
Abstract
Salinomycin, a polyether antibiotic produced by Streptomyces albus, is widely used in animal husbandry as an anticoccidial drug and growth promoter. Situated within the salinomycin biosynthetic gene cluster, slnR encodes a LAL-family transcriptional regulator. The role of slnR in salinomycin production in S. albus was investigated by gene deletion, complementation, and overexpression. Gene replacement of slnR from S. albus chromosome results in almost loss of salinomycin production. Complementation of slnR restored salinomycin production, suggesting that SlnR is a positive regulator of salinomycin biosynthesis. Overexpression of slnR in S. albus led to about 25 % increase in salinomycin production compared to wild type. Quantitative RT-PCR analysis revealed that the expression of most sal structural genes was downregulated in the ΔslnR mutant but upregulated in the slnR overexpression strain. Electrophoretic mobility gel shift assays (EMSAs) also revealed that SlnRDBD binds directly to the three intergenic regions of slnQ-slnA1, slnF-slnT1, and slnC-slnB3. The SlnR binding sites within the three intergenic regions were determined by footprinting analysis and identified a consensus-directed repeat sequence 5′-ACCCCT-3′. These results indicated that SlnR modulated salinomycin biosynthesis as an enhancer via interaction with the promoters of slnA1, slnQ, slnF, slnT1, slnC, and slnB3 and activates the transcription of most of the genes belonging to the salinomycin gene cluster but not its own transcription.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Streptomyces are Gram-positive filamentous bacteria that can produce a wide variety of bioactive secondary metabolites. Applications for such metabolites include their uses in antimicrobial or anticancer treatments as well in other agricultural or veterinary end uses (Challis and Hopwood 2003). The biosynthesis of secondary metabolites involves strict regulation at a number of stages including those at global, pleiotropic, and pathway-specific levels (Chen et al. 2008). Pathway-specific regulators only affect the biosynthetic pathway of a single, specific antibiotic. These pathway-specific regulatory genes are usually found within the respective antibiotic biosynthesis gene clusters. In several cases, the overexpression of positive pathway-specific regulatory genes has resulted in elevated antibiotic production (Bibb 2005; Martin and Liras 2010).
The first pathway-specific regulatory proteins to be described in actinomycetes belonged to the protein family known as Streptomyces antibiotic regulatory proteins (SARPs) (Wietzorrek and Bibb 1997). These are characterized by the presence of an OmpR-like DNA-binding domain (Mizuno and Tanaka 1997). The SARP family includes TylS and TylT from the tylosin pathway (Bate et al. 2002), RedD of the undecylprodigiosin pathway (Narva and Feitelson 1990), and ActII-orf4 of the actinorhodin pathway (Arias et al. 1999), among others. Recently, a novel family of transcriptional regulators, the large ATP-binding regulators of the LuxR (LAL) family, was identified (Wilson et al. 2001). The LAL family is characterized by the unusually large size of its members 1′ Walker A or Walker B motif and a conserved helix-turn-helix (HTH) DNA binding motif near the C-terminal end (Walker et al. 1982). So far, several regulators of the LAL family have been identified in Streptomyces antibiotic gene clusters. These include PikD from the pikromycin pathway in S. venezuelae (Wilson et al. 2001), RapH from the rapamycin pathway in S. hygroscopicus (Molnar et al. 1996; EnejKuscer et al. 2007), AveR from the avermectin pathway in S. avermitilis (Guo et al. 2010), and GdmRI and GdmRII from the geldanamycin pathway in S. hygroscopicus 17997 (He et al. 2008). The LAL family regulator can directly and indirectly control the expression of multiple genes (Chao et al. 2015), and many LAL family pathway-specific regulators play positive roles in the production of secondary metabolites (Arias et al. 1999; Mo et al. 2012). The LuxR family regulators usually contain less than 250 residues, constituting two functional domains, a signal-binding domain, and a DNA-binding domain. The C-terminal region contains the DNA-binding domain with a classical HTH domain. There are many examples of LuxR members in streptomycetes which are operon/cluster-specific regulators. For instance, PAS-LuxR protein (PimM in S. natalensis) binding specificity rely on the DBD domain and regulation mechanism is pretty well-studied (Santo-Aberturas et al. 2011). In addition, a subfamily of LuxR-regulators, named, LAL family, consists of members of unusual size (∼900 amino acids), with an ATP/GTP-binding motif close to the N-terminal end and an HTH domain at the C-terminus. Up to now, the reports on the regulation mechanism of LAL family regulators are limited.
Salinomycin from S. albus is a polyether ionophore antibiotic that selectively binds K+ ions and transports them across cell membranes, leading to cell death (Riddell 2002). It is widely used in animal husbandry as an anticoccidial drug and growth promoter (Jiang et al. 2012). Recently, salinomycin has been found to act in the countering of resistance and in the termination of cancer stem cells (Gupta et al. 2009). This provides new therapeutic strategies and opportunities for cancer treatments (Antoszczak and Huczynski 2015). The salinomycin (sal) biosynthetic gene cluster has been sequenced (S. albus XM211, GeneBank JN033543.1 and S. albus DSM 41398, GeneBank HE586118.1), and the coding for the putative regulatory factors involved in salinomycin biosynthesis has been seen to slnR in S.albus XM211 (Jiang et al., 2012), which has another name salJ in s.albus DSM41398 (Yurkovich et al., 2012). Actually, salJ and slnR are two completely homologous genes. SlnR is located downstream of the salinomycin biosynthetic gene cluster (Fig. 1a). Analysis using the NCBI BLASTP program revealed that SlnR is highly homologous to many transcriptional regulators of the LAL family. In the papers that have been reported, members of this family all act as positive pathway-specific regulators to their corresponding biosynthetic clusters (Guo et al. 2010; Mo et al. 2012, Wilson et al. 2001).

Amino acid sequence alignment of SlnR with different proteins of the LAL family. a Gene organization of the salinomycin gene cluster (b) Walker A NTP-binding motif in the N terminal of LAL-family regulators. c LuxR-type HTH DNA-binding motif at the C-terminal of LAL family regulator. The NCBI database accession numbers of the sequences used in this analysis are as follows: AEZ53964.1(SlnR), BAA84600.1(AveR), AAC68887.1(PikD), AAM88362.1(NbmM), AAC38065.1(RapH), and AAY28238.1(HbmRI)
Earlier sequence analysis showed that the salinomycin gene cluster in s.albus DSM41398 is completely homologous with s.albus XM211 (GeneBank JN033543.1 and HE586118.1). It reveals that the sequence of the salinomycin gene cluster is highly conserved. Further studies discovered biosynthesis pathway for salinomycin. The polyketide chain of salinomycin is synthesized by nine sal PKS genes (slnA1 to slnA9) which are collinearly arranged in the cluster (Fig. 1a). These PKS genes are required for the biosynthesis of the salinomycin polyketide skeleton. Upstream of the PKS genes, salN and salO are two defined putative regulatory genes belonging to MarR family. The salP and salQ genes are involved in the formation of the butyrate extender unit for salinomycin biosynthesis (Yurkovich et al., 2012). Downstream of the PKS genes, the cluster is taken to include two genes encoding discrete (type II) thioesterases (slnDI, slnDII), acytochrome P450 (slnF), and associated ferredoxin (slnE); two putative export genes (slnTI, slnTII); three epoxide hydrolases/cyclases (slnBI, slnBII, slnBIII); an epoxidase (slnC); and a putative regulatory gene (slnR) (Jiang et al. 2012). Proposed biosynthesis pathway for salinomycin was shown in Fig. S1.
Although the biosynthesis pathway for salinomycin has been elucidated, the regulation mechanism in salinomycin production remains unknown. The aim of this study is to evaluate the general role of the slnR gene and to aid in the understanding of the pathway-specific regulatory mechanisms of slnR in S. albus. Our results provide evidence for SlnR as a pathway-specific activator for salinomycin biosynthesis. We demonstrate that SlnR serves as a transcriptional activator of the sal biosynthetic cluster, controlling up to 21 of the 25 genes within the sal cluster. We also show that SlnR regulates multiple gene expressions by directly binding to three sites of the intergenic regions of the sal biosynthetic cluster. Overexpression of slnR enhanced salinomycin production in the wild-type S. albus strain by approximately 25 %.
Materials and methods
Strains, plasmids, and growth conditions
S. albus (wild-type strain) was obtained from Zhejiang ShenghuaBiok Biological Company (China). It was grown at 28°C and used as a host strain for gene propagation and gene disruption. ISP4 medium (Difco™, BD) was used for sporulation, and YEME (Kieser et al. 2000) (0.3 % yeast extract, 0.3 % malt extract, 0.5 % tryptone, 4 % glucose) was used for the growth of mycelia for the purposes of extracting DNA. Tryptic soybean broth (3 % TSB) was used as seed medium, and 0.4 % yeast extract, 1 % malt extract, and 0.4 % glucose (YMG) and 10 % soybean oil or industrial medium were used as the fermentation medium. Spores cultivated on ISP4 plates for 7–8 days were added to 250-ml flasks containing 30-ml seed medium. The cultures were then incubated for 28 h at 33 °C on a rotary shaker (220 rpm). Four milliliters of the culture was then transferred to 250-ml flasks containing 40 ml of fermentation medium. The cultures were grown for 10 days at 33 °C on a rotary shaker (245 rpm).
E. coli DH5α and E. coli BL21 (DE3) (Novagen) were used as the cloning host and expression host, respectively. E. coli ET12567 (dam dcmhsds) was used to propagate non-methylated DNA for transformation in S. albus. Bacillus subtilis was used as indicator strain for salinomycin bioassays. E. coli strains were grown at 37 °C in Luria-Bertani (LB) medium. When necessary, the media were supplemented with antibiotics (100 μg/ml for ampicillin; 50 μg/ml each for apramycin, kanamycin, and thiostrepton; and 25 μg/ml for chloromycetin). pIJ8600 was used for the complementation test. pIJ8660, which contains the constitutive and strong expression promoter ermEp*, was used to construct a vector for constitutive slnR expression.
DNA manipulation and sequence analysis
DNA manipulation in E. coli and Streptomyces was performed according to standard procedures of Kieser et al. (2000). All the plasmids and strains used in this study are listed in Table 1, and primers are listed in Tables S1 and S2. S. albus genomic DNA was isolated using the standard procedure (Liu et al. 2015). A homologous sequence database search and multiple alignment analysis were executed using BLASTP, ClustalX, and DNAMAN software. A protein secondary structure analysis was performed using Predict Protein (Rost et al. 2004).
Construction of slnR-deletion mutants
The construction of the slnR::aac(3)IV-oriT mutant was done according to Gust et al. (2003). The fosmid (7G12), harboring the slnR gene, was screened out from the fosmid genomic library of S. albus by PCR amplification using the primer pair PT1/PT2, located 928 nucleotide (nt) upstream and 817 nt downstream of the slnR gene, respectively. The disruption cassette aac(3)IV-oriT (apramycin resistance gene and oriT) was amplified using PCR with the long primer pair tar-F/tar-R (59 nt/58 nt), each of which having at the 5′end a 39-nt matching sequence adjacent to the slnR gene. A HindIII/EcoRI fragment carrying the apramycin cassette aac (3) IV-oriT, which was digested from pIJ773, served as template DNA. The mutagenized 7G12 with an slnR deletion was created by electrotransforming the PCR fragment containing the amplified disruption cassette into E. coli BW25113/pKD46 (with the genes encoding the λRed system) containing fosmid 7G12. Gene deletion was verified by PCR analysis (primers test-F/test-R). The resulting mutated fosmid was first transformed into the non-methylating E. coli ET12567 using plasmid pUZ8002 and then transferred into S. albus by intergeneric conjugation. Apramycin-resistant exconjugants were selected, and then slnR-knockout mutants were confirmed by PCR using primer pair test-F/test-R and sequence analysis.
Complementation of the slnR mutant
Using the genomic DNA of S. albus as the template, a 3260-bp fragment carrying the complete slnR gene and its possible promoter was amplified by PCR using the primers 8600-F/8600-R. The PCR product was then digested by XbaI/KpnI. The 3.2-kb XbaI/KpnI digested fragment was cloned into the integrative vector pIJ8600 to give the complementation plasmid pIJ8600-slnR. By intergenic conjugation, the plasmid pIJ8600-slnR was introduced into the slnR mutant in an S. albus background. This resulted in a thiostrepton-resistant exconjugant. The correct integration in the exconjugants was confirmed by Southern hybridization and PCR analysis.
Overexpression of the slnR gene in S.albus
A 2718-bp fragment carrying the full-length slnR gene was amplified by PCR using the primers 8660-F/8660-R. The PCR fragment was digested with NdeI/KpnI and then inserted into the corresponding sites in an integrative vector pIJ8661 to produce the recombinant vector pIJ8661-slnR. The resultant plasmid was introduced by intergenic conjugation into S. albus. This generated the apramycin-resistant exconjugants. The genotype of the exconjugants was confirmed by PCR analysis.
Expression and purification of protein SlnR-DBD
The DNA-binding domain (DBD) region (residues 803–906) of SlnR was amplified using the primers 2T-F/2T-R. It was then cloned into expression plasmid pGEX2T to generate the plasmid pGEX2T::DBD (pGEX2T-DBD). The overnight culture of E. coli BL21 (DE3), containing either expression vector pGEX2T or pGEX2T-DBD, was inoculated at 1:100 concentration in 200-ml LB with 100 μg/ml ampicillin. Isopropyl-β-d-thiogalactopyranoside (IPTG) was then added to a final concentration of 0.1 mM when the culture was grown to OD600 0.4–0.6 at 37 °C. After a further 18 h of incubation at 16 °C, the cells were harvested by centrifugation and disrupted by sonication on ice. The supernatant was then recovered by centrifugation (4 °C, 13,000×g for 30 min). Soluble GST and GST-SlnRDBD were purified with GST-binding resin, respectively, as described by the manufacturer’s protocol (Novagen, USA).
Electrophoretic mobility gel shift assays
Promoter regions were amplified by PCR (the primers are listed in Table S1), then cloned into a pTA2 vector and verified by sequence analysis. Biotin-labeled probes used for electrophoretic mobility gel shift assays (EMSAs) were obtained by PCR using the biotin-labeled T3/T7 universal primer pair and were then gel-purified. A standard binding reaction contained 1 ng of labeled DNA probe and incubated with 0, 0.2, 0.4, 0.8, and 1 μM of purified GST-SlnRDBD, respectively, at 30 °C for 30 min in a binding buffer (10 mM Tris–Cl (pH 8.0), 100 mM Na2HPO4, 50 μg/ml sheared sperm DNA). EMSA analysis then followed as previously described (Liu et al. 2015).
DNase I footprinting assays
DNase I footprinting assays were performed using a fluorescence labeling procedure (Zianni et al. 2006). DNA fragments were prepared by PCR using the fluorescence-labeled primer 28 forward and primer 29 reverse (Table S1) and were purified from the agarose gel. Labeled DNA fragments (70 ng) and purified GST-SlnRDBD protein (8 μM) were added to a final reaction mixture volume of 50 μl and incubated at 25 °C for 30 min. DNase I digestions (0.01 U of DNaseI with 10 mM MgCl2 and 1 mM CaCl2) were carried out for 1 min at 25 °C and stopped with 50 μl of 0.1 M EDTA (pH 8.0). After phenol-chloroform extraction and ethanol precipitation, the samples were loaded into an ABI 3130 sequencer and electropherograms were analyzed using the Genemapper v4.0 software (Applied Biosystems, USA) to align and determine the protected region. A DNA sequencing ladder was prepared as previously described (He et al. 2010; Liu et al. 2015). The alignment and consensus of the binding sequences was obtained by clustalX software.
RNA extraction and quantitative RT-PCR analysis
The total RNA of S. albus strains was prepared from mycelium grown in YMG liquid cultures using an RNA extraction kit (Ailaide, China) following the manufacturer’s instructions. To avoid chromosomal DNA contamination, the RNA sample was treated with DNaseI (Takara, Japan) and subsequently confirmed by PCR using three different primer pairs. The RNA concentration was subsequently determined by measuring the A260 using a spectrophotometer (Thermo ND LITE, USA). The relative expression level was calculated using the comparative threshold cycle (C t ) method. Results were normalized to the expression of hrdB. Each experiment was performed in triplicate.
The DNase I-treated RNA sample (2 μg) was used as a template for complementary DNA (cDNA) preparation using M-MLV reverse transcriptase (Takara, Japan). Quantitative real-time PCR (qRT-PCR) was carried out using a SYBR Premix Ex Taq kit (Takara, Japan) according to the manufacturer’s instructions. The synthesized cDNA was then used for qRT-PCR using sequence-specific primers (listed in Table S1). The hrdB gene, which encodes the major σ factor in Streptomyces, was used as the internal control. The fold changes of target gene expression were quantified with the 2−ΔΔct method, and the mRNA level of the genes in the WT strain was assigned a value of 1.
Salinomycin bioassay and HPLC analysis
Salinomycins, produced by either S. albus or the mutant strains, were measured using a disk agar diffusion method with B. subtilis as the indicator strain. For HPLC analysis, the fermentation broth (5 ml) was extracted with 45 ml of methanol for 30 min and centrifuged at 12,000×g for 10 min. The supernatant was directly applied to a high-performance liquid chromatography (HPLC) system (Aglient 1260 infinity) with a C18 column (Eclipse XDB-C18, 4.6 × 150 mm) developed with methanol (91 %) and 0.2 M NaAC (pH 5.8) (9 %) at a flow rate of 1.0 ml/min. Salinomycin was detected by UV absorption at 210 nm where authentic samples of salinomycin were used as internal standards.
Results
SlnR, a cluster-situated regulator, positively regulates salinomycin production
The entire sal biosynthetic gene cluster contains 25 ORFs spanning a distance of 104 kb (Yurkovich et al., 2012; Jiang et al. 2012) (Fig. 1a; Fig. S1). Computer-assisted analysis of the slnR gene for the putative regulator SlnR has revealed that slnR contains 2721 nucleotides and encodes a protein of 906 amino acids (with a predicted molecular mass of 97 kDa). Initial analysis using the NCBI BLASTP program showed that this protein is homologous to many other putative transcriptional regulators of the LAL family. Further analysis of SlnR was performed using the SwissProt protein motif identification software. Query returns revealed a hypothetical N-terminal NTP-binding domain (a.a. 3–127) and Walker A motif (GXXGXGKT) (a.a.32–39) (Fig. 1b), as well as a possible C-terminal HTH DNA-binding domain (a.a.843–893) belonging to the LuxR family (Evelyne Richet and Olivier Raibaud 1989; Wilson et al. 2001) (Fig. 1c). Members of this family almost always act as positive pathway-specific regulators to their corresponding biosynthetic clusters (Wilson et al. 2001; Widdick et al. 2003).
In order to begin probing SlnR’s function, the slnR gene was deleted from the chromosome using an apramycin resistant gene (aac(3)IV) replacement cassette (Fig. 2a). The slnR deletion mutant strain (named S. albus slnRDM) was identified by PCR (Fig. S2). To confirm that the deletion of slnR was the sole reason for the loss of salinomycin production, a 3.2-kb fragment containing slnR and its own promoter region was reintroduced into slnRDM by the pIJ8600 plasmid. The complementation strain (named S. albus slnRCOM) restored salinomycin production (Fig. S4), suggesting that the slnR gene is required for salinomycin biosynthesis. Meanwhile, morphological observation revealed that the morphological characteristics of slnRDM strain are identical to wild type of S. albus when grown on solid media (Fig. S3). It means that SlnR, as a pathway-specific regulator, cannot affect the morphological differentiation of S. albus.

Overexpressing slnR improves salinomycin production (a) time course of salinomycin production in WT, slnR-deletion strain (slnRDM), and slnR overexpression strain (slnROE). b Growth curves in liquid culture
Salinomycin bioassays using Bacillus subtilis as an indicator strain lacked any inhibition zone in the slnRDM strain in contrast to the wild type where an obvious inhibition zone was observed (Fig. S4). Furthermore, HPLC detection of salinomycin production during fermentation revealed a lack of salinomycin peaks in the fermentation culture of the slnRDM strain. In contrast, the wild-type strain, slnRCOM, and slnR overexpression strain (named S. albus slnROE) produced obvious salinomycin peaks (Fig. S5). It indicated that salinomycin production level was restored in slnRCOM strain.
Many LAL family pathway-specific positive regulators play key roles in the production of secondary metabolites. For instance, PikD of the pikromycin cluster of S. venezuelae (Wilson et al. 2001) is the pathway-specific activator in that particular biosynthetic gene cluster. AveR of the avermectin cluster in S. avrmitilis (Guo et al. 2010) is also described as a positive pathway-specific regulator. SlnR, which has been assigned to the LAL family, has also now been discovered to positively regulate salinomycin production in this work.
SlnR protein binds specifically to the intergenic region of slnQ-slnA1, slnF-slnT1, and slnC-slnB3
The recombinant soluble GST-SlnRDBD was expressed in E. coli BL21 (DE3) and has been purified by GST binding resin (Fig. S6a). In addition, soluble GST protein has also been purified and used as control protein (Fig. S6b). In electrophoretic mobility shift assays (EMSAs), we designed eight biotin-labeled probes (probes 1–8) containing all possible promoter regions for the EMSAs (Fig. 3a). Of the eight probes tested, probe 3 (308 bp of the slnQ-slnA1 intergenic region), probe 4 (248 bp of the slnF-slnT1 intergenic region), and probe 5 (240 bp of the slnC-slnB3 intergenic region) all gave retarded signals (Fig. 3b). However, no retarded signals occurred with probes 1, 2, 6, 7, 8, or 9 (pTA2 vector sequence, as a negative control) (Fig. S7). Moreover, the GST protein also did not bind to any of the promoters tested (Fig. S8). It confirms the binding specificity of SlnR to the three promoter regions.

Gel electrophoretic mobility shift assay (EMSA) for determination of SlnR binding sites. a Schematic representation of the probes used for EMSAs. The lengths and positions of the eight probes are shown. b EMSAs with probes 3, 4, and 5. Each lane contained 1 ng probe. Lanes 1 to 5 contain 0, 0.2, 0.4, 0.8, and 1 μM of purified GST-SlnRDBD, respectively
To determine the SlnRDBD binding sequence, the intergenic regions shown above to be retarded in EMSAs were studied by DNase I protection analysis. GST-SlnRDBD protein (8 μM) was tested using 5′-end FAM-labeled DNA fragment. The DNase I footprinting assay revealed that there were one binding sites for probe 3, probe 4, and probe 5 (Fig. 4a, c, e) Nucleotide sequences of the slnQ-slnA1, slnF-slnT1, and slnC-slnB3 intergenic regions and the SlnR-binding sites are showed in Fig. 4b, d, f. Nucleotide sequence consensus of three binding sites is shown in Fig. 4g. These sites were analyzed and identified a consensus-directed repeat sequence 5′-ACCCCT-3′. In order to validate the consensus binding site, three mutant DNA probes were constructed with the repeat sequence (5′-ACCCCT-3′) deleted. The binding assay was then studied by EMSAs. Interestingly, the mutant probes cannot form a complex with the SlnR protein (Fig. S9). This result validates the proposed binding sites.


DNase I footprinting assay for SlnR binding site determination 5′-FAM-labeled probe3 amplified from pTA2-A1 (a), 5′-FAM-labeled probe 4 amplified from pTA2-F (c), and 5`-FAM-labeled probe 5 amplified from pTA2-B3 (e) were used in the DNase I footprinting assay with or without purified GST-SlnRDBD (8 μM), respectively. The protected region is underlined. Nucleotide sequences of the slnQ-slnA1 intergenic region (b), slnF-slnT1intergenic region (d), and slnC-slnB3intergenic region (f); the SlnR-binding sites are overlined and the initiation codons are marked with arrows. g Nucleotide sequence consensus of three binding sites. A consensus-directed repeat sequence 5′-ACCCCT-3′ was displayed with underline
Therefore, the SlnR binding sites were seen to be located at the slnQ-slnA1, slnF-slnT1, and slnC-slnB3 intergenic regions, and the hypothetical regulatory pathway of SlnR related to salinomycin production in S. albus was shown in Fig. 6. In addition, SlnR cannot bind to probe 6 or probe 7 which infers that SlnR cannot regulate its own expression.
There have been few previous reports of soluble LAL regulators which have been expressed in the E. coli system for its large molecular weight and complex structures. Understanding the regulation mechanism of this novel regulatory protein family by EMSA is therefore quite a challenge. Only one example of a truncated form of AveR, containing the C-terminal HTH domain (AveRc) fused to a his6-tag at its N-terminus, has been previously noted to have been expressed as an inclusion body (Guo et al. 2010; Kitani et al. 2009). As the inclusion body has no native protein structure, it is very difficult to verify the function of its DNA-binding domain by EMSA. In this study, a soluble GST-SlnRDBD (residues 803-906) was successfully expressed and EMSAs have been used here to prove the direct binding of the SlnRDBD to the multiple binding sites in its target promoters.
Regulation of SlnR on expression of salinomycin biosynthetic genes
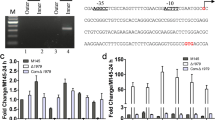
The abolished production of salinomycin in the slnR-deletion mutant (slnRDM) and the overproduction of salinomycin in the slnR-overexpression strain (slnROE) both indicate the possible positive regulatory roles of SlnR in the pathway. To test whether SlnR regulates salinomycin production through affecting the transcriptional levels of sal biosynthetic genes, we performed quantitative RT-PCR (qRT-PCR) analysis. As compared to those of the wild-type strain, the transcript levels of most of the genes (slnQ, slnA1, slnF, slnT1, slnB3 and slnC), with the exception of slnN, slnO, and slnD2, were greatly decreased in the slnRDM strain (Fig. 5a). In contrast, most of the genes’ expression levels (including slnO, slnQ, slnA1, slnF, slnT1, slnB3, slnC, slnR, and slnD2) were obviously increased in the slnROE strain (Fig. 5b). However, slnN transcript level has little change whether in the slnRDM or slnROE strains.

Compared to wild-type strain (S. albus), fold changes of relative mRNA expression of salinomycin biosynthetic genes in different strains by qRT-PCR. The mRNA transcription level of genes in WT was assigned a value of 1 (a) slnR-deletion strain (slnRDM) and WT; b slnR-overexpressed strain (slnROE) and WT
The above results demonstrate the correlation between qRT-PCR and three binding sites. From the hypothetical scheme of the regulatory pathway of SlnR (Fig. 6), we can see the transcription direction of all genes in sal cluster. The three binding sites, which were located in the three intergenic regions, just situated in three important DNA regions. The promoters in three intergenic regions can control most of gene transcription in sal cluster according to the transcriptional direction.

Hypothetical scheme of the regulatory pathway of SlnR related to salinomycin production in S. albus
Overexpression of slnR leads to overproduction of salinomycin
In general, overexpression of a pathway-specific transcriptional activator leads to increased production of the corresponding antibiotic (Pan et al. 2011). To test this possibility, the slnR gene was cloned into the integration vector (pIJ8661) containing the aac3 (IV) gene and ermE*p while empty pIJ8661 also as control. The slnR-over expression plasmid pIJ8661-slnR and empty pIJ8661 were then introduced into the wild-type strain. The slnR overexpressed strain (slnROE) was confirmed by quantitative RT-PCR analysis and analyzed for its effects on cell growth, bioassay, and salinomycin productivity in the fermentation medium. The production levels of salinomycin in slnROE were performed by comparing antibacterial activity using an agar diffusion assay with B. subtilis as the indicator strain (Chen et al. 2008). The results showed that samples from the pIJ8661-slnR transformant had a much larger growth inhibition zone than did that of the wild-type strain (Fig. S4).
Shake-flask fermentation followed by HPLC analysis showed that the overexpression strain had raised salinomycin production by 10 % (at 4 days) to 25 % (at 7 days) as compared to the wild type (Fig. 2a). The cell concentration of the overexpression strain, and the wild type was measured every 24 h during the fermentation process. No apparent difference in the dry cell weight between the overexpression strain and wild type was observed (Fig. 2b). However, the salinomycin productions of two control strains which contain empty pIJ8600 or pIJ8661 have no significant change compared with the WT strain (Fig. S10).
Discussion
This study provides a valuable initial understanding of the first pathway-specific regulatory gene, slnR, in S. albus. We demonstrated that a LAL family regulator, SlnR, has a positive effect on the salinomycin production. EMSA results indicated that the SlnR protein could bind three intergenic regions and control up to 21 of the 25 genes within the sal cluster. The binding sites also have been identified by DNaseI footprinting assay. Analysis of the SlnR-binding region revealed consensus-directed repeat sequence (5′-ACCCCT-3′); it may be a feature of the SlnR-binding target. In addition, the significantly enhanced production of salinomycin (up to 25 %) in the genetically engineered strain slnROE in which the positive regulatory gene was overexpressed in the background of wild-type strain demonstrates the potential of this approach for strain development (Pan et al. 2011). It may be possible to upregulate the entire salinomycin antibiotic pathway to further increase the production of known metabolites.
Up to now, numerous regulatory proteins involved in antibiotic production have been identified in Streptomyces. Pathway-specific regulators specifically regulate the transcription of certain biosynthetic genes (Wietzorrek and Bibb 1997). Many pathway-specific regulators belong to SARP family proteins (e.g., ActII-orf4, RedD, and TylS), and the mechanism of action is very clear (Arias et al. 1999; Bate et al. 2002 ). On the other hand, the LAL family proteins of pathway-specific regulators have been reported limited in recent years (Wilson et al. 2001; Kitani et al. 2009), and little is known about their precise mechanisms of regulation. The large size of LAL family regulator is a “bottleneck” in high-level expression and purification of intact LAL protein (Guo et al. 2010; Santos-Aberturas et al. 2011). This makes it more difficult to understand the regulation mechanism of this novel regulatory family. In this study, GST-SlnRDBD fusion proteins were successfully expressed and used for EMSAs. The DNA-binding domain (SlnRDBD) was found to bind with high affinity to the three intergenic regions, thus suggesting that binding specificity is dependent of the DBD domain. However, the effect of N-terminal NTP-binding motif of SlnR in regulation of salinomycin production is known little. The ATPase/GTPase activity of SlnR N-terminus may affect the DNA-binding of SlnR and its target promoters; it also needs further research and exploration. This report provides important clues toward understanding the pathway-specific regulation of salinomycin biosynthesis and provides a useful means for controlling the expression of genes involved in the salinomycin biosynthesis.
References
Antoszczak M, Huczynski A (2015) Anticancer activity of polyether lonophore-salinomycin. Anti Cancer Agents Med Chem 5:575–591
Arias P, Fernandez-Moreno MA, Malpartida F (1999) Characterization of the pathway-specific positive transcriptional regulator for actinorhodin biosynthesis in Streptomyces coelicolor A3(2) as a DNA-binding protein. J Bacteriol 181:6958–6968
Bate N, Stratigopoulos G, Cundliffe E (2002) Differential roles of two SARP-encoding regulatory genes during tylosin biosynthesis. Mol Microbiol 43:449–458
Bibb M (2005) Regulation of secondary metabolism in Streptomyces. Curr Option Microbiol 8:208–215
Challis GL, Hopwood DA (2003) Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci U S A 100:14555–14561
Chen Y, Wendt E, Shen B (2008) Identification and utility of FdmR1 as a Streptomyces antibiotic regulatory protein activator for fredericamycin production in Streptomyces griseus ATCC49344 and heterologous hosts. J Bacteriol 190:5587–5596
Guo J, Zhao J, Li L, Chen Z, Wen Y, Li J (2010) The pathway-specific regulator AveR from Streptomyces avermitilis positively regulates avermectin production while it negatively affects oligomycin biosynthesis. Mol Gen Genomics 283:123–133
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES (2009) Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 4:645–659
Gust B, Challis GL, Fowler K, Kieser T, Chater KF (2003) PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A 4:1541–1546
He W, Lei J, Liu Y, Wang Y (2008) The LuxR family members GdmRI and GdmRII are positive regulators of geldanamycin biosynthesis in Streptomyces hygroscopicus 17997. Arch Microbiol 189:501–510
He X, Li R, Pan Y, Liu G, Tan H (2010) SanG,a transcriptional activator, controls nikkomycin biosynthesis through binding to the sanN-sanO intergenic region in Streptomyces ansochromogenes. Microbiology 156:828–837
Jiang C, Wang H, Kang Q, Liu J, Bai L (2012) Cloning and characterization of the polyether salinomycin biosynthesis gene cluster of Streptomyces albus XM211. Appl Environ Microbiol 4:994–1003
Kieser TBM, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich
Kitani S, Ikeda H, Sakamoto T, Noguchi S, Nihira T (2009) Characterization of a regulatory gene, aveR, for the biosynthesis of avermectin in Streptomyces avermitilis. Appl Microbiol Biotechnol 82:1089–1096
Kuscer E, Coates N, Challis J, Gregory M, Wilkinson B, Sheridan R, Petkovie H (2007) Role of rapH and rapG in positive regulation of rapamycin biosynthesis in Streptomyces hygroscopicus. J Bacteriol 189:4756–4763
Liu S-P, Yu P, Yuan P-H, Zhou Z-X, Bu Q-T, Mao X-M, Li Y-Q (2015) Sigma factor WhiGch positively regulates natamycin production in Streptomyces chattanoogensis L10. Appl Microbiol Biotechnol 99:2715–2726
Martin JF, Liras P (2010) Engineering of the regulatory cascades and networks controlling antibiotic biosynthesis in Streptomyces. Curr Option Microbiol 13:263–273
Mizuno T, Tanaka I (1997) Structure of the DNA-binding domain of the OmpR family of response regulators. Mol Microbiol 24:665–667
Mo SJ, JiYoo Y, Ban YH, Lee S-K, Kim E, Suh J-W, Yoon YJ (2012) Roles of fkbN in positive regulation and tcs7 in negative regulation of FK506 biosynthesis in Streptomyces sp. strain KCTC 1604BP. Appl Environ Microbiol 7:2249–2255
Molnar J, Aparicio JF, Haydock SF, Khaw LE, Schwecke T, konig A, Staunton J, Leadlay PF (1996) Organisation of the biosynthetic gene cluster for raamycin in Streptomyces hygroscopicus: analysis of genes flanking the polyketide synthase. Gene 169:1–7
Narva KE, Feitelson JS (1990) Nucelotide sequence and transcriptional analysis of the redD locus of Streptomyces coelicolor A3(2). J Bacteriol 172:326–333
Pan Y, Wang A, He X, Tian Y, Liu G, Tan H (2011) SabR enhances nikkomycin production via regulating the transcriptional level of sang, a pathway-specific regulatory gene in Streptomyces ansochromogenes. BMC Microbiol 11:164–170
Richet E, Raibaud O (1989) MalT, the regulatory protein of the Escherichia coli maltose system, is an ATP-dependent transcriptional activator. EMBO J 8:981–987
Riddell FG (2002) Structure, conformation, and mechanism in the membrane transport of alkali metal ions by ionophoric antibiotics. Chirality 14:121–125
Rost B, Yachday G, Liu J (2004) The PredictProtein server. Nucleic Acids Res 32:W321–W326
Santo-Aberturas J, Vicente CM, Guerra SM, Payero TD, Martin JF, Aparicio JF (2011) Molecular control of polyene macrolide biosynthesis direct binding of the regulator PimM to eight promoters of pimaricin genes and identification of binding boxes. J Biol Chem 286:9150–9161
Santos-Aberturas J, Payero TD, Vicente CM, Guerra SM, Cañibano C, Martín JF, Aparicio JF (2011) Functional conservation of PAS-LuxR transcriptional regulators in polyene macrolide biosynthesis. Metab Eng 13:756–767
Sun J, Kelemen GH, Fernández-Abalos JM, Bibb MJ (1999) Green fluorescent protein as a reporter for spatial and temporal gene expression in Streptomyces coelicolor A3(2). Microbiology 145(9):2221–2227
Walker JE, Saraste M, Runswick MJ, Gay NJ (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951
Widdick DA, Dodd HM, Barraille P, White J, Stein TH, Chater KF, Gasson MJ, Bibb MJ (2003) Cloning and engineering of the cinnamycin biosynthetic gene cluster from Streptomyces cinnamoneus DSM40005. Proc Natl Acad Sci U S A 7:4316–4321
Wietzorrek A, Bibb M (1997) A novel family of proteins that regulates antibiotic production in stretomycetes appears to contain an OmpR-like DNA-binding fold. Mol Microbiol 25:1177–1184
Wilson DJ, Xue Y, Reynolds KA, Sherman DH (2001) Characterization and analysis of the PikD regulatory factor in the pikromycin biosynthetic pathway of streptomyces venezuelae. J Bacteriol 183:3468–3475
Xie C, Deng J-J, Wang H-X (2015) Identification of AstG1, a LAL family regulator that positively controls ansatrienins production in Streptomyces sp.XZQH13. Curr Microbiol 70:859–864
Yurkovich ME, Tyrakis PA, Hong H, Sun Y, Samborskyy M, Kamiya K, Leadlay PF (2012) A late-stage intermediate in salinomycin biosynthesis is revealed by specific mutation in the biosynthetic gene cluster. Chembiochem 13:67–71
Zianni M, Tessanne K, Merighi M, Laguna R, Tabita FR (2006) Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J Biomol Tech 17:103–113
Acknowledgments
This study was supported by the National Basic Research Program of China (No. 2012CB721005), National High Technology Research and Development Program of China (No. 2012AA022107), and Zhejiang Provincial Natural Science Foundation of China (Nos. LY16C010001, LZ12C01001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(PDF 962 kb)
Rights and permissions
About this article

Cite this article
Zhu, Z., Li, H., Yu, P. et al. SlnR is a positive pathway-specific regulator for salinomycin biosynthesis in Streptomyces albus . Appl Microbiol Biotechnol 101, 1547–1557 (2017). https://doi.org/10.1007/s00253-016-7918-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7918-5