
Abstract
The function of the regulatory protein AveR in Streptomyces avermitilis was examined. An aveR deletion mutant abolished avermectin production and produced more oligomycin, and its phenotype was complemented by a single copy of the aveR gene. Removal of the C-terminal HTH domain of AveR abolished avermectin biosynthesis, indicating the importance of HTH domain for AveR function. Promoter titration and promoter probe assays suggested that the transcription of aveA1, encoding polypeptide AVES1 of avermectin PKS, was activated by AveR. Chromatin immunoprecipitation (ChIP) assay showed that the predicted promoter regions of both the ave cluster and the olm cluster were target sites of AveR, and the DNA-binding activity of AveR was dependent on its HTH domain. RT-PCR analysis revealed that the transcriptions of ave structural genes were dependent on AveR, but that of olm structural genes and putative pathway-specific regulatory genes increased in the aveR mutants. Consistent with these observations, overexpression of aveR successfully increased avermectin production. These results indicated that aveR encodes a pathway-specific activator essential for avermectin biosynthesis and it also negatively affects oligomycin biosynthesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Streptomycetes are Gram-positive, filamentous soil bacteria with complex life cycle and ability to produce a wide variety of secondary metabolites, including many commercially important antibiotics. The production of secondary metabolites involves the expression of clustered biosynthetic genes that are activated in a growth phase-dependent manner. This expression pattern is often controlled by pathway-specific regulatory genes (Bibb 2005). Analysis of these regulatory genes is crucial for understanding the mechanism of regulation and for construction of overproducing strains.
Avermectins, a series of potent anthelmintic and insecticidal macrolide antibiotics (A1a, A1b, A2a, A2b, B1a, B1b, B2a, and B2b) produced by Streptomyces avermitilis, are used widely in the medical, veterinary, and agricultural fields (Burg et al. 1979; Ikeda and Omura 1997). S. avermitilis also produces small amount of a separate family of macrolide antibiotics called oligomycins, which are strongly toxic compounds (Pinna et al. 1967; Omura et al. 2001). Although the S. avermitilis genome has been sequenced (Omura et al. 2001; Ikeda et al. 2003) and the avermectin biosynthetic pathway has been elucidated (Ikeda and Omura 1997; Ikeda et al. 1999, 2001), most of the regulatory factors and mechanisms in avermectin production remain unknown. Genes coding for regulatory factors involved in avermectin biosynthesis include aveR (Ikeda et al. 1993, 1999; Kitani et al. 2009), aveR1/aveR2 (Stutzman-Engwall and Price 2001), orfX (Hwang et al. 2003), afsK-av (Rajkarnikar et al. 2006), and aveI (Chen et al. 2008). aveR, located at the far left of the avermectin biosynthetic gene cluster (Fig. 1), encodes a putative pathway-specific regulator. aveR mutants generated by Tn4560 mutagenesis did not produce avermectins and failed to convert any intermediates after aglycon formation (Ikeda et al. 1993). Sequence analysis revealed that the product of aveR was a LAL family regulator (large ATP-binding regulators of the LuxR family) (Wilson et al. 2001; Kitani et al. 2009), which contains an N-terminal NTP binding domain and a C-terminal LuxR-like helix-turn-helix DNA-binding domain. The role of potential regulatory protein AveR regarding the onset of the avermectin is recently described to be positive by gene disruption and feeding experiments, while a higher amount of aveR resulted in complete loss of avermectin (Kitani et al. 2009). In this paper, evidence for AveR as a pathway-specific activator for avermectin biosynthesis as well as a negative regulator for oligomycin biosynthesis is given, and in contrast to the findings of Kitani et al. (2009) overexpression of aveR enhanced avermectin production in wild-type strain S. avermitilis ATCC31267.

Organization of the gene cluster for avermectin biosynthesis. Direction of transcription and relative sizes of ORFs are indicated (Ikeda et al. 1999); BI–BVIII: aveBI–aveBVIII. Numbers indicate putative promoter regions identified in the present study
Materials and methods
Strains, plasmids, and growth conditions
Streptomyces avermitilis ATCC31267 (wild-type strain) was grown at 28°C and used as a host strain for gene propagation and gene disruption. Solid YMS medium (Ikeda et al. 1988) and liquid YEME (Kieser et al. 2000) medium with 25% sucrose were used for sporulation and growth of mycelia for the purposes of extracting DNA and preparing protoplasts, respectively. Seed medium and fermentation medium (Chen et al. 2007) were used for avermectin production. Soluble fermentation medium II (5% soluble starch, 1.2% yeast extract, 0.05% K2HPO4·3H2O, 0.05% MgSO4·7H2O, 0.4% KCl, 0.0005% CoCl2·6H2O) was used to cultivate mycelia for growth, ChIP, and RT-PCR analysis. RM14 (MacNeil and Klapko 1987) was used for regeneration of protoplasts and for selection of transformants. E. coli DH5α and E. coli BL21 (DE3) (Novagen, Shanghai, China) were used as cloning host and expression host, respectively. E. coli ET12567 (dam dcm hsdS) (MacNeil and Klapko 1987) was used to propagate non-methylated DNA for transformation in S. avermitilis. E. coli strains were grown at 37°C in Luria–Bertani (LB) medium, and transformed as described by Sambrook et al. (1989). S. lividans TK54 was grown at 30°C on YMS agar and used as a host strain for promoter probe assay. Antibiotics used in this study were described previously (Zhao et al. 2007).
Plasmid pIJ2925 (Janssen and Bibb 1993) was used for routine cloning and subcloning experiments. pKC1139 (Bierman et al. 1992) was used to construct gene disruption mutants via homologous recombination. pSET152 (Bierman et al. 1992) was used to introduce a single copy of aveR into S. avermitilis. pIJ963 (Kieser et al. 2000) was used to provide hygromycin resistance gene (hyg) for constructing gene replacement vector. Streptomyces high copy number (50–100) vector pIJ486 (Kieser et al. 2000) containing reporter gene neo was used to construct plasmids for promoter titration and promoter probe assay. Vector pIJ4090 (Kieser et al. 2000) was used to provide the Streptomyces strong constitutive promoter ermE*p. pET-28a (+) (Novagen), a cloning/expression vector for conferring N-terminal His6 tag on expressed proteins, was used for gene overexpression in E. coli. TA vectors pMD18-T (TaKaRa, Dalian, China) and pGEM®-T Easy (Promega, Beijing, China) were used for cloning PCR products.
Construction of aveR deletion mutants
Using genomic DNA of ATCC31267 as template, a 0.5-kb fragment upstream of the aveR start codon was amplified by PCR with primers 93 (CTGAAGCTTACGGCATCACGCTCACTGC) and 94 (CTGAGATCTCACGAGTGGGATG CGCAGT) containing engineered sites HindIII and BglII, and a 0.5-kb fragment downstream of the aveR stop codon was amplified with primers 95 (AGTCTGCAGCGTCTTCCGCCTCACGACATG) and 96 (ACTGAATTCACACCCTGTTCGCAGAAGTCGAG) containing PstI and EcoRI. The two PCR fragments were digested with HindIII/BglII and PstI/EcoRI, respectively. The 1.7-kb hygromycin resistance gene (hyg) was excised from pIJ963 by PstI/BglII digestion. These three fragments were simultaneously ligated into HindIII/EcoRI-digested pKC1139 to generate pDIaveR (Fig. 2a).

Schematic representation of the strategy used for deletion of the entire aveR gene (a), and its 3′-terminus (b), respectively. Long broad arrows indicate genes and their directions. Short small arrows indicate positions of primers used for cloning exchange regions and confirming gene deletions, as described in “Materials and methods”. Double-crossover recombination led to replacement of aveR with hygromycin resistance gene hyg, and in-frame deletion of the 3′-terminus of aveR, which encodes the C-terminal HTH domain of AveR. The aveR deletion mutant was named as AveR-D19. The fusion in the truncated aveR used the original stop codon for aveR
Transformation of pDIaveR and selection of double-crossover recombination strains were performed as described previously (Zhao et al. 2007). The aveR-deleted mutants were confirmed by PCR analysis using primers 117 (GGTCACCGTGATCGTCACG), hhz (CCATCCCAGCTCGGCAAG), 118 (CTGGTGGCCGTTCACTACG) and hqf (CGGGATCGCCAATCTCTAC) (Fig. 2a). Primers 117 and 118 flank the exchange regions, while primers hhz and hqf are specific for hyg. When primer pairs 117/hhz and 118/hqf, which are specific for replacement of aveR with hyg, were used for PCR analysis of putative aveR deletion mutants, a 0.64-kb band and a 0.95-kb band appeared, respectively, whereas such bands were not detected when genomic DNA of wild-type strain ATCC31267 was used as the template. In contrast, when primers 1 (AACCATATGCAGGGAGTTTCCTGTC) and 2 (GCGGAATTCATGTCGTGAGGCGGAAG) located within the deletion region of aveR were used, only the wild-type strain produced 2.9-kb PCR fragment as predicted (data not shown). These results indicate that aveR deletion mutants (S. avermitilis AveR-D19) were obtained in which the aveR gene was completely replaced with hyg by double-crossover recombination.
Complementation of aveR deletion mutants
A 3.1-kb DNA fragment carrying the promoter and coding region of aveR was amplified with primers 1* (ACGGATGTCTCCAGGAAGG) and 2* (GGTCAGTTGGCCTGGTCCG) by PCR using genomic DNA of ATCC31267 as the template, and was then cloned into pGEM®-T Easy vector to produce pCZ10. The sequence of aveR in pCZ10 was verified by nucleotide sequencing. The 3.1-kb SphI/SpeI fragment of aveR was excised from pCZ10 and inserted into SphI/XbaI-digested pIJ2925 to produce pCZ11. The 3.1-kb BglII fragment of aveR from pCZ11 was cloned into the BamHI site of pSET152 to give complementation plasmid pCZ13, which was integrated into the chromosome of S. avermitilis after transformation. The 3.1-kb EcoRI/HindIII fragment containing aveR from pCZ11 was cloned into the corresponding sites of pKC1139 to produce pCZ12 (Fig. S1A), which was used to introduce multi-copies of aveR into S. avermitilis.
Deletion of the 3′-terminal portion of aveR
A 0.5-kb DNA fragment upstream of the coding region for C-terminal HTH domain of AveR was amplified by PCR with primers 71 (ATCAAGCTTCCGCGACTTCCTCACCG, HindIII) and 72 (ACTGAGCTCGGACTCGCTCAGCAG, SacI). A 0.5-kb DNA fragment downstream of the HTH coding region was also amplified with primers 73 (ACTGAGCTCCGACACCTCGCGGACCAG, SacI) and 74 (GCTGAATTCTGTTCAGGATCAATGTGCG, EcoRI). These two PCR fragments were then digested with HindIII/SacI and SacI/EcoRI, respectively, and were simultaneously ligated into HindIII/EcoRI-digested pKC1139 to generate pDHTH (Fig. 2b). The deletion has been controlled by DNA sequencing, which confirmed the desired in-frame deletion of 51 codons and the usage of the original aveR stop codon.
pDHTH was transformed into ATCC31267, and the expected deletion mutants were selected using the same strategy as for selection of aveR deletion mutants and confirmed by PCR analysis. When primers 69 (GACGCCAAGGAGTGCGC) and 70 (ACACCCTGTTCGCAGAAGTCGAG) flanking the exchange regions were used, the mutant strains generated a 1.35-kb PCR product (Fig. 2b), whereas the wild-type strain generated a 1.5-kb PCR product (data not shown). These results indicate that an internal 150-bp fragment of aveR, which encodes the C-terminal HTH domain, was deleted in these mutants.
Construction of plasmids for promoter titration and promoter probe assay as well as for overproduction of AveR
A 327-bp DNA fragment containing the putative aveA1 promoter was amplified by PCR with primers 6 (ATCATGGTCGGGAACCTCC) and 7 (TCACCGCTAGGCAATGCTC), and was cloned into pMD18-T to produce pJL1311. The 327-bp fragment was cut from pJL1311 with BamHI/HindIII and inserted into BamHI/HindIII-digested pIJ486 to generate pJL62, in which the 327-bp aveA1 promoter was located upstream of the reporter gene neo. The recombinant plasmid pJL62 was transformed into protoplasts of ATCC31267 for promoter titration assay, and was also used in promoter probe assay to detect activity of aveA1 promoter.
In order to enhance expression of AveR in Streptomyces, the plasmid pJL66 was constructed. There is a rare TTA codon 1390–1392 nucleotides downstream of the start codon of aveR. To change the TTA leucine codon to a CTG leucine codon, site-directed mutagenesis was performed in vitro by PCR. Two primer pairs were designed: primers 22 (TCGGCGGCCGCTTCTACCAG, NotI) and 23 (GACGAGCAGAGGGACGAG GGCCGTCTGATGGCCGGTC); and primers 24 (GACCGGCCATCAGACGGCC
GGCCGTCTGATGGCCGGTC); and primers 24 (GACCGGCCATCAGACGGCC CTCGTCCCTCTGCTCGTC) and 25 (GTGTGCGGCCGCCTCGCTG, NotI). Primer 23 and primer 24 were complementary to opposite strands of the TTA codon region of aveR, and contained the desired mutation as indicated by shading above. PCR products amplified with primers 22/23 and 24/25, respectively, were mixed and then used as template for PCR with primers 22/25 again. The resulting 0.9-kb PCR product was cloned into pMD18-T to give pCTG. The inserted 0.9-kb DNA fragment in pCTG was sequenced to confirm that it contained the designed TTA → CTG mutation without other mutation. The 0.9-kb NotI fragment from pCTG was subsequently ligated with NotI-digested pCZ11 to give pCZ11*, which contained mutated aveR (called aveR*). The aveR* fragment was excised from pCZ11* with BglII/HindIII, and cloned into the corresponding sites of pIJ4090 to generate pJL63. pJL63 was digested with BglII, and the resulting 3.2-kb fragment containing strong constitutive promoter ermE*p and aveR* was ligated with BamHI-digested pSET152 to give AveR-high expression plasmid pJL66 (Fig. S1B), in which aveR gene was controlled by strong promoter ermE*p and contained TTA → CTG mutation.
CTCGTCCCTCTGCTCGTC) and 25 (GTGTGCGGCCGCCTCGCTG, NotI). Primer 23 and primer 24 were complementary to opposite strands of the TTA codon region of aveR, and contained the desired mutation as indicated by shading above. PCR products amplified with primers 22/23 and 24/25, respectively, were mixed and then used as template for PCR with primers 22/25 again. The resulting 0.9-kb PCR product was cloned into pMD18-T to give pCTG. The inserted 0.9-kb DNA fragment in pCTG was sequenced to confirm that it contained the designed TTA → CTG mutation without other mutation. The 0.9-kb NotI fragment from pCTG was subsequently ligated with NotI-digested pCZ11 to give pCZ11*, which contained mutated aveR (called aveR*). The aveR* fragment was excised from pCZ11* with BglII/HindIII, and cloned into the corresponding sites of pIJ4090 to generate pJL63. pJL63 was digested with BglII, and the resulting 3.2-kb fragment containing strong constitutive promoter ermE*p and aveR* was ligated with BamHI-digested pSET152 to give AveR-high expression plasmid pJL66 (Fig. S1B), in which aveR gene was controlled by strong promoter ermE*p and contained TTA → CTG mutation.
Overexpression and purification of the recombinant AveR
A DNA fragment encoding the 394 carboxy-terminal amino acids of AveR was obtained by PCR with primers 68 (CATATGACAGCGGCCTTGTGGGC, NdeI) and 2 (containing EcoRI site). The PCR fragment digested with NdeI/EcoRI was inserted into the corresponding sites in pET-28a (+) to generate pET-AveRc (Fig. S1C), which was further confirmed by DNA sequencing, and then introduced into E. coli BL21 (DE3) for protein overexpression.
After induction by IPTG, the recombinant protein His6-AveRc was expressed as inclusion body in E. coli. Cells containing His6-AveRc were harvested, washed twice with PBS buffer, and resuspended in 5 ml PBS buffer. The cell suspension was treated with lysozyme on ice, and then disrupted by sonication. After centrifugation, the supernatant was removed, and the pellet was washed three times with 4 M urea containing 1% Triton X-100. 8 M urea was added to solubilize inclusion bodies in the pellet, and the solution was centrifuged. The supernatant was dialyzed against a stepwise decrease in urea concentration solution (4 M, 2 M, 1 M urea and PBS buffer); each dialysis was performed over a period of 24 h. The purified protein was diluted with PBS buffer to 1 mg ml−1 for use in antibody induction.
Preparation of antibodies against AveR protein
The purified recombinant protein His6-AveRc was mixed with Freund’s complete adjuvant and injected into a rabbit. After 2 weeks, the antigen was injected into the same rabbit with Freund’s incomplete adjuvant. Further booster immunizations were given at 2-week intervals. The rabbit was bled 2 weeks after each boost, and serum was prepared. Each serum was stored at 4°C, and its potency checked by ELISA. After several booster immunizations, the immune serum reached a high potency and was used as a source of anti-AveR antibodies for subsequent Western blotting and ChIP assay.
Chromatin immunoprecipitation (ChIP) assay
Streptomyces avermitilis cultures grown in fermentation medium II for 40 h were cross-linked by addition of formaldehyde (final concentration 1%) for 30 min at room temperature. 125 mM glycine (final concentration) was added to stop the reaction, and incubation was continued for 5 min with gentle shaking. Mycelia were collected, washed twice with cold PBS buffer, and stored in 300 mg (wet weight) aliquots at −80°C until use. 300 mg frozen mycelia was ground into a fine powder in liquid nitrogen, resuspended in 3 ml immunoprecipitation (IP) buffer (Grainger et al. 2004) containing 1 mM PMSF, and subjected to sonication, which broke down the DNA to 500–1,000 bp fragments. After centrifugation (10 min, 12,000 rpm, 4°C), the supernatant was removed and used as input sample (positive control) in immunoprecipitation experiments.
An 800-μl input sample was pre-cleared with 50 μl 50% protein G agarose/salmon sperm (SS) DNA (Upstate, catalog no. 16-201) for 60 min at 4°C with gentle rotation. Agarose was spun down, supernatant was transferred to a new tube, and 80 μl anti-AveR antibodies was added. After incubation overnight with gentle rotation, 70 μl 50% protein G agarose/SS DNA was added, and incubation continued 4 h at 4°C. An immunoprecipitation experiment without antibody was run as negative control. The protein G agarose beads were washed as described by Grainger et al. (2004). To elute immune complexes, the beads were incubated in 300 μl freshly made elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature for 15 min with rotation. A total of 12 μl 5 M NaCl was added to the eluate or to 300 μl positive-control sample, and cross-links were reversed by incubation at 65°C for 5 h. The positive-control sample was subsequently treated in the same manner as the experimental sample. Residual protein was degraded by addition of 200 μg ml−1 Proteinase K at 45°C for 1 h, followed by phenol/chloroform extraction. The DNA was precipitated with ethanol and resuspended in 30 μl TE, whereas the positive-control sample was resuspended in 100 μl TE. To determine the identity of the immunoprecipitated DNA, PCR using primer sets shown in supplementary Table S1 was performed on 2 μl immunoprecipitated DNA or control DNA for 25–30 cycles.
Western blotting and RT-PCR analysis
After grown in fermentation medium II for 40 h, mycelia of S. avermitilis were collected, flash-frozen in liquid nitrogen, and ground into a fine powder. For Western blotting analysis, cells were suspended in ice-cold extraction buffer containing protease inhibitors (50 mM HEPES, pH 7.4, 137 mM NaCl, 10% glycerol, 1 mM PMSF, 1 μg ml−1 leupeptin, 1 μg ml−1 pepstatin A). Cell debris was removed by centrifugation, and the supernatant was used as total protein extract. 100 μg total protein of each sample was separated in 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis, after which the proteins were transferred onto PVDF membrane. Polyclonal antiserum raised against AveR was used at a dilution of 1:1,500. Western blots were developed using an ECL detection system (Amersham, Beijing, China).
For RNA preparation, total RNA was isolated from the ground mycelial paste using Trizol reagent (Invitrogen, Shanghai, China) according to the manufacturer’s instructions, and the sample was treated with DNase I (Promega) to remove the contaminating chromosomal DNA. RT-PCR was conducted with TaKaRa One Step RNA PCR Kit. As a negative control, the DNase-treated RNA sample was used as a template for PCR, to confirm that the amplified products were not derived from chromosomal DNA. For semiquantitative analysis, samples were taken at four-cycle intervals between cycles 24 and 35 to compare non-saturated PCR product formation. Data were verified in three independent experiments. hrdB gene, which encodes the major sigma factor in Streptomyces, was used as positive control for RT-PCR assay. For oligonucleotides, see supplementary Table S2.
Fermentation and HPLC analysis of products
Fermentation of S. avermitilis ATCC31267 and its mutants was performed as described previously (Chen et al. 2007). Avermectins and oligomycins in fermentation culture were identified by HPLC analysis as described by Chen et al. (2007).
Results
Deletion of aveR abolished avermectin biosynthesis
The entire ave gene cluster contains 18 ORFs spanning a distance of 82-kb (Ikeda et al. 1999) (Fig. 1). The aveR gene for the putative regulator AveR contains 2,850 nucleotides and encodes a protein of 949 amino acids (predicted molecular mass, 101 kDa) including an N-terminal NTP-binding domain (a.a. 17–233), and a C-terminal HTH DNA-binding domain (a.a. 892–941) belonging to the LuxR family (Wilson et al. 2001). To study its function, an aveR deletion strain was constructed (Fig. 2a) and designated as S. avermitilis AveR-D19. AveR-D19 displayed normal growth on YMS solid medium (data not shown). HPLC analysis revealed no avermectin peaks in fermentation culture of AveR-D19 (Fig. 3c). In contrast, wild-type strain ATCC31267 produced all eight avermectin components (Fig. 3a).

HPLC analysis of fermentation products from a wild-type strain ATCC31267; c aveR deletion mutant AveR-D19; d aveR truncation mutant AveR-DHTH. b Standards for oligomycins. A1a, A1b, A2a, A2b, B1a, B1b, B2a, B2b eight components of avermectin; OlmA oligomycin A
To confirm that deletion of aveR was the sole reason for loss of avermectin production, a 3.1-kb DNA fragment containing aveR and its promoter region was reintroduced into AveR-D19 through the pSET152-based complementation plasmid pCZ13. The complementation strain restored avermectin production, but at a level of ca. 50% of wild-type production (data not shown). This result suggests that aveR is required for avermectin biosynthesis, and that aveR function may depend on position in the S. avermitilis chromosome.
The HTH domain of AveR is essential for avermectin productivity
As in other members of the LAL family, an HTH domain was found in the C-terminal region of AveR. To evaluate the function of this domain, a 150-bp internal fragment in the 3′-terminal portion of aveR encoding the HTH domain was deleted (Fig. 2b). The aveR truncation mutant was designated as AveR-DHTH. Growth of AveR-DHTH was similar to that of wild-type strain. HPLC analysis showed that no avermectin was produced by AveR-DHTH (Fig. 3d). Thus, the C-terminal HTH domain is essential for AveR function, and is probably responsible for DNA binding.
In vivo titration of AveR with aveA1 promoter
Generally, pathway-specific regulators directly affect expression of linked structural genes via binding to their promoter regions. aveA1, which encodes the polyketide synthase AVES1 required for avermectin aglycon biosynthesis, might be the target of AveR. To test this, a multi-copy promoter titration plasmid pJL62 carrying the aveA1 promoter region was constructed and transformed into ATCC31267. The avermectin production was reduced significantly in the resulting transformants (data not shown). A plausible explanation of this finding is that titration of AveR with the 327-bp aveA1 promoter region at high copy number affected interaction of AveR with the corresponding chromosomal region, and that AveR is a transcriptional activator of aveA1.
Analysis of AveR activity as transcriptional activator by promoter probing
The promoter probe plasmid pIJ62, in which the reporter neo gene is under control of the aveA1 promoter, was transformed into S. avermitilis ATCC31267 and aveR deletion mutant AveR-D19. The resulting transformants were cultured on YMS agar containing 5 μg ml−1 kanamycin. No growth of these transformants was observed after 10 days incubation, possibly because the aveA1 promoter in pIJ62 was very weak due to limited number of AveR regulator in cells. Therefore, the AveR-high expression plasmid pJL66 was constructed and co-transformed with pJL62 into S. lividans TK54 instead. The co-transformants TK54 (pIJ62 + pIJ66) grew well even on YMS agar containing 150 μg ml−1 kanamycin, whereas the control transformants TK54 (pJL62 + pSET152) did not grow on YMS agar containing 50 μg ml−1 kanamycin (Fig. 4b, c), indicating that aveA1 promoter is inactive without AveR. Both TK54 (pIJ62 + pIJ66) and TK54 (pJL62 + pSET152) grew on thiostrepton-containing YMS plates (Fig. 4a) because pJL62 contains thiostrepton-resistant gene tsr. The same growth patterns were obtained for ATCC31267 (pIJ62 + pIJ66) and ATCC31267 (pJL62 + pSET152) grown on YMS agar containing corresponding antibiotics (data not shown). Thus, promoter probe assay confirmed that AveR positively regulates the activity of aveA1 promoter.

Growth of S. lividans TK54 transformed with promoter probe plasmid pJL62. Strains were grown for 7 days on YMS agar containing thiostrepton (a 50 μg ml−1) or kanamycin (b 50 μg ml−1, c 150 μg ml−1). 1 S. lividans TK54 transformed with pJL62 and pJL66; 2 S. lividans TK54 transformed with pJL62 and pSET152
Identification of AveR target promoters by ChIP assay
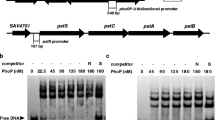
To screen the DNA targets of AveR in ave cluster, we established a ChIP assay using S. avermitilis strains treated with formaldehyde to cross-link AveR to its DNA targets. For the wild-type strain, avermectins were detected by HPLC after 36-h growth in fermentation medium II, suggesting that AveR may interact with its targets and thereby inducing avermectin production prior to this time point. We therefore cross-linked S. avermitilis cells with formaldehyde after 40-h growth in fermentation medium II, extracted and fragmented the cross-linked DNA by sonication, and performed immunoprecipitation with anti-AveR antibodies to select DNA fragments attached to AveR, using a sample without antibody as a negative control. PCR assays were conducted using primers that flank the predicted promoter regions. Based on sequence analysis, the ave gene cluster contains six major putative transcriptional units: aveD-aveF, aveA1-aveA2-aveC, aveA4-aveA3-aveE, orf1-aveBI, aveBIII-aveBII, and aveBVIII-aveBVII-aveBVI-aveBV. Four promoter regions were therefore chosen at intergenic regions between divergently transcribed genes in the ave gene cluster: 327-bp aveD-aveA1 intergenic region; 260-bp aveA4-orf1 intergenic region; 126-bp aveBIII-aveBIV intergenic region; and 238-bp region immediately upstream of aveBVIII (Fig. 1). Primer pairs used for the four promoter regions are shown in Table S1. PCR products with correct size were obtained with these primers when immunoprecipitated DNA or positive control DNA from wild-type strain was used as template (Fig. 5a). In contrast, these primers amplified DNA fragments only from positive control DNA, not from anti-AveR-immunoprecipitated DNA of mutant strain AveR-DHTH (Fig. 5b). No correct PCR bands were obtained using all the primer pairs when negative control DNA without antibody was used as template (Fig. 5). These results demonstrate that the four promoter regions were selectively enriched as a result of ChIP using anti-AveR antibodies, and that AveR binds specifically to these regions with its C-terminal HTH domain.

ChIP analysis of AveR binding to ave promoter regions (a, b) and olm promoter regions (c, d) in vivo. Anti-AveR antibodies were used to immunoprecipitate AveR/DNA complexes from cells (a, c wild-type strain; b AveR-DHTH; d AveR-D19) treated with formaldehyde. PCR was performed with primers flanking putative promoter regions (Table S1). DNA used for PCR was total DNA prior to immunoprecipitation (positive control; lanes “+”), immunoprecipitated DNA (experimental sample; lanes “S”), and negative control DNA without antibody (lanes “−”). hrdB promoter region was used as a control
Gene expression analysis of the ave cluster by RT-PCR
To screen the structural genes controlled by aveR, transcripts from the ave cluster were analyzed by one-step RT-PCR applied to total RNA obtained from S. avermitilis wild-type strain after the onset of avermectin production, and concurrently from aveR deletion mutant AveR-D19. Most transcripts were visualized using 25 cycles of PCR, but some required 30 cycles. In negative controls, containing DNA polymerase but lacking reverse transcriptase, no PCR product was detected with any of the primers when fewer than 35 PCR cycles were used. RT-PCR showed that all structural genes of the ave gene cluster in wild-type strain were transcribed (Fig. 6). Corresponding transcripts were not observed for mutant strain AveR-D19. This gene expression pattern shows that AveR is required for the transcription of all avermectin biosynthetic genes.

RT-PCR analysis of the ave cluster. Analysis was carried out in S. avermitilis wild-type strain (+) and AveR-D19 (−). In general, 25 cycles of PCR were used (a); in case this generated no apparent product, analysis was repeated at 30 cycles in attempt to detect low level of transcripts (b)
Overexpression of aveR leads to overproduction of avermectin
As a general rule, overexpression of a pathway-specific transcriptional activator is associated with increased production of the corresponding antibiotic. To test this possibility, we introduced the AveR-high expression vector pJL66, in which aveR gene was controlled by ermE*p and contained TTA → CTG mutation, into wild-type strain, and analyzed its effects on cell growth and avermectin productivity in soluble fermentation medium II. Enhanced expression of aveR resulted in increased avermectin production, but growth of the transformant was similar to that of wild-type strain (Fig. 7a, b). Overexpression of aveR was confirmed by both Western blotting and semiquantitative RT-PCR analysis, comparing the wild-type strain with its transformant ATCC31267/pJL66 and aveR deletion mutant AveR-D19 (Fig. 7c). In addition, multi-copy plasmid pCZ12 (based on pKC1139) and integrative plasmid pCZ13 (based on pSET152) containing aveR with its own promoter were transformed into wild-type strain, respectively. The resulting transformants and their host strain ATCC31267 as well as vector control strains were separately cultured in fermentation medium for 10 days. HPLC analysis of fermentation products showed that transformant containing only pSET152 produced nearly the same amount of avermectin as host strain, while introduction of empty pKC1139 caused a small reduced avermectin production. In contrast, transformation of plasmid pCZ12 and pCZ13 containing aveR led to enhanced avermectin production (Fig. 7d), indicating that the increased production was due to the overexpression of aveR gene. These results further demonstrated the role of aveR as an important pathway-specific activator gene for avermectin biosynthesis.

Effect of enhanced expression of aveR on growth (a) and avermectin production (b) of S. avermitilis grown in soluble fermentation medium II. Filled triangles wild-type strain ATCC31267; open triangles transformant of wild-type strain containing control plasmid pSET152 (ATCC31267/pSET152); filled squares transformant of wild-type strain containing AveR-high expression vector pJL66 (ATCC31267/pJL66). c Western blotting and semiquantitative RT-PCR analysis of aveR in wild-type stain (WT), AveR-D19 and ATCC31267/pJL66. The same batch of cells was used for both Western blotting and RT-PCR analysis. d Comparison of avermectin production in various S. avermitilis strains grown in fermentation medium. Fermentation medium is usually used for avermectin production; however, it contains insoluble yeast meal. So, we used soluble fermentation medium II in a and b for analysis of cell growth as well as avermectin production in the same batch of cells. Avermectin production is lower in soluble fermentation medium II than that in fermentation medium
Deletion of aveR enhanced oligomycin biosynthesis
The aveR mutants lost the ability to produce avermectins, but produced oligomycins at a higher level than wild-type strain (Fig. 3c, d). In complementation experiments, DNA fragment containing aveR and its promoter region restored avermectin production as well as reduced oligomycin production to the level of wild-type strain in the mutant AveR-D19 (data not shown). To test whether aveR affects the transcription of oligomycin biosynthetic genes, semiquantitative RT-PCR analyses were performed to detect the expression of two selected polyketide synthase (PKS) genes (olmA1 and olmA4) and two putative pathway-specific regulatory genes (olmRI and olmRII) in olm cluster. As shown in Fig. 8, all the detected olm genes were upregulated in the aveR mutants. In ChIP assays, the predicted promoter regions of the olm genes were amplified from both positive control DNA and anti-AveR-immunoprecipitated DNA (Fig. 5c). Since AveR, OlmRI and OlmRII are all members of the LAL family, antibodies raised against one might cross-react with the others. To investigate whether AveR antibodies immunoprecipitated OlmRI and/or OlmRII, the ChIP experiment was repeated in aveR null mutant AveR-D19 and showed that the olm promoter DNA was no longer immunoprecipitated (Fig. 5d).These data indicate that AveR has negative effects on oligomycin biosynthesis, and represses transcription of olm genes.

Semiquantitative RT-PCR analyses of the olmA1, olmA4, olmRI, and olmRII transcript levels. Samples were collected from S. avermitilis wild-type stain, AveR-D19 and AveR-DHTH grown in fermentation medium II for 40 h
Discussion
We describe here a detailed functional analysis of the aveR gene, and demonstrate that aveR encodes a pathway-specific positive regulator required for transcription of all the avermectin biosynthetic genes by direct interaction with ave promoters. We also showed that aveR exerts a negative role on oligomycin production, but does not affect the growth of S. avermitilis. A very recent paper of Kitani et al. (2009) performed a number of similar experiments. As described in the following section only some results could be confirmed here.
Attempts to express high level of full-length AveR protein for purification were unsuccessful in both E. coli and S. avermitilis. The aveR gene was cloned in a high-copy-number vector under control of a strong promoter: T7 promoter in E. coli to express His6-tagged AveR or tipA promoter in S. avermitilis to express native AveR. Both approaches yielded a very low level of the target protein, and MBP (maltose-binding protein) fusion protein system (BioLabs) in E. coli also failed. These results could not be explained by the presence of unusual codon TTA in the aveR gene, since the replacement of aveR gene in the expression vector with mutated gene aveR* containing TTA-to-CTG mutation did not lead to production of enough AveR protein for purification. Another possibility is that the large size of the AveR protein is a “bottleneck” in the translation process. We are not aware of any examples of intact LAL regulators in Streptomyces purified through overexpression. This makes it more difficult to understand the regulation mechanism of this novel regulatory protein family. A high level of truncated form of AveR containing the C-terminal HTH domain (AveRc), fused to a hexahistidine tag at its N-terminus, was therefore expressed as inclusion body, and was used for preparation of anti-AveR antibodies for ChIP assay. ChIP assay identified four AveR target promoter regions, all located within the ave cluster. The finding that AveR without HTH domain is unable to bind ave promoters demonstrated the DNA-binding activity of AveR HTH domain. The promoter regions (intergenic regions) identified in this study were predicted to contain divergent promoters. Sequence alignment of the promoter regions bound by AveR did not predict a consensus AveR-binding sequence; more experiments are needed to reveal the binding sites of AveR.
Overexpression studies with an extra copy of aveR containing TTA-to-CTG mutation, expressed under the control of strong constitutive promoter ermE*p in wild-type S. avermitilis ATCC31267, demonstrated a positive effect of the aveR gene on avermectin production. This finding was further confirmed by the facts that multi-copies of aveR as well as an extra copy of aveR with its native promoter promoted avermectin production in ATCC31267. Our results are in contrast to the findings of Kitani et al. (2009), who reported that a higher amount of aveR resulted in complete loss of avermectin in wild-type strain KA320 and its spontaneous mutant K139. These authors disrupted aveR by gene replacement in S. avermitilis, and the resulting mutant lost avermectin productivity and was unable to convert an avermectin intermediate to any avermectin derivatives. They concluded that AveR is a positive regulator for avermectin biosynthesis, but they postulated a maximum threshold concentration of aveR for the production of avermectin. The differing behavior of aveR may reflect differences in the parental strains used and the growth media adopted in the respective studies. They described KA320 as a wild-type strain isogenic to MA-4680 and since ATCC31267 is designated MA-4680, there should be no obvious difference in genetic background between strain KA320 and ATCC31267. However, they described that strain KA320 is phenotypically unstable and frequent reisolation is necessary; thus the spontaneous mutant K139 from KA320 is used as the major wild-type strain in their study, whereas the strain ATCC31267 was not so unstable during our experiments. The linear chromosome of Streptomyces easily undergoes large rearrangement, such as large deletions, circularization, arm replacement, and amplifications in terminal regions. We therefore analyzed the chromosomal structure of ATCC31267 by pulsed-field gel electrophoresis (PFGE) (Fig. S2) and showed that its AseI restriction patterns were consistent with the published data (http://avermitilis.ls.kitasato-u.ac.jp/physicalmap), suggesting no large rearrangement occurred in the chromosome of our strain. We sequenced the entire aveR gene and its promoter region of ATCC31267 again, but did not find any difference with the published sequence. As KA320 was so unstable, we are not sure if chromosomal rearrangement occurred in this strain and its mutant K139 used by Kitani et al. (2009). The discrepancies among these results still remain to be studied.
Like aveR, olmRI and olmRII are two putative pathway-specific regulatory genes belonging to the LAL family. Disruption of olmRI or olmRII resulted in loss of oligomycin production (our unpublished data), suggesting that OlmRI and OlmRII are positive regulators for oligomycin biosynthesis. The aveR mutants enhanced yield of oligomycin, and the transcript levels of olm structural and regulatory genes were consistent with the production of oligomycin, suggesting the possibility that the negative effect of aveR on oligomycin biosynthesis is mediated by regulatory gene olmRI and olmRII. However, ChIP results indicated that the AveR protein could bind putative olmA1 and olmA4 promoters as well as olmRI and olmRII promoters. Several tetratricopeptide repeats (TPRs), which are implicated in protein–protein interactions, were predicted in AveR, OlmRI, and OlmRII. It seems likely that the various LAL regulators present in the same bacterial cell may communicate with each other and form a complex regulatory network. Thus, AveR may repress olm genes by interacting with other proteins, such as OlmRI and OlmRII. Another possibility is that AveR affects the expression of olm genes through interacting directly with their promoters. More experiments will be needed to clarify the regulatory mechanism of AveR on oligomycin biosynthesis. Furthermore, we still could not rule out the possibility that carbon flux contributes to the increased oligomycin production. Avermectin and oligomycin belong to type I polyketides, and both require acetate and propionate extender units for synthesis of polyketide backbone (Ikeda et al. 1999, 2001; Omura et al. 2001). Therefore, loss of avermectin production in aveR mutants might allow additional acetate and propionate to enter the biosynthetic pathway of oligomycin and stimulate oligomycin production.
aveR is the first pathway-specific regulatory gene characterized in S. avermitilis, and the present work also showed the pleiotropic effects of aveR on oligomycin biosynthesis. These results will contribute to elucidation of other pathway-specific regulatory factors and regulatory mechanisms of secondary metabolism in S. avermitilis. This knowledge will be useful in improving and modifying methods for production of secondary metabolites, including avermectin and oligomycin, by this industrially important bacterial strain.
References
Bibb MJ (2005) Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol 8:208–215
Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE (1992) Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49
Burg RW, Miller BM, Baker EE, Birnbaum J, Currie SA, Hartman R, Kong YL, Monaghan RL, Olson G, Putter I, Tunac JB, Wallick H, Stapley EO, Oiwa R, Omura S (1979) Avermectins, new family of potent anthelmintic agents: producing organisms and fermentation. Antimicrob Agents Chemother 15:361–367
Chen Z, Wen J, Song Y, Wen Y, Li JL (2007) Enhancement and selective production of avermectin B by recombinants of Streptomyces avermitilis via intraspecific protoplast fusion. Chin Sci Bull 52:616–622
Chen L, Lu Y, Chen J, Zhang W, Shu D, Qin Z, Yang S, Jiang W (2008) Characterization of a negative regulator AveI for avermectin biosynthesis in Streptomyces avermitilis NRRL8165. Appl Microbiol Biotechnol 80:277–286
Grainger DC, Overton TW, Reppas N, Wade JT, Tamai E, Hobman JL, Constantinidou C, Struhl K, Church G, Busby SJ (2004) Genomic Studies with Escherichia coli MelR protein: applications of chromatin immunoprecipitation and microarrays. J Bacteriol 186:6938–6943
Hwang YS, Kim ES, Biro S, Choi CY (2003) Cloning and analysis of a DNA fragment stimulating avermectin production in various Streptomyces avermitilis strains. Appl Environ Microbiol 69:1263–1269
Ikeda H, Omura S (1997) Avermectin biosynthesis. Chem Rev 97:2591–2610
Ikeda H, Kotaki H, Tanaka H, Omura S (1988) Involvement of glucose catabolism in avermectin production by Streptomyces avermitilis. Antimicrob Agents Chemother 32:282–284
Ikeda H, Takada Y, Pang CH, Tanaka H, Omura S (1993) Transposon mutagenesis by Tn4560 and applications with avermectin-producing Streptomyces avermitilis. J Bacteriol 175:2077–2082
Ikeda H, Nonomiya T, Usami M, Ohta T, Omura S (1999) Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis. Proc Natl Acad Sci USA 96:9509–9514
Ikeda H, Nonomiya T, Omura S (2001) Organization of biosynthetic gene cluster for avermectin in Streptomyces avermitilis: analysis of enzymatic domains in four polyketide synthases. J Ind Microbiol Biotechnol 27:170–176
Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S (2003) Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol 21:526–531
Janssen GR, Bibb MJ (1993) Derivatives of pUC18 that have BglII sites flanking a multiple cloning site and that retain ability to identify recombinant clones by visual screening of Escherichia coli colonies. Gene 124:133–134
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich
Kitani S, Ikeda H, Sakamoto T, Noguchi S, Nihira T (2009) Characterization of a regulatory gene, aveR, for the biosynthesis of avermectin in Streptomyces avermitilis. Appl Microbiol Biotechnol 82:1089–1096
MacNeil DJ, Klapko LM (1987) Transformation of Streptomyces avermitilis by plasmid DNA. J Ind Microbiol 2:209–218
Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M (2001) Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc Natl Acad Sci USA 98:12215–12220
Pinna LA, Lorini M, Moret V, Siliprandi N (1967) Effect of oligomycin and succinate on mitochondrial metabolism of adenine nucleotides. Biochim Biophys Acta 143:18–25
Rajkarnikar A, Kwon HJ, Ryu YW, Suh JW (2006) Catalytic domain of AfsKav modulates both secondary metabolism and morphologic differentiation in Streptomyces avermitilis ATCC 31272. Curr Microbiol 53:204–208
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Stutzman-Engwall KJ, Price BS (2001) Streptomyces avermitilis regulatory genes for increased avermectin production. US Patent 6,197,591
Wilson DJ, Xue Y, Reynolds KA, Sherman DH (2001) Characterization and analysis of the PikD regulatory factor in the pikromycin biosynthetic pathway of Streptomyces venezuelae. J Bacteriol 183:3468–3475
Zhao JL, Wen Y, Chen Z, Song Y, Li JL (2007) An adpA homologue in Streptomyces avermitilis is involved in regulation of morphogenesis and melanogenesis. Chin Sci Bull 52:623–630
Acknowledgments
We thank Prof. Gang Liu (Chinese Academy of Science) and Prof. Linquan Bai (Shanghai Jiaotong University, China) for their critical reading of the manuscript. This work was supported by grants from the National Basic Research Program of China (Grant No. 2009CB118905) and the National High Technology Research and Development Program (Grant No. 2006AA10A209).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Klug.
J. Guo and J. Zhao contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Guo, J., Zhao, J., Li, L. et al. The pathway-specific regulator AveR from Streptomyces avermitilis positively regulates avermectin production while it negatively affects oligomycin biosynthesis. Mol Genet Genomics 283, 123–133 (2010). https://doi.org/10.1007/s00438-009-0502-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-009-0502-2