896. doi:10.1128/CVI.05671-11
Darrieux M, Moreno AT, Ferreira DM, Pimenta FC, de Andrade AL, Lopes AP, Leite LCC, Miyaji EM (2008) Recognition of pneumococcal isolates by antisera raised against PspA fragments from different clades. J Med Microbiol 57:273–278. doi:10.1099/jmm.0.47661-0
Frace AM, Klimov AI, Rowe T, Black RA, Katz JM (1999) Modified M2 protein produce heterotypic immunity against influenza A virus. Vaccine 18:2237–2244
Goulart C, Darrieux M, Rodriguez D, Pimenta FC, Brandileone MCC, Andrade ALSS, Leite LCC (2011) Selection of family 1 PspA molecules capable of inducing broad-ranging cross-reactivity by complement deposition and opsonophagocytosis by murine peritoneal cells. Vaccine 29:1634–1642. doi:10.1016/j.vaccine.2010.12.074
Haughney A, Petersen LK, Schoofs AD, Ramer-Tait AE, King JD, Briles DE, Wannemuehler MJ, Narasimhan B (2013) Retention of structure, antigenicity and biological function of pneumococcal surface protein A (PspA) released from polyanhydride nanoparticles. Acta Biomater 9:8262–8271. doi:10.1016/j.actbio.2013.06.006
Hollingshead SK, Becker R, Briles DE (2000) Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect Immun 68:5889–5900
Horta ACL, Silva AJ, Sargo CR, Gonçalves VM, Zangirolami TC, Giordano RC (2011) Robust artificial intelligent tool for automatic start-up of the supplementary medium feeding in recombinant E. coli cultivations. Bioprocess Biosyst Eng 34(7):891–901. doi:10.1007/s00449-011-0540-0
Horta ACL, Sargo CR, Silva AJ, Gonzaga MC, Santos MP, Gonçalves VM, Zangirolami TC, Giordano RC (2012) Intensification of high cell-density cultivations of rE. coli for production of S. pneumoniae antigenic surface protein, PspA3, using model-based adaptive control. Bioprocess Biosyst Eng 35:1269–1280. doi:10.1007/s00449-012-0714-4
Horta ACL, Silva AJ, Sargo CR, Vélez AM, Gonzaga MC, Gonçalves VM, Giordano RC, Zangirolami TC (2014) A supervision and control tool based on artificial intelligence for high cell density cultivations. Braz J Chem Eng 31:457–468
Hou EW, Prasad R, Beard WA, Wilson SH (2004) High-level expression and purification of untagged and histidine-tagged HIV-1 reverse transcriptase. Protein Expr Purif 34:75–86. doi:10.1016/j.pep.2003.10.018
Huang WC, Kim JD (2013) Cationic surfactant-based method for simultaneous harvesting and cell disruption of a microalgal biomass. Bioresour Technol 149:579–581. doi:10.1016/j.biortech.2013.09.095
Jedrzejas MJ, Hollingshead SK, Lebowitz J, Chantalat L, Briles DE, Lamani E (2000) Production and characterization of the functional fragment of pneumococcal surface protein A. Arch Biochem Biophys 373:116–125. doi:10.1006/abbi.1999.1544
Jedrzejas MJ, Lamani E, Becker RS (2001) Characterization of selected strains of pneumococcal surface protein A. J Bio Chem 276:33121–33128. doi:10.1074/jbc.M103304200
Krause M, Neubauer A, Neubauer P (2016) The fed-batch principle for the molecular biology lab: controlled nutrient diets in ready-made media improve production of recombinant protein in Escherichia coli. Microb Cell Factories 15:110–123. doi:10.1186/s12934-016-0513-8
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:682–685
Lamani E, McPherson DT, Hollingshead SK, Jedrzejas MJ (2000) Production, characterization, and crystallization of truncated forms of pneumococcal surface protein A from Escherichia coli. Protein Expr Purif 20:379–388. doi:10.1006/prep.2000.1320
Lander RJ, Winters MA, Meacle FJ, Buckland BC, Lee AL (2002) Fractional precipitation of plasmid DNA from lysate by CTAB. Biotechnol Bioeng 79:776–784. doi:10.1002/bit.10335
Lowry OH, Rosenbrough NJ, Farr RH (1951) Protein measurement with Folin phenol reagent. J Bio Chem 193:265–275
Lu W, Sun Z, Tang Y, Chen J, Tang F, Zhang J, Liu J (2011) Split intein facilitated tag affinity purification for recombinant proteins with controllable tag removal by inducible auto-cleavage. J Chromatogr A 1218:2553–2560. doi:10.1016/j.chroma.2011.02.053
Ludwig C, Wear MA, Walkinshaw MD (2010) Streamlined, automated protocols for the production of milligram quantities of untagged recombinant human cyclophilin-A (hCyoA) and untagged human proliferating cell nuclear antigen (hPVNA) using ÄKTAxpress™. Protein Expr Purif 71:54–61
McCool TL, Cate TR, Moy G, Weiser JN (2002) The immune response to pneumococcal proteins during experimental human carriage. J Exp Med 195:359–365
Mirza S, Hollingshead SK, Benjamin WH Jr, Briles DE (2004) PspA protects Streptococcus pneumoniae from killing by apolactoferrin, and antibody to PspA enhances killing of pneumococci by apolactoferrin [corrected]. Infect Immun 72:5031–5040. doi:10.1128/IAI.72.9.5031-5040.2004
Moreno AT, Oliveira ML, Ferreira DM, Ho PL, Darrieux M, Leite LC, Ferreira JM Jr, Pimenta FC, Andrade AL, Miyaji EM (2010) Immunization of mice with single PspA fragments induces antibodies capable of mediating complement deposition on different pneumococcal strains and cross-protection. Clin Vaccine Immunol 17:439–446. doi:10.1128/CVI.00430-09
Oliveira MLS, Miyaji EN, Ferreira DM, Moreno AT, Ferreira PCD, Moreno AT, Ferreira PCD, Lima FA, Santos FL, Sakauchi MA, Takata CS, Higashi HG, Raw I, Kubrusly FS, Ho PL (2010) Combination of pneumococcal surface protein A (PspA) with whole cell pertussis vaccine increases protection against pneumococcal challenge in mice. PLoS One 5(5):e10863
Panda AK, Chakraborty AK (1998) Interaction of mixed surfactants with bacterial lipopolysaccharide. J Colloid Interface Sci 203:260–264. doi:10.1006/jcis.1998.5505
Pichon B, Ladhani SN, Slack MP, Seconds-Pichon A, Andrews NJ, Waight PA, Miller E, George R (2013) Changes in molecular epidemiology of Streptococcus pneumoniae causing meningitis following introduction of pneumococcal conjugate vaccination in England and Wales. J Clin Microbiol 51:820–827. doi:10.1128/JCM.01917-12
PROTEIN CALCULATOR V3.4 (2013) available at <http://protcalc.sourceforge.net/> Accessed 01 June 2016
Qian J, Yao K, Xue L, Xie G, Zheng Y, Wang C, Shang Y, Wang H, Liu L, Li C, Ji W, Wang Y, Xu P, Yu S, Tang YW, Yang Y (2012) Diversity of pneumococcal surface protein A (PspA) and relation to sequence typing in Streptococcus pneumoniae causing invasive disease in Chinese children. Eur J Clin Microbiol Infect Dis 31:217–223. doi:10.1007/s10096-011-1296-9
Rathore AS, Velayudhan A (2003) Scale-up and optimization in preparative chromatography. principles and biopharmaceutical applications. Marcel Gekker Inc., New York
Ren B, McCroy MA, Pass C, Bullard DC, Ballantyne CM, Xu Y, Briles DE, Szalai AJ (2004) The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J Imunol 173:7506–7512
Senkovich O, Cook WJ, Mirza S, Hollingshead SK, Protasecich II, Briles DE, Chattopadhay D (2007) Structure of a complex of human lactoferrin N-lobe with pneumococcal surface protein A provides insight into microbial defense mechanism. J Mol Biol 370:701–713. doi:10.1016/j.jmb.2007.04.075
Shi C, Tarimala A, Meng Q, Wood DW (2014) A general purification platform for toxic proteins based on intein trans-splicing. Appl Microbiol Biotechnol 98:9425–9435. doi:10.1007/s00253-014-6080-1
Sreerama N, Woody RW (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal Biochem 287:252–260. doi:10.1006/abio.2000.4880
Studier FW (2005) Protein production by auto-induction in high density shaking culture. Protein Expr Purif 41:313–322. doi:10.1016/j.pep.2005.01.016
Tomanee P, Hsu JT, Yoichiro I (2004) Fraction of protein, RNA and plasmid DNA in centrifugal precipitation chromatography using cationic surfactant CTAB containing inorganic salts NaCl and NH4Cl. Biotechnol Bioeng 88:52–59. doi:10.1002/bit.20203
Vélez AM, Silva AJ, Horta AC, Sargo AC, Campani G, Gonçalvez GS, Lima RCG, Zangirolami TC (2014) High-throughput for penicillin G acylase production in rE. coli fed-batch cultivations. BMC Biotechnol 14:6. doi:10.1186/1472-6750-14-6
Walker CLW, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O’Brien KL, Campbell H, Blackt RE (2013) Global burden of childhood pneumonia and diarrhea. Lancet 381:1405–1416. doi:10.1016/S0140-6736(13)60222-6
Wear MA, Pattserson A, Malone K, Dunsmore C, Turner NJ, Walkinshaw (2005) A surface plasmon resonance-based assay for small molecule inhibitors of human cyclophilin A. Anal Biochem 345:214–226. doi:10.1016/j.ab.2005.06.037
Whitmore L, Wallace BA (2004) DICHROWEB: an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32:668–673. doi:10.1093/nar/gkh371
World Health Organization (2009) Recommendations to assure the quality, safety and efficacy of pneumococcal conjugate vaccines. WHO press, World Health Organization, Geneva, Switzerland. Available at http://www.who.int/biologicals/publications/trs/areas/vaccines/pneumo/en/
Wu J, Filutowicz M (1999) Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim Pol 46:591–599
Yoshihisa H (1999) Process for producing vaccine for bacterial toxin belonging to RTX toxin family. Canadian patent number CA 02109233
Zhang D, Pan W (2005) Evaluation of three Pichia pastoris-expressed Plasmodium falciparum merozoite proteins as a combination vaccine against infection with blood-stage parasites. Infect Immun 73:6530–6536. doi:10.1128/IAI.73.10.6530-6536.2005
Acknowledgments
The authors would like to thank Milena Apetito Akamatsu and Bianca Pereira Carvalho Holanda for performing endotoxin quantification and the Sao Paulo Research Foundation for financial support (FAPESP grant numbers 2012/04858-7, 2015/06255-6 and 2015/10.291-8). Douglas B. Figueiredo receives a scholarship from CNPq (140573/2015-1). Gilson Campani Jr. received a scholarship from FAPESP (2011/16605-3). Stefanie Krashowetz, Rafaela T. Zanardo, Mauricio P. Santos, and Gabriel G. Silva received scholarships from CAPES.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Figueiredo, D.B., Carvalho, E., Santos, M.P. et al. Production and purification of an untagged recombinant pneumococcal surface protein A (PspA4Pro) with high-purity and low endotoxin content. Appl Microbiol Biotechnol 101, 2305–2317 (2017). https://doi.org/10.1007/s00253-016-7983-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7983-9