
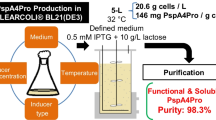
Abstract
Pneumococcal surface protein A (PspA) is essential for Streptococcus pneumoniae virulence and its use either as a novel pneumococcal vaccine or as carrier in a conjugate vaccine would improve the protection and the coverage of the vaccine. Within this context, the development of scalable production and purification processes of His-tagged recombinant fragment of PspA from clade 3 (rfPspA3) in Escherichia coli BL21(DE3) was proposed. Fed-batch production was performed using chemically defined medium with glucose or glycerol as carbon source. Although the use of glycerol led to lower acetate production, the concentration of cells were similar at the end of both fed-batches, reaching high cell density of E. coli (62 g dry cell weight/L), and the rfPspA3 production was higher with glucose (3.48 g/L) than with glycerol (2.97 g/L). A study of downstream process was also carried out, including cell disruption and clarification steps. Normally, the first chromatography step for purification of His-tagged proteins is metal affinity. However, the purification design using anion exchange followed by metal affinity gave better results for rfPspA3 than the opposite sequence. Performing this new design of chromatography steps, rfPspA3 was obtained with 95.5% and 75.9% purity, respectively, from glucose and glycerol culture. Finally, after cation exchange chromatography, rfPspA3 purity reached 96.5% and 90.6%, respectively, from glucose and glycerol culture, and the protein was shown to have the expected alpha-helix secondary structure.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pneumococcal surface protein A (PspA) is an essential virulence factor of S. pneumoniae, a pathogenic bacterium responsible for pneumonia, meningitis, otitis media, sinusitis, bacteremia and sepsis, causing millions of death around the world, mainly of children and elderly. All current pneumococcal vaccines are based on the protection given by capsular polysaccharides (PS), alone or conjugated to a carrier protein. PS are T cell independent antigens which do not provide protection for children younger than 2 years old, nonetheless, the covalent linkage of PS to a carrier protein through chemical conjugation alters the immune response against PS, as a result, T cell-dependent response is achieved, booster effect is observed, and children are protected (Bogaert et al. 2004). More than 90 pneumococcal serotypes are known, each one corresponding to a different PS on bacterial capsule, and approximately 20 of them have been responsible for severe infections (Kamerling 2000). Due to the relatively low coverage of the available pneumococcal conjugate vaccines, serotype replacement has been observed in all countries that introduced these vaccines in their immunization programs (Mera et al. 2008; Ansaldi et al. 2010). Existing conjugate vaccines are being produced with proteins not related to the pneumococcal disease, such as nontoxic diphtheria protein CRM197. An alternative to broaden the coverage of conjugate vaccines is to use a pneumococcal protein as carrier (Briles et al. 2000b).
PspA was shown to be indispensible for S. pneumoniae virulence (Abeyta et al. 2003; Miyaji et al. 2003; Silva et al. 2007) and to give high cover after immunization (Briles et al. 1997; Briles et al. 2000a), which led PspA to be considered a potential candidate for a protein vaccine as well as a good carrier candidate. Based on the alignment of amino acids sequence of PspA from distinct S. pneumoniae strains, this protein was classified in three families and six clades (Hollingshead et al. 2000). Antibody cross-recognition occurs into the same family and around 94% strains so far isolated from patients belong to families 1 and 2 (Brandileone et al. 2004; Miyaji et al. 2002; Silva et al. 2007), and a clinical trial with healthy adults immunized with a PspA from clade 2 showed cross-reactivity with PspA belonging to clades 1 to 5 from different strains (Nabors et al. 2000). If PspA were used as carrier, this new conjugate vaccine should reach much wider coverage than the current 13-valent conjugate pneumococcal vaccine. PspA has been also proposed as vaccine alone or together with other pneumococcal proteins (Baril et al. 2006; Ferreira et al. 2009; Ochs et al. 2008; Oliveira et al. 2010).
Escherichia coli has been one of the most used platform for producing recombinant proteins at large scale, since E. coli is a well-characterized prokaryote (Shiloach and Fass 2005) and able to reach high cell densities (Choi et al. 2006). Fed-batch culture is one of the most performed strategies to reach high cell densities of E. coli and consequently high recombinant protein productivities (Babaeipour et al. 2008; Goyal et al. 2009; McDonald et al. 1996; Son et al. 2010). Chemically defined medium is commonly applied in this production strategy because the cultivation parameters can be easier controlled during the feeding phase and avoid inhibition by medium compounds, for instance, carbon source accumulation (Gnoth et al. 2008).
Although glucose is the most commonly used and preferential carbon source for E. coli, its use may produce high concentration of acetate, which can provoke negative effects on cell growth and protein production. Investigations on cultivation conditions, medium compositions, and genetic approaches have been used in order to minimize acetate formation (De Mey et al. 2007). The use of different carbon sources such as glycerol has been shown to reduce acetate formation (Hansen and Eriksen 2007; Martinez et al. 2008) and also improve the activity of the products (Hansen and Eriksen 2007).
In general, recombinant protein purification for vaccine purposes requires high purity and a process able to maintain bioactivity of the protein at the end of the downstream process; thereby, expensive and more complex purification technologies are required, which lead to increased costs of the process (Geng and Wang 2008). Liquid chromatography is a largely well-established tool in industrial scale purification (Noble 2001). In addition, a great number of possibilities for the purification strategy are available nowadays, and the choice of the strategy can influence time and cost of total purification process (Shukla et al. 2007). All these facts demonstrate the need of purification process optimization along with right process selection in order to achieve the highest efficiency with a minimum number of steps (Ansenjo and Andrews 2008).
Developing a whole production process of a specific product requires not only efficient but also economical up- and downstream processing, especially when this production is aimed to be scaled-up for mass vaccination (Hewitt and Nienow 2007). Studies of up- and downstream processing are usually performed separately (Hortsch and Weuster-Botz 2010; Goyal et al. 2009; Azevedo et al. 2010); however, if such studies were performed in an integrated manner, they can provide suitable results in order to develop more effective process (Balbas 2001; Gottschalk 2008; Roush and Lu 2008). Here, the influence of distinct carbon sources, glucose, and glycerol, at high cell density of E. coli BL21(DE3) was compared for production and purification of the recombinant fragment of PspA3 in an attempt to obtain an effective process amenable to industrial scale-up.
Materials and methods
Microorganism and plasmid
The recombinant N-terminal fragment of pspA gene from clade 3 of family 2 (GenBank accession n. AY082389.1) was obtained from S. pneumoniae strain St 259/98, deposited at Nucleo de Colecao de Microrganismos, Instituto Adolfo Lutz (Sao Paulo, SP, Brazil). The fragment without the signal peptide, containing the amino acids 17 to 360, was cloned into pET37b + plasmid vector (Department of Microbial Genetics, National Institute of Genetics Shizuoka-ken, Japan), with a polyhistidine tag in the amino terminal and inserted into E. coli BL21(DE3) (Invitrogen, Carlsbad, CA, USA). The clone was kindly provided by Dr. Eliane Miyaji from Centro de Biotecnologia, Instituto Butantan (São Paulo, Brazil).
E. coli cultivation for rfPspA3 production
The cell production was performed in three distinct phases: inoculum, batch, and fed-batch. Two different carbon sources, glycerol, or glucose were used to compare the growth and the protein production. The glycerol concentration was 20 g/L for inoculum preparation, 40, and 696 g/L in batch and feeding media, respectively, and the glucose concentration was 10 g/L for inoculum preparation, 20 and 600 g/L in batch and feeding media, respectively. The composition of chemically defined media was adapted from Seeger et al. (1995). For inoculum and batch cultivation, the medium contained 13.3 g/L KH2PO4, 4.0 g/L (NH4)2HPO4, 1.2 g/L MgSO4·7H2O, 1.7 g/L citric acid, 14.1 mg/L EDTA, 2.5 mg/L CoCl2·6H2O, 15.0 mg/L MnCl2·4H2O, 1.5 mg/L CuCl2·2H2O, 3.0 mg/L H3BO3, 2.1 mg/L Na2MoO4·2H2O, 33.8 mg/L Zn(CH3COO)2·2H2O, 100.8 mg/L Fe(III)citrate, 4.5 mg/L thiamine hydrochloride, 25 mg/L kanamicyn sulfate. The feeding medium was composed of 20.0 g/L MgSO4·7H2O, 13.0 mg/L EDTA, 4.0 mg/L CoCl2·6H2O, 23.5 mg/L MnCl2·4H2O, 2.3 mg/L CuCl2·2H2O, 4.7 mg/L H3BO3, 4.0 mg/L Na2MoO4·2H2O, 16.0 mg/L Zn(CH3COO)2·2H2O, 40.0 mg/L Fe(III)citrate, 4.5 mg/L thiamine hydrochloride, 25 mg/L kanamycin sulfate.
Inoculum
Two stages of inoculum preparation were performed in flasks. One isolated colony from LB/kanamycin plate was inoculated into a flask of 100 mL containing 10 mL of chemically defined medium and, after growing to optical density at 600 nm (OD600nm) of approximately 3.0, 2 mL aliquot from this culture was added to 200 mL of fresh medium in flasks of 2 L. An orbital shaker Innova 44 (New Brunswick Scientific, USA) was used to maintain both cultures at 200 rpm and 37°C. The second culture was grown until OD600nm = 5.0 and inoculated into the reactor obtaining an initial OD600nm of 0.1, i.e., 0.04 gDCW/L.
Batch cultivation
Five liters bioreactor Bioflo 2000 (New Brunswick Scientific, USA) was used to perform the cultivation. The automatic control of the culture was set at 30°C for temperature and 30% saturation of dissolved oxygen (DO) by agitation and adding pure oxygen gas. The pH was controlled at 6.7 by addition of ammonia solution (24% w/w). Polypropylene glycol (PPG) was used as antifoam agent when it was necessary. The cultivation started with 4.5 L of medium to which 0.5 L of 20 g/L lactose was added for induction.
Fed-batch cultivation
The initial volume was 3.5 L of medium and, after complete consumption of the carbon source of batch phase (same bioreactor and conditions as batch cultivation), shown by abrupt DO increase, the feeding was started using the fed-batch medium. The pH was adjusted to 6.9, and the exponential feeding followed the equation proposed by Seeger et al. (1995). The yield coefficients were calculated from previous batches on glucose and glycerol. The specific cell growth rate set (μ set) during the fed-batch phase was 0.12 and 0.10 h–1, respectively, for glucose and glycerol, and the μ set during the induction phase was 0.05 h–1 for both cultures.
Induction
The induction was performed by a mix of isopropyl-β-d-thiogalactopyranoside (IPTG, Sigma-Aldrich, USA) and 20 g/L of lactose for 4 h for both batch and fed-batch cultivations. The IPTG concentration was 0.1 mM for induction of batch culture and 0.5 mM for fed-batch culture. The rfPspA3 was produced as soluble intracellular protein, and after the cultivation process, the cell separation was performed by centrifugation at 17,969×g for 30 min at 4°C. The supernatant was discarded, and the cells were frozen at −20°C.
Cell disruption
The suspension for cell disruption was prepared with 100 g of wet cell pellet resuspended in 1 L of lysis buffer, 25 mM Tris pH 8.0 + 0.1% of Triton X-100 + 1.0 mM of phenylmethanesulfonylfluoride (PMSF-protease inhibitor). A mixer X520 (CAT Ingenieurbüro, Germany) was used before lysing to homogenize the cell suspension. The homogenized suspension was disrupted using a continuous high-pressure APV 60 Homogenizer (APV Gaulin, MA, USA) in a close loop for 10 min at 600 bars. The homogenizer has a jacketed reservoir and a tube-and-shell heat exchanger in the inlet and outlet, respectively, to maintain the temperature during the disruption at about 12°C.
The efficiency of the homogenizer was tested with cell suspensions from both cultures, with glucose or glycerol as carbon source. Disruption samples were collected at intervals of 1 min from a total of 10 min. The samples were divided in three parts: (1) one aliquot was used for measurement of OD600nm; (2) other aliquot was serially diluted and plated on LB/agar for measurement of colony forming-units (CFU); (3) 2.0 mL was centrifuged at 20,817×g for 60 min, and the supernatant was used to quantify total protein and release of rfPspA3 by SDS-PAGE densitometry.
Clarification
Three different techniques were tested: centrifugation, microfiltration with 0.1 μm of cut-off, and ultrafiltration with 750 kDa of cut-off.
The cell disruption fraction was centrifuged at 17,696×g for 2 h at 4°C (Avanti J-25i, Beckman, CA, USA). After that, the supernatant was separated from the pellet and filtered in 0.45 μm.
The filtration tests were performed with hollow fibers of polyestersulfone: (1) with a pore of 0.1 μm and 1,200 cm2 of filter area (CFP-1-E-5A); 2) with 750 kDa of cut-off and 850 cm2 of filter area (UFP-750-E-4X2MA), both membranes purchased from GE Health Care, USA. In both cases, the cell disruption fraction was concentrated to one third of the initial volume and then the concentrate was washed with six volumes of 25 mM acetate buffer pH 6.0. The transmembrane pressure was kept constant in 5 psi.
Chromatography steps
The chromatographic steps were operated by Äkta Explorer system and monitored and controlled by Unicorn 3.21 software. Metal affinity resin, 500 mL of IMAC-Sepharose, and 250 mL of anion exchange Q-Sepharose were packed into XK50 columns, while 75 mL of cation exchange SP-Sepharose was packed into XK26 column. A flow-rate of 150 cm/h was applied for all chromatography steps. All chromatography equipment and resins were purchased from GE Healthcare Life Sciences, USA.
Previous studies of adsorption and desorption were performed in test tubes to choose the best concentration of eluents during the process. The initial conditions were chosen based on the theoretical pI (4.74) of the rfPspA3. After that, a study of the sequence of chromatography steps was performed using cells from production with glucose as carbon source. Finally, after the definition of the best sequence, the whole purification process was performed with cell disruption fractions from cultures with both carbon sources, and the results were compared to study the integration of production and purification processes. The chromatography steps were performed as described below.
IMAC-Sepharose was previously charged with 500 mM NiSO4, washed with water, and equilibrated with 25 mM Tris buffer pH 8.0. Protein adsorption was done with 5 mM imidazole in 25 mM Tris pH 8.0, and elution of the protein of interest was performed with 200 mM of imidazole in the same Tris buffer. In order to obtain higher purity, a washing step with 7 mM imidazole was introduced for the material from glycerol production and with 15 mM imidazole for the material from glucose production. The column was cleaned with 500 mM imidazole and 300 mM NaCl in 25 mM Tris buffer pH 8.0. Ethylenediaminetetraacetic acid (EDTA) (100 mM) was used to strip off the metal ion and prior to resin regeneration with NiSO4.
Q-Sepharose was equilibrated with acetate buffer (25 mM sodium acetate, pH 6.0) with 50 mM NaCl. The same buffer was used for washing and elution steps. The washing step was performed with 100 mM NaCl and the elution step with 300 mM NaCl. The cleaning of this resin was performed with 500 mM NaCl in 25 mM Tris buffer pH 8.0. The regeneration was performed with 100 mM NaOH.
SP-Sepharose was equilibrated with histidine buffer (20 mM l-histidine hydrochloride, pH 6.0), and the rfPspA3 was eluted with 30 mM phosphate buffer pH 7.0. The cleaning was performed with 25 mM Tris buffer pH 8.0 with 500 mM NaCl.
Analytical methods
Biomass concentration
Sampling of the cultures was done every hour. The spectrophotometer U-1800 (Hitach High-Tech, Japan) was used at 600 nm of wavelength for OD reading, after appropriated sample dilution. To determine the dry cell weight, 10 mL of each sample were centrifuged in previously weighed tubes at 17,800×g for 15 min, and the supernatant was discarded. The pellets were dried at 60°C until constant weight. A linear correlation was obtained between OD600nm and cell concentration in grams per liter.
Concentration of glucose, glycerol, and acetic acid
The concentration of glucose, glycerol, and acetic acid was analyzed by HPLC (Shimadzu, Japan). Aminex HPX-87H column (Biorad Life Sciences, USA) was used for this analysis. The mobile phase was 5 mM H2SO4, and the running was performed at 60°C for 30 min at 0.6 mL/min. The refraction index detection (RID) was used for glycerol and glucose analysis and UV detection at 210 nm for acetic acid.
Plasmid stability
The vector stability test was performed in the inoculum, before the induction and at the end of the production process. A sample of cell suspension was serially diluted in 0.85% NaCl and 100 μL of each dilution were plated in LB agar. After 24 h, 70 colonies were replicated on two plates with and without 25 mg/L kanamycin. The stability was recorded as the percentage of kanamycin resistant colonies.
Protein quantification
Total protein was quantified by Lowry's method using Folin phenol reagent (Lowry et al. 1951) and bovine serum albumin as standard. Samples with interfering chemical compounds were previously dialyzed against 20 mM phosphate buffer pH 7.0 for 24 h.
Gel electrophoresis
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to Laemmli et al. (1970) with 12% acrylamide gel. Samples with high concentration of NaCl or other interfering chemicals were previously dialyzed for 24 h as described above. Samples from cell suspension were adjusted to OD600nm = 5.0 and heated at 100°C for 15 min in presence of SDS and beta-mercaptoethanol to disrupt the cells and 20 μL was applied to the gel. Diluted samples from the purification process were first concentrated by centrifugal vacuum concentrator Centrivap (Labconco, USA). Gels were stained by Comassie Blue R-250.
Relative purity of rfPspA3
Two methods were used to determine the relative purity of rfPspA3: densitometry of SDS-PAGE bands using a densitometer GS-800 with Quantity One 4.6.3 software (Biorad Life Sciencs, USA) and high performance size exclusion chromatography (HPSEC) in TKSgel G2000SWXL (Tosoh Bioscience, Japan) with UV detection at 280 nm. In the densitometry, the relative purity was calculated as the percentage of the intensity of rfPspA3 band against the sum of intensity of all other bands in the same lane and, in HPSEC, as the percentage of rfPspA3 peak area against the sum of all peaks areas in the chromatogram.
Circular dichroism
The method was performed according to Kelly et al. (2005). Sample (60 μL) at approx. 8.5 μM rfPspA3 was used for analysis. The polarimeter spectrum Jasco J-810 was used to perform the analysis with Software Spectra Manager (Jasco, Germany) in a wavelength range from 185 to 260 nm. The analysis was performed at room temperature.
Results
E. coli cultivation
Two carbon sources were used to produce rfPspA3 and evaluate their influence on E. coli growth, recombinant protein production, and also on the purification of rfPspA3 along the process.
Kinetic parameters were similar in batch culture mode (data not shown) and during the batch phase of fed-batch mode performed with the same carbon source. The yield coefficient on biomass was the same (0.3 gDCW/g carbon source), while the maximum specific growth rate (μ max) was higher with glucose (μ max = 0.54 h–1) than with glycerol (μ max = 0.41 h–1). The lower acid acetic production in the glycerol culture resulted in higher cell concentration at the end of batch. As the maximum cell concentration was higher with glycerol, but the cultivation time was longer due to slower growth rate, the same cell productivity was achieved in batch culture mode with both carbon sources (Table 1).
Comparing fed-batch phase of both cultures, the growth profiles were similar for them and cell concentration reached 62 g/L in both cases. During the process, higher acetate production was obtained using glucose as carbon source, it peaked at 4.5 g/L at the end of batch phase and its production was between 1.0 and 2.5 g/L during fed-batch phase, while using glycerol acetate production peaked at 0.58 g/L at the end of batch phase and its production was near zero during fed-batch phase. In spite of higher acetate production, the fed-batch strategy permitted to achieve the same final cell concentration with glucose and glycerol and, as the cell production was faster with glucose, slightly higher cell productivity was obtained when compared to that with glycerol (Table 1).
Similar cell productivities were obtained in batch mode using both carbon sources; however, it was achieved sixfold higher cell production and almost threefold higher cell productivity in fed-batch mode when compared to simple batch mode with both carbon sources (Table 1). Regarding rfPspA3 production, an increase of productivity was also obtained in fed-batch mode compared to simple batch, 1.6-fold higher rfPspA3 productivity was achieved with glucose, and 1.4-fold higher with glycerol (Table 1).
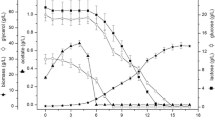
During the induction phase of fed-batch cultures, lactose was totally consumed by the microorganism after 4 h (Fig. 1) and the production of rfPspA3 increased with the time of induction (Fig. 2). It can be also observed in Fig. 2 that besides rfPspA3, many other proteins were produced in high cell density cultivation. These by-products can vary according to the environment that E. coli is exposed to and can also interfere in the downstream process; for this reason, it is important to integrate up- and downstream processes.

E. coli BL21(DE3) fed-batch culture for rfPspA3 production using two different carbon sources, glucose (A) and glycerol (B), and combined induction with 0.5 mM of IPTG and 20 g/L of lactose. Acetate (stars), cell (squares), lactose (circles), glucose or glycerol (triangles)

SDS-PAGE of rfPspA3 production in fed-batch cultures with chemically defined medium using glucose (left side) or glycerol (right side) as carbon source. BI—before induction; 1—after 1 h of induction; 2—after 2 h of induction; 3—after 3 h of induction; 4—after 4 h of induction. A cell suspension of each sample was adjusted to OD = 5.0 and 20 μL of the suspension was applied to all lanes
Cell disruption
Cells pellets, around 100 g from both cultures, were thawed for disruption, which was performed in a close loop using a high-pressure continuous homogenizer for 10 min. The profile of cell disruption and rfPspA3 release showed that 5–6 min were enough for maximum disruption (Fig. 3).

Profile of cell disruption by high-pressure continuous homogenizer of E. coli BL21(DE3) produced with glucose (solid line and symbols) or glycerol (dashed line and open symbols). OD600nm (triangles), CFU/mL (circles), concentration of rfPspA3 released into the supernatant (squares)
According to Fig. 3, the values of CFU and OD600nm were near zero after 6 min, indicating complete cell lysis. The cells from the glycerol culture resisted the freezing and thawing process better than cells from glucose. Initial cell viability of the culture was 9.8 × 104 CFU/mL with glycerol and 4 × 104 CFU/mL with glucose (Fig. 3, circles), while the initial concentration of rfPspA3 in the supernatant was 1.0 mg/mL with glycerol and 1.6 mg/mL with glucose (Fig. 3, squares). After 10 min, the cell disruption yielded 2.9 ± 0.1 and 3.3 ± 0.1 mg/mL of rfPspA3 from fed-batch cultures with glucose and glycerol as carbon source, respectively. The rfPspA3 produced here was shown to be soluble: after 2 h of centrifugation at 20,817×g, all rfPspA3 was in the supernatant and none in the insoluble fraction (data not shown).
Efficiency of cell lysis was also compared to a control sample where cells were treated with 100 mM NaOH and a complete reduction in OD600nm was observed, representing 100% efficiency of disruption. The efficiency of the disruption process by high-pressure continuous homogenizer, measured as reduction of OD600nm, was 96.7 ± 4.0% and 92.7 ± 3.8% for culture with glucose and glycerol, respectively.
Although higher production of rfPspA3 was obtained in fed-batch with glucose, higher rfPspA3 concentration from the cultivation with glycerol was obtained during cell disruption (Fig. 3). This result cannot be attributed to the efficiency of the disruption based on OD600nm reduction, since it was similar for material from glucose and glycerol cultures, but it could be related to the cryoprotector effect of glycerol, which could also have a positive consequence avoiding rfPapA3 degradation by proteases released from E. coli during disruption. Regarding rfPspA3 released from cell disruption compared to the amount produced in fed-batch cultures (3.48 and 2.97 g/L, Table 1), the yield was higher for material from glycerol (about 3.3 mg/mL of rfPspA3 from glycerol and 2.9 mg/mL from glucose culture, Fig. 3).
Clarification
The performance of mechanical disruption provides different sizes of cell debris in the homogenate, which can be related to the choice of the clarification method in this work. No purification was observed after centrifugation and the tangential filtration resulted in 1.4-fold (microfiltration) and 1.3-fold (ultrafiltration) of purification (results analyzed by HPSEC); however, they became unfeasible due to the lower yield of rfPspA3 obtained in both (43.9% and 62.5%, respectively) comparing to the centrifugation (100%). Probably, debris obstructed the pores of membranes forming secondary layers which retained the rfPspA3. The low recovery in these purification steps would mean a loss of 9.5 g rfPspA3 for microfiltration and 6.4 g for ultrafiltration of all protein produced in 5 L reactors (17 g). Thus, high value of recovery was prioritized in the clarification step, since other purification steps were included and provided the required purity.
Chromatography steps
Two chromatography steps were pre-selected based on physicochemical features of rfPspA3: the anion exchange Q-Sepharose, and the metal affinity IMAC-Sepharose. Starting from the same centrifuged fraction, two sequences were tested: anion exchange followed by metal affinity and the opposite sequence. Both sequences resulted in similar outcomes (Table 2).
The first sequence was selected because it yielded a slightly better purification and recovery results. Besides, anion exchange resins are cheaper than metal affinity resins and can be loaded with high amounts of proteins from a very impure raw material. Hence, the first sequence should increase the life-time of metal affinity, which will be used with a pre-purified fraction. Thereby, the centrifuged material from fed-batch culture with glycerol as carbon source was also purified using the sequence anion exchange followed by metal affinity chromatography (Table 3).
Another screening of liquid chromatography was performed with size exclusion, hydrophobic, and strong cation exchange chromatography. Only cation exchange chromatography (SP-Sepharose) yielded good purification factor. Despite the theoretical pI of rfPspA3 is 4.74, the protein was adsorbed to the resin at pH 6.0, i.e., theoretically with the same charge of the cation exchange. As PspA has two well-defined regions with opposite charges (Jedrzejas et al. 2001), this allowed the protein to adsorb to the SP-Sepharose at pH 6.0.
The whole process was applied for purification of rfPspA3 from fed-batch cultures with glucose and glycerol. As it can be seen in Table 3, the performance of each individual chromatography differed according to the carbon source used to produce the raw material, but the final results were relatively similar. The anion exchange worked better for the material from glucose culture, resulting in rfPspA3 with higher purity and recovery than that produced with glycerol. This may be a consequence of higher amount (3.48 g/L, Table 1) and relative purity of rfPspA3 in the centrifuged fraction from glucose culture (22.1%, Table 3) than from glycerol (2.97 g/L, Table 1 and 17.7%, Table 3, respectively for amount and purity). Besides, the conditions of anion exchange were established using material from fed-batch with glucose and after applied for purification of PspA from glycerol culture.
The metal affinity chromatography, on the opposite, seemed to work better for the fraction from glycerol culture, resulting in higher step recovery than that from glucose (70% against 39%, Table 3) and higher purification factor (2.55 against 1.45, Table 3). Finally, the purification factor of cation exchange was higher in the fraction from glycerol than that from glucose (1.19 against 1.01, Table 3), while it occurred the opposite with the recovery, which reached 62% from glucose and only 34% from glycerol (Table 3).
In order to visualize the efficiency of downstream process, the Fig. 4 presents the fractions obtained in each purification step from fed-batch cultures with glucose or glycerol as carbon sources. The increase of purity can be clearly noted. A large amount of impurities were removed in the first and mainly in the second chromatography steps. The rfPspA3 purity after cation exchange was high in both process, but the recovery was low in the fraction obtained from glycerol culture, therefore an improvement of this last step in an attempt to obtain higher rfPspA3 recovery might be necessary.

SDS-PAGE of fractions from downstream steps. CDR—cell disruption, CC—clarification by centrifugation, AEC—anion exchange chromatography, MAC—metal affinity chromatography, CEC—cation exchange chromatography. The following amount of total protein was applied to each lane: CDR and CC—15 μg, AEC and MAC—5 μg, CEC—3 μg
Circular dichroism
At the end of the purification process, the purified rfPspA3 retained the secondary structure as shown by the circular dichroism (CD) spectrum (Fig. 5). The CD spectrum profile showed α-helical secondary structure without β-sheets (Kelly et al. 2005). This profile is similar to the N-terminal fragments of PspA from different strains obtained by Jedrzejas et al. (2001). According to this author, all PspA constructions had α-helical secondary structure content, adopting a coiled-coil structure with an elongated rod-like shape, and no β-sheet was detected.

CD spectrum of rfPspA3 purified from fed-batch culture
Discussion
Batch cultures were firstly performed in order to obtain the kinetic parameters to apply them later in the fed-batch cultures to reach high cell densities. The cell production was similar for both carbon sources at the end of the fed-batch, approximately 62 g/L, but the cell productivity was slightly lower in the glycerol culture, due to lower specific growth rate during the fed-batch phase and consequently longer cultivation time (Table 1).
The culture with glucose produced 4.5 g/L acetate at the end of batch phase, but only 0.58 g/L with glycerol. During the fed-batch phase, the acetate was consumed and a plateau of about 1.2 g/L was reached with glucose, while it remained near zero with glycerol. According to De Mey et al. (2007), the presence of acetate in concentrations above 1 g/L can be harmful to the cell growth and also to recombinant protein production. The comparison of these carbon sources has been reported by different authors (Martinez et al. 2008; Hansen and Eriksen 2007), and they have also found a decrease of acetate formation using glycerol. Hansen and Eriksen (2007) obtained 40% higher product activity using glycerol. However, in our case, the final cell concentration was higher only in simple batch culture mode with glycerol, but it did not lead to higher protein production (Table 1). In fed-batch culture mode, the specific growth rate used during the fed-batch phase was more important to the final results than the acetate produced, certainly because the acetate level was below the critical value in both cases and the overflow mechanism did not occur. Maybe the lower protein production observed using glycerol was related to the higher maintenance coefficient on glycerol than on glucose (Neijssel et al. 1999), which means that more energy would be required for growth and protein synthesis, and the cultivation conditions have to be optimized for glycerol.
This higher protein production in fed-batch with glucose could be also explained by the lac operon repression system, which avoids the recombinant protein production before induction (Narang 2009). Glycerol does not exert carbon catabolite repression effect on E. coli, as can be observed in Fig. 2. Besides that, the plasmid stability was lower during production with glycerol (88%), while it was 100% stable during all process with glucose.
Concerning the sequence of chromatography steps, metal affinity certainly presented higher PF values than anion exchange chromatography, and both sequences yielded similar final results (Table 2). In almost all articles on recombinant protein purification, the sole chromatography step used is metal affinity and, when more than one step is necessary, metal affinity is the first step in the large majority of reports. Very few articles report the use of ion exchange before metal affinity and, in general, they did not discuss the reasons for adopting this sequence. Zhao et al. (2010) states that cation exchange is efficient for LPS removal and Schmidt et al. (2007) attributes the low recovery of His-tagged protein to a change in oxidation state or to a complex formation between the Ni2+ resin and yeast intracellular contents when large volumes of crude yeast cell lysate is applied to Ni2+ resin. Here, the sequence anion exchange followed by metal affinity was chosen since the binding capacity of IMAC-Sepharose was greatly diminished after five performances as first chromatography step (not shown); this probably could be caused by the high load of impurities into the column. Therefore, choosing the anion exchange as the first chromatography step could decrease the total cost of the process, since Q-Sepharose resin has about 70% lower prices than IMAC-Sepharose (supplier information) and longer life-time.
The highest final purity reached when glucose was used for the upstream processing could be explained by the difference between the rfPspA3 relative purity in the initial fractions: 22.1% in centrifuged fraction from glucose culture and 17.7% from glycerol, which was a consequence of the higher specific production of rfPspA3 on glucose than on glycerol, 56 and 48 mg/gDCW, respectively. Higher specific production was also reached using glucose in batch than using glycerol, 106 against 86 mg/gDCW.
Studying a large scale production and purification of recombinant fibroblast, three chromatography steps were necessary to reach 90.6% of purity (McDonald et al. 1996), but only two steps were necessary to purify recombinant thermostable chitinase up to 12.7 purification factor; however, non-homogeneous product with incorrectly folded proteins was obtained in this process (Christodoulou et al. 2001). Here, rfPspA3 with the same CD profile as previous study (Jedrzejas et al. 2001) was obtained after three chromatography steps with no damage to the secondary structure (Fig. 4). According to Table 3, the rfPspA3 could be obtained with 36% of recovery and 95.5% of purity from the glucose fed-batch culture after only two chromatographic steps. As it is already a quite high purity, the feasibility of a procedure with two instead of three chromatography steps will depend on the criteria of purity that would be required by regulatory agencies for licensing the vaccine product.
In conclusion, the carbon sources used in fed-batch cultures exerted the expected influence on acetate production, although the higher acetate amount produced with glucose did not affect the rfPspA3 production and the fed-batch permitted to reach the same final cell concentration of E. coli BL21(DE3) than that obtained with glycerol. The higher rfPspA3 specific production on glucose culture seemed to influence downstream processing, since higher purity was obtained using the protein produced with glucose as carbon source. Finally, a scalable process for production of PspA with high yield in high cell density fed-batch culture was developed as well as a purification process consisting of cell disruption by high-pressure continuous homogenizer, centrifugation, and three steps of liquid chromatography, which allowed to obtain the rfPspA3 with high purity and right conformation.
References
Abeyta M, Hardy GG, Yother J (2003) Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus pneumoniae. Infect Immun 71:218–225. doi:10.1128/IAI.71.1.218-225.2003
Ansaldi F, De Florentiis D, Canepa P, Durando P, Bassetti M, Icardi G (2010) Serotype replacement in Streptococcus pneumoniae after conjugate vaccine introduction: impact, doubts and perspective for new vaccines. Rev Med Microbiol 21:56–64. doi:10.1097/MRM.0b013e32833a345f
Ansenjo JA, Andrews BA (2008) Protein purification using chromatography: selection of type, modelling and optimization of operating conditions. J Mol Recognit 22:65–76. doi:10.1002/jmr.898
Azevedo AM, Gomes AG, Borlido L, Santos IF, Prazeres DM, Aires-Barros MR (2010) Capture of human monoclonal antibodies from a clarified cell culture supernatant by phenyl boronate chromatography. J Mol Recognit 23:569–576. doi:10.1002/jmr.1068
Babaeipour V, Shojaosadati SA, Khalilzadeh R, Maghsoudi N, Tabandeh F (2008) A proposed feeding strategy for the overproduction of recombinant proteins in Escherichia coli. Biotechnol Appl Biochem 49:141–147. doi:10.1042/BA20070089
Balbas P (2001) Understanding the art of producing protein and nonprotein molecules in Escherichia coli. Mol Biotechnol 19:251–267. doi:10.1385/MB:19:3:251
Baril L, Dietemann J, Essevaz-Roulet M, Béniguel L, Coan P, Briles DE, Guy B, Cozon G (2006) Pneumococcal surface protein A (PspA) is effective at eliciting T cell mediated responses during invasive pneumococcal disease in adults. Clin Exp Immunol 145:277–286. doi:10.1111/j.1365-2249.2006.03148.x
Bogaert D, Hermans PW, Adrian PV, Rumke HC, de Groot R (2004) Pneumococcal vaccines: an update on current strategies. Vaccine 22:2209–2220. doi:10.1016/j.vaccine.2003.11.038
Brandileone MC, Andrade AL, Teles EM, Zanella RC, Yara TI, Di Fabio JL, Hollingshead SK (2004) Typing of pneumococcal surface protein A (PspA) in Streptococcus pneumoniae isolated during epidemiological surveillance in Brazil: towards novel pneumococcal protein vaccines. Vaccine 22:3890–3896. doi:10.1016/j.vaccine.2004.04.009
Briles DE, Hollingshead SK, Swiatlo E, Brooks-Walter A, Szalai A, Virolainen A, McDaniel LS, Benton KA, White P, Prellner K, Hermansson A, Aerts PC, Van Dijk H, Crain MJ (1997) PspA and PspC: their potential for use as pneumococcal vaccines. Microb Drug Resist 3:401–408. doi:10.1089/mdr.1997.3.401
Briles DE, Hollingshead SK, Brooks-Walter A, Nabors GS, Ferguson L, Schilling M, Gravenstein S, Braun P, King J, Swift A (2000a) The potential to use PspA and other pneumococcal proteins to elicit protection against pneumococcal infection. Vaccine 18:1707–1711. doi:10.1016/S0264-410X(99)00511-3
Briles DE, Hollingshead SK, Nabors GS, Paton JC, Brooks-Walter A (2000b) The potential for using protein vaccines to protect against otitis media caused by Streptococcus pneumoniae. Vaccine 19:S87–S95. doi:10.1016/S0264-410X(00)00285-1
Choi JH, Keum KC, Lee SY (2006) Production of recombinant proteins by high cell density culture of Escherichia coli. Chem Eng Sci 61:876–885. doi:10.1016/j.ces.2005.03.031
Christodoulou E, Duffner F, Vorgias CE (2001) Overexpression, purification, and characterization of a thermostable chitinase (Chi40) from Streptomyces thermoviolaceus OPC-520. Protein Expr Purif 23:97–105. doi:10.1006/prep.2001.1490
De Mey M, De Maeseneire S, Soetaert W, Vandamme E (2007) Minimizing acetate formation in E. coli fermentations. J Ind Microbiol Biotechnol 34:689–700. doi:10.1007/s10295-007-0244-2
Ferreira DM, Darrieux M, Silva DA, Leite LCC, Ferreira JMC Jr, Ho PL, Miyaji EN, Oliveira MLS (2009) Characterization of protective mucosal and systemic immune responses elicited by pneumococcal surface protein PspA and PspC nasal vaccines against a respiratory pneumococcal challenge in mice. Clin Vaccine Immunol 16:636–645. doi:10.1128/CVI.00395-08
Geng X, Wang L (2008) Liquid chromatography of recombinant proteins and protein drugs. J Chromatogr B Anal Technol Biomed Life Sci 866:133–153. doi:10.1016/j.jchromb.2008.01.041
Gnoth S, Jenzsch M, Simutis R, Lubbert A (2008) Control of cultivation processes for recombinant protein production: a review. Bioprocess Biosyst Eng 31:21–39. doi:10.1007/s00449-007-0163-7
Gottschalk U (2008) Bioseparation in antibody manufacturing: the good, the bad and the ugly. Biotechnol Prog 24:496–503. doi:10.1021/bp070452g
Goyal D, Sahni G, Sahoo DK (2009) Enhanced production of recombinant streptokinase in Escherichia coli using fed-batchculture. Bioresour Technol 100:4468–4474. doi:10.1016/j.biortech.2009.04.008
Hansen R, Eriksen NT (2007) Activity of recombinant GST in Escherichia coli grown on glucose and glycerol. Process Biochem 42:1259–1263. doi:10.1016/j.procbio.2007.05.022
Hewitt CJ, Nienow AW (2007) The scale-up of microbial batch and fed-batch fermentation processes. Adv Appl Microbiol 62:105–135. doi:10.1016/S0065-2164(07)62005-X
Hollingshead SK, Becker R, Briles DE (2000) Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect Immun 68:5889–5900
Hortsch R, Weuster-Botz D (2010) Growth and recombinant protein expression with Escherichia coli in different batch cultivation media. Appl Microbiol Biotechnol 90:69–76. doi:10.1007/s00253-010-3036-y
Jedrzejas MJ, Lamani E, Becker RS (2001) Characterization of selected strains of pneumococcal surface protein A. J Biol Chem 276:33121–33128. doi:10.1074/jbc.M103304200
Kamerling JP (2000) Pneumococcal polysaccharides: a chemical view. In: Tomasz A (ed) Streptococcus pneumoniae molecular biology and mechanisms of disease. Mary Ann Liebert Publishers Inc., New York, pp 81–114
Kelly SM, Jess TJ, Price NC (2005) How to study proteins by circular dichroism. Biochim Biophys Acta 1751:119–139. doi:10.1016/j.bbapap.2005.06.005
Laemmli UK, Beguin F, Gujer-Kellenberger G (1970) A factor preventing the major head protein of bacteriophage T4 from random aggregation. J Mol Biol 47:69–74. doi:10.1016/0022-2836(70)90402-X
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Martinez K, de Anda R, Hernandez G, Escalante A, Gosset G, Ramirez OT, Bolivar FG (2008) Coutilization of glucose and glycerol enhances the production of aromatic compounds in an Escherichia coli strain lacking the phosphoenolpyruvate: carbohydrate phosphotransferase system. Microb Cell Fact 7:1. doi:10.1186/1475-2859-7-1
McDonald JR, Ong M, Shen C, Parandoosh Z, Sosnowski B, Bussell S, Houston LL (1996) Large-scale purification and characterization of recombinant fibroblast growth factor-saporin mitotoxin. Protein Expr Purif 8:97–108. doi:10.1006/prep.1996.0079
Mera R, Miller LA, Fritsche TR, Jones RN (2008) Serotype replacement and multiple resistance in Streptococcus pneumoniae after the introduction of the conjugate pneumococcal vaccine. Microb Drug Resist 14:101–107. doi:10.1089/mdr.2008.0782
Miyaji EN, Ferreira DM, Lopes AP, Brandileone MC, Dias WO, Leite LC (2002) Analysis of serum cross-reactivity and cross-protection elicited by immunization with DNA vaccines against Streptococcus pneumoniae expressing PspA fragments from different clades. Infect Immun 70:5086–5090. doi:10.1128/IAI.70.9.5086-5090.2002
Miyaji EN, Dias WO, Tanizaki MM, Leite LC (2003) Protective efficacy of PspA (pneumococcal surface protein A)-based DNA vaccines: contribution of both humoral and cellular immune responses. FEMS Immunol Med Microbiol 37:53–57. doi:10.1016/S0928-8244(03)00108-1
Nabors GS, Braun PA, Herrmann DJ, Heise ML, Pyle DJ, Gravenstein S, Schilling M, Ferguson LM, Hollingshead SK, Briles DE, Beckera RS (2000) Immunization of healthy adults with a single recombinant pneumococcal surface protein A (PspA) variant stimulates broadly cross-reactive antibodies to heterologous PspA molecules. Vaccine 18:1743–1754
Narang A (2009) Quantitative effect and regulatory function of cyclic adenosine 5′-phosphate in Escherichia coli. J Biosci 34(3):445–463
Neijssel OM, Teixeira de Mattos MJ, Tempest DW (1999) Growth yield and energy distribution. In: Neidhardt FC (ed) Escherichia coli and Salmonella: cellular and molecular biology, chapter 107, 2nd edn. ASM, Washington, p 10
Noble JB (2001) Scale-up considerations. In: Roe SD (ed) Protein purification techniques, 2nd edn. Oxford University Press, New York, p 262
Ochs MM, Bartlett W, Briles DE, Hicks B, Jurkuvenasa A, Lau P, Ren B, Millar A (2008) Vaccine-induced human antibodies to PspA augment complement C3 deposition on Streptococcus pneumoniae. Microb Pathog 44:204–214. doi:10.1016/j.micpath.2007.09.007
Oliveira MLS, Miyaji EN, Ferreira DM, Moreno AT, Ferreira PCD, Lima FA, Santos FL, Sakauchi MA, Takata CS, Higashi HG, RawI KFS, Ho PL (2010) Combination of pneumococcal surface protein A (PspA) with whole cell pertussis vaccine increases protection against pneumococcal challenge in mice. PLoS One 5(5):e10863. doi:10.1371/journal.pone.0010863
Roush DJ, Lu Y (2008) Advances in primary recovery: centrifugation and membrane technology. Biotechnol Prog 24:488–495. doi:10.1021/bp070414x
Schmidt SA, Williams SJ, Wang CA, Sornaraj P, James B, Kobe B, Dodds PN, Ellis JG, Anderson PA (2007) Purification of the M flax-rust resistance protein expressed in Pichia pastoris. Plant J 50:1107–1117. doi:10.1111/j.1365-313X.2007.03104.x
Seeger A, Schneppe B, McCarthy JEG, Deckwer WD, Rinas U (1995) Comparison of temperature-induced and isopropyl-beta-d-thiogalacto-pyranoside-induced synthesis of basic fibroblast growth-factor in high-cell-density cultures of recombinant Escherichia coli. Enzyme Microb Technol 17:947–953. doi:10.1016/0141-0229(94)00123-9
Shiloach J, Fass R (2005) Growing E. coli to high cell density: a historical perspective on method development. Biotechnol Adv 23:345–357. doi:10.1016/j.biotechadv.2005.04.004
Shukla AA, Hubbard B, Tressel T, Guhan S, Low D (2007) Downstream processing of monoclonal antibodies: application of platform approaches. J Chromatogr B Anal Technol Biomed Life Sci 848:28–39. doi:10.1016/j.jchromb.2006.09.026
Silva M, Cabrera-Crespo J, Sbrogio-Almeida ME, Miyaji EN, Ho PL, Leite LC, Lopes AP (2007) Optimizing expression of Streptococcus pneumoniae surface protein A, PspA: serocross-reactivity within families of antisera induced against clades 1 and 3. Mol Biotechnol 37:146–154. doi:10.1007/s12033-007-0043-z
Son YJ, Bae JY, Chong SH, Lee HS, Mo SH, Kim TY, Choe H (2010) Expression, high cell density culture and purification of recombinant EC-SOD in Escherichia coli. Appl Biochem Biotechnol 162:1585–1598. doi:10.1007/s12010-010-8940-1
Zhao M, Wu M, Guo L, Jiang J, Huang W, Lin X, Zhang Z, Xiang D, Lu H, Zhu S, Yu Y, Moldenhauer A, Han W (2010) Expression, purification, and characterization of a novel soluble form of human delta-like-1. Appl Biochem Biotechnol 160:1415–1427. doi:10.1007/s12010-009-8603-2
Acknowledgements
We would like to thank Dr. Eliane N. Myiaji for kindly providing the microorganism and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Apoio à Pesquisa do Estado de São Paulo (FAPESP) for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Carvalho, R.J., Cabrera-Crespo, J., Tanizaki, M.M. et al. Development of production and purification processes of recombinant fragment of pneumococcal surface protein A in Escherichia coli using different carbon sources and chromatography sequences. Appl Microbiol Biotechnol 94, 683–694 (2012). https://doi.org/10.1007/s00253-011-3649-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3649-9