
Abstract
The brown seaweed, Sargassum myriocystum associated with heterotrophic bacterium, Bacillus subtilis MTCC 10407 (JF834075) exhibited broad-spectra of potent antibacterial activities against pathogenic bacteria Aeromonas hydrophila, Vibrio vulnificus, and Vibrio parahaemolyticus. B. subtilis MTCC 10407 was found to be positive for polyketide synthetase (pks) gene, and therefore, was considered to characterize secondary metabolites bearing polyketide backbone. Using bioassay-guided fractionation, two new antibacterial O-heterocyclic compounds belonging to pyranyl benzoate analogs of polyketide origin, with activity against pathogenic bacteria, have been isolated from the ethyl acetate extract of B. subtilis MTCC 10407. In the present study, the secondary metabolites of B. subtilis MTCC 10407 with potent antibacterial action against bacterial pathogens was recognized to represent the platform of pks-1 gene-encoded products. Two homologous compounds 3 (3-(methoxycarbonyl)-4-(5-(2-ethylbutyl)-5,6-dihydro-3-methyl-2H-pyran-2-yl)-butyl benzoate) and 4 [2-(8-butyl-3-ethyl-3,4,4a,5,6,8a-hexahydro-2H-chromen-6-yl)-ethyl benzoate] also have been isolated from the ethyl acetate extract of host seaweed S. myriocystum. The two compounds isolated from ethyl acetate extract of S. myriocystum with lesser antibacterial properties shared similar structures with the compounds purified from B. subtilis that suggested the ecological and metabolic relationship between these compounds in seaweed-bacterial relationship. Tetrahydropyran-2-one moiety of the tetrahydropyrano-[3,2b]-pyran-2(3H)-one system of 1 might be cleaved by the metabolic pool of seaweeds to afford methyl 3-(dihydro-3-methyl-2H-pyranyl)-propanoate moiety of 3, which was found to have no significant antibacterial activity. It is therefore imperative that the presence of dihydro-methyl-2H-pyran-2-yl propanoate system is essentially required to impart the greater activity. The direct involvement of polarisability (Pl) with the target bioactivity in 2 implied that inductive (field/polar) rather than the steric effect (parachor) appears to be the key factor influencing the induction of antibacterial activity. The present work may have a footprint on the use of novel O-heterocyclic polyketide products from seaweed-associated bacterium for biotechnological, food, and pharmaceutical applications mainly as novel antimicrobial secondary metabolites.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Seaweeds provide nutrient-rich living substrata for microbial inhabitants, and they are an inevitable component of coastal ecosystem (Ali et al. 2012). Recent scientific reports indicated that the surface-associated bacteria might play a role in seaweed chemical defenses against surface fouling due to its unique inhibitory metabolite production (Kumar et al. 2011). Even though seaweeds and their associated bacteria are actively being engaged in chemically mediated interactions by exchanging minerals, secondary metabolites, and other nutrients, studies on these interactions lag behind that of other marine eukaryotes (Hollants et al. 2012; Goecke et al. 2010). Marine microbial symbionts were the true producers or active participant in the biosynthesis of active natural products from the eukaryotic hosts, and the seaweed species were reported for low levels of microbial infection (Kubanek et al. 2003; Zhao et al. 2011). The emerging diverse compounds with wide antimicrobial spectrum reported from seaweeds and its associated bacterial flora make them a treasure trove of biotechnological research (Chakraborty et al. 2014). Although examples are rare, it is believed that marine eukaryotes may use their surface-associated bacteria as a source of antimicrobial chemical defenses in competition and in the protection of the host (Penesyan et al. 2009). Pluralities of culture-dependent and culture-independent studies on sponges and their associated microbial population corroborated this hypothesis (Zhang et al. 2009). The surfaces of seaweeds were reported to attract various surface colonizing micro and macro-organisms of opportunistic nature. However, the defensive compounds produced by the seaweed-associated microorganisms can serve as a powerful tool to resist the surface colonizers. The earlier study of seaweed-associated bacteria with polyketide synthase genes proposed the application of polyketides as potential antibacterial pharmacophores to strive against the pathogenic microorganisms (Chakraborty et al. 2014).
In the current study, heterotrophic Bacillus subtilis MTCC 10407 associated with brown seaweed Sargassum myriocystum was isolated by a culture dependent method from the Gulf of Mannar in the southeast coast of India to isolate O-heterocycle pyran derivatives of polyketide origin based on bioactivity against human opportunistic food pathogenic bacteria. The antimicrobial potential of the isolated secondary metabolites were examined with polymerase chain reaction by using the amplifying gene encodings for polyketide synthetase (pks). Seaweed-bacterial interactions were studied by comparing the antibacterial metabolites of colonizing bacteria with its host-derived seaweed secondary metabolites. The pks-assisted biosynthetic pathway of the O-heterocycle pyrans was proposed and the ecological interactions of seaweed and its associated bacterial metabolites have been demonstrated based on codetection of bacterial metabolites in the host extracts.
Experimental
General
Human pathogenic bacterial strains, such as Vibrio vulnificus MTCC 1145, Vibrio harveyi MTCC 3438 were acquired from Institute of Microbial Technology in India, whereas Vibrio parahaemolyticus ATCC®17802™ was obtained from American Type Culture Collection (Manassas, VA 20110) in the USA. The 1D and 2D (correlation spectroscopy 1H–1H COSY, nuclear Overhauser effect spectroscopy (NOESY), heteronuclear multiple-bond correlation spectroscopy (HMBC), heteronuclear single-quantum correlation spectroscopy (HSQC)) nuclear magnetic resonance (NMR) spectra (500 MHz; Bruker Avance DPX 500), Fourier-transform infrared (FTIR; Perkin-Elmer 2000 FTIR), ultra-violet (UV; Varian Cary 50 UV-vis spectrometer, USA), and mass spectra (Applied Biosystems QTrap 2000, Darmstadt, Germany) were acquired according to the earlier report of Chakraborty et al. (2014). All reagents and solvents were obtained from Merck, Germany.
Seaweed samples and associated antibacterial isolates
The brown seaweed S. myriocystum was gathered from intertidal zone of the Gulf of Mannar in the southeast coast (9° 17′ 0″ N, 79° 7′ 0″ E) of India via scuba diving. The bacterial strains associated with seaweeds were isolated as described by Quevrain et al. (2014) and assayed for their antagonistic properties against pathogenic bacteria, such as Aeromonas hydrophilla (MTCC646), V. parahaemolyticus (ATCC 17802), V. vulnificus (MTCC1145), Vibrio alginolyticus (MTCC4439), and V. parahaemolyticus (MTCC451) as described previously (Chakraborty et al. 2014) by spot-over lawn technique (Kumar et al. 2011). The antagonistic bacteria were perceived by employing biochemical methods, bacterial membrane fatty acid analysis, and 16S ribosomal RNA (rRNA) gene sequencing using the primers AGAGTTTGATCCTGGCTCAG (forward) and ACGGCTACCTTGTTACGACTT (reverse) (Armstrong et al. 2001). The bacterial isolate under this study was B. subtilis MTCC 10407 from S. myriocystum, and this candidate strain was subjected to metabolite gene (pks) screening using the primers RTRGAYCCNCAGCAICG (forward) and VGTNCCNGTGCCRTG (reverse) as reported earlier (Kumar et al. 2011).
Bioassay-guided purification of antibacterial compound from B. subtilis MTCC 10407 associated with S. myriocystum
B. subtilis (MTCC 10407) was cultured, and the preparation followed by the recovery of secondary metabolites from the bacterium was accomplished by surface-culturing over plates coated with nutrient agar. The generated bacterial secondary metabolites were consequently homogenized with ethyl acetate (Arrow Engineering Inc., Pennsylvania Ave, USA) followed by refluxing. The culture volume (4 L) was thereafter extracted by solvent ethyl acetate (EtOAc) to yield ethyl acetate extract (5.56 g) after evaporation (Heidolph, Schwabach, Germany). The ethyl acetate extract (5 g) was fractionated through flash liquid chromatography (Biotage SP1-B1A, Sweden) on a silica gel column (Biotage no. 25+M 0489-1, 230–400 mesh; Biotage, Sweden) at a collection UV wavelength of 246 nm, and with a stepwise gradient of dichloromethane-methanol (CH2Cl2/MeOH) as solvent system (CH2Cl2/MeOH; 0–100 % MeOH) to yield F1–14 as 14 pooled fractions. Fraction F3 (106 mg) was further fractionated through vacuum liquid chromatography using adsorbent silica gel (180–230 mesh), with a stepwise gradient of n-hexane to ethyl acetate as solvent system to yield 20 fractions (15 mL each). The similar fractions were mixed together to afford seven pooled fractions (F3–1–F3–7) after thin layer chromatography (TLC) methods (n-hexane/EtOAc, 9:1 (v/v)). The fraction F3–4 (52 mg) was further separated by chromatography on silica coated on a preparatory thin layer plate (0.5 % MeOH/CH2Cl2) to afford 2-(7-(2-ethylbutyl)-2,3,4,4a,6,7-hexahydro-2-oxopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate (1, ~98 % purity, 6.3 mg). The TLC (GF254) using 3 % EtOAc/n-hexane supported its purity. The active fraction F4 (542.3 mg) eluted at 4 % MeOH/CH2Cl2 was fractionated by chromatography with a stepwise gradient of CH2Cl2/MeOH (0–100 % MeOH) to provide subfractions F4–1 through F4–4 (45 mL each). The subfraction F4–1 (23 mg) was chromatographed over preparatory TLC (PTLC) using MeOH/CH2Cl2 (0.5:95.5, v/v) to afford 2-((4Z)-2-ethyl-octahydro-6-oxo-3-((E)-pent-3-enylidene)-pyrano-[3,2b]-pyran-7-yl)-ethyl benzoate (2, ~99 % purity by 1 % MeOH/CHCl3 on TLC, 5.9 mg).
2-(7-(2-Ethylbutyl)-2,3,4,4a,6,7-hexahydro-2-oxopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate (1)
Yellowish oil; UV (MeOH) λ max (log ε), 248 nm (3.14); TLC (Si gel GF254 15 mm; EtOAc/n-hexane (3:17, v/v) R f, 0.16; GC R t, 14.45 min; IR (KBr, cm−1) ν max, 814.06 (aromatic C–Hδ), 1312.76 (C–Oν), 1378.29 (C–Hρ), 1648.94 (C=Cν), 1690.28 (C–COν), 1738.96 (C=Oν), 2923.22 (alkane C–Hν), and 3010.12 (aromatic C–Hν); 1H NMR (500 MHz, CDCl3 δ in ppm) δ 7.78–7.65 (m, 2H), 7.57–7.43 (m, 3H), 5.34 (d, 1H), 4.31 (t, J = 7.2 Hz, 2H), 4.23 (dq, J = 6.7 Hz, 1H), 4.09 (d, J = 6.67 Hz, 2H), 2.38 (m, 1H), 2.04 (m, 1H), 1.68 (t, 2H), 1.72 (m, 2H), 1.53 (m, 2H), 1.46 (m, 1H), 1.26 (m, 2H), 1.01 (m, 2H), 0.92 (t, 3H), and 0.88 (m, 3H); 13C NMR (125 MHz, CDCl3 δ in ppm) δ 179.99 (C-11), 167.72 (C-7), 138.77 (C-15), 132.51 (C-1), 131.32 (C-2), 130.75 (C-6), 128.84 (C-3), 128.11 (C-4), 128.71 (C-5), 123.12 (C-16), 72.41 (C-14), 71.79 (C-18), 65.57 (C-8), 43.76 (C-10), 38.05 (C-17), 30.58 (C-9), 27.73 (C-13), 27.73 (C-19), 22.7 (C-20), 19.76 (C-23), 19.16 (C-21), 14.12 (C-22), and 13.12 (C-24); 1H–1H COSY and HMBC data, see Table 1; high-resolution mass spectrometry with electrospray ionization (HRMS-ESI) m/e calcd for C23H31O5 387.2172, found 387.2315 [M + H]+.
2-((4Z)-2-ethyl-octahydro-6-oxo-3-((E)-pent-3-enylidene)-pyrano-[3,2b]-pyran-7-yl)-ethyl benzoate (2)
Yellowish oil; UVMeOH λ max (log ε), 254 nm (2.83); TLC (Si gel GF254; CHCl3/MeOH 9:1, v/v) R f, 0.50; GC R t, 16.01 min; IR (KBr, expressed in cm−1) ν max, 727.22 (C–Hρ), 814.16 (aromatic C–Hδ), 1371.26 (C–Hρ), 1526.21 (aromatic C=Cν), 1652.42 (C=Cν), 1690.21 (C–CO–Oν), 1726.86 (C=Oν), 2923.22 (alkane C–Hν), and 3346 (broad O–Hν); 1H NMR (500 MHz, d-chloroform, δ in ppm) δ 7.69–7.61 (m, 3H), 7.50–7.42 (m, 2H), 5.80 (m, 1H), 5.27 (dd, 1H), 4.97 (dd, 1H), 4.23 (t, J = 6.7 Hz, 2H), 4.08 (dt, 1H), 3.91 (m, 1H), 3.59 (m, 1H), 2.34 (m, 1H), 1.97 (m, 2H), 1.62 (t, 2H), 1.67 (m, 1H), 1.48 (m, 2H), 1.45 (d, 2H), 0.87 (d, 3H), 0.81 (t, 3H); 13C NMR (125 MHz, d-chloroform, δ in ppm) δ 173.52 (C-11), 167.73 (C-7), 142.78 (C-16), 132.3 (C-1), 130.9 (C-6), 130.87 (C-2), 130.14 (C-18), 129.95 (C-3), 128.84 (C-4), 128.84 (C-5), 123.78 (C-20), 114.63 (C-21), 69.06 (C-13), 65.57 (C-8), 62.16 (C-17), 53.61 (C-12), 34.84 (C-10), 30.58 (C-9), 30.12 (C-14), 29.38 (C-15), 27.4 (C-19), 19.8 (C-23), 14.14 (C-22), and 13.81 (C-24); 1H–1H COSY and HMBC data, refer to Table 1; HRMS (ESI) m/e calcd for C24H31O5 399.2172, found 399.2268 [M + H]+.
Bioassay-guided chromatographic purification of the ethyl acetate extract of S. myriocystum
The seaweed (S. myriocystum) sample were air-dried and ground to obtain seaweed powder (400 g), which was extracted by MeOH (2000 mL × 3) at an elevated temperature (~45 °C) for 3 h. The MeOH extract was filtered to obtain a filtrate before being concentrated at about 50 °C in vacuo to a volume measuring one third of the initial filtrate. The resulting concentrate was partitioned with solvents n-hexane (150 mL × 3), CH2Cl2 (150 mL × 3) and EtOAc (150 mL × 3) in succession (Raola and Chakraborty 2016). Evaporation of the solvents from these fractions under reduced pressure furnished n-hexane (3.6 g), CH2Cl2 (4.24 g), and EtOAc (7.5 g) fractions. The crude EtOAc extract (7.5 g) of S. myriocystum was fractionated through vacuum liquid chromatography into a glass column (90 × 4 cm) using silica gel (60–120 mesh, 50 g), with a stepwise gradient of n-hexane to ethyl acetate as solvent system (n-hexane/EtOAc, 99:1 to 1:19, v/v) to yield PG1–9 as nine pooled fractions (30 mL each) after TLC analysis (n-hexane/EtOAc, 9:1, v/v). The fraction PG4 (eluted at 5:1, n-hexane/EtOAc, v/v) was a mixture of compounds. This fraction was further fractionated by flash chromatography on a silica gel column (230–400 mesh, Biotage no. 25+M 0489-1) using a step gradient of ethyl acetate/n-hexane (0–20 % EtOAc) at a collection UV wavelength of 242 nm to furnish a total of 95 fractions (9 mL each). The similar fractions were mixed together to afford four pooled fractions (PG4–1-PG4–4) after TLC methods (8 % n-hexane-EtOAc, v/v). The subfraction PG4–3 as eluted at EtOAc/n-hexane (3:17, v/v) was again separated by chromatography on silica gel (GF254 PTLC) coated on a preparatory thin-layer plate using a stepwise gradient system from 0.5 % MeOH/CH2Cl2 yielding compound 3, i.e., 2-(8-butyl-3-ethyl-3,4,4a,5,6,8a-hexahydro-2H-chromen-6-yl)-ethyl benzoate (6.2 mg). The fractions PG7 and PG8 as eluted with n-hexane/EtOAc (3:7 and 1:4, v/v, respectively) were pooled together before being chromatographed on a silica gel column (180–230 mesh, 3.5 × 15cm) using CHCl3/MeOH (100:0 to 9:1, v/v) to yield 53 fractions (10 mL each), and patterns were pooled (TLC, n-hexane/EtOAc, 9:1, v/v) together yielding five pooled fractions (PG7–1 through PG7–5). The subfraction PG7–4 eluted with CHCl3/MeOH (23:2, v/v) was fractionated over preparatory TLC on silica with a stepwise gradient system from 3 % MeOH/CH2Cl2 to obtain four subfractions (PG7–4–1–PG7–4-4). Repeated preparatory thin-layer chromatographic separation (silica gel GF254) of PG7–4–4 on adsorbent using CH2Cl2/MeOH (97:3, v/v) afforded 3-(methoxycarbonyl)-4-(5-(2-ethylbutyl)-5,6-dihydro-3-methyl-2H-pyran-2-yl)-butyl benzoate (4, 6.15 mg) as major pure component.
3-(Methoxycarbonyl)-4-(5-(2-ethylbutyl)-5,6-dihydro-3-methyl-2H-pyran-2-yl)-butyl benzoate (3)
White crystalline solid; m.p. 123 °C; UVMeOH λ max (log ε), 235 nm (3.84); TLC (Si gel GF254; CHCl3/MeOH 99:1, v/v) R f, 0.45; GC R t, 8.6 min; IR (KBr, expressed in cm−1) ν max, 729.29 (C–Hρ), 1376.26 (C–Hρ), 1454.38 (C–Hδ), 1466.91 (C–Hδ), 1506 (aromatic C–Hν), 1642 (C=Cν), 1736.96 (C=Oν), and 2854.7 (C–Hν); 1H NMR (500 MHz, d-chloroform, δ in ppm) δ 7.77–7.69 (m, 2H), 7.55 (dq, J = 6.8 Hz, 3H), 5.38 (d, J = 9.3 Hz, 2H), 4.33 (t, J = 6.7 Hz, 2H), 4.24 (t, J = 5.9 Hz, 1H), 4.11 (d, J = 6.7 Hz, 1H), 3.69 (s, 3H), 2.34 (m, 1H), 2.16 (s, 3H), 2.02 (m, 1H), 1.72 (m, 2H), 1.61 (m, 2H), 1.52 (m, 2H), 1.44 (m, 1H), 1.26 (m, 2H), 1.03 (m, 2H), 0.92 (m, 3H), and 0.88 (m, 2H); 13C NMR (125 MHz, d-chloroform, δ in ppm) δ 173.3 (C-11), 167.7 (C-7), 132.45 (C-1), 130.9 (C-6), 130.87 (C-2), 129.95 (C-3), 129.93 (C-16), 128.8 (C-4), 128.84 (C-5), 114.05 (C-15), 71.79 (C-18), 68.16 (C-14), 65.57 (C-8), 51.43 (C-12), 38.74 (C-17), 34.13 (C-10), 30.37 (C-9), 29.7 (C-13), 24.89 (C-23), 22.99 (C-24), 22.69 (C-19), 22.63 (C-20), 19.05 (C-21), 14.96 (C-22), and 13.96 (C-25); 1H–1H COSY, and HMBC data, refer to Table 2; HRMS (ESI) m/e calcd for C25H37O5 417.2642, found 417.2768 [M + H]+.
2-(8-butyl-3-ethyl-3,4,4a,5,6,8a-hexahydro-2H-chromen-6-yl)-ethyl benzoate (4)
Amorphous yellow powder; m.p. 107 °C; UVMeOH λ max (log ε), 229 nm (3.40); TLC (Si gel GF254; CHCl3/MeOH, 99:1, v/v) R f, 0.86; GC R t,17.22 min; IR (KBr, expressed in cm−1) ν max, 731.29 (C–Hρ), 812.06 (aromatic C–Hδ), 1376.26 (C–Hρ), 1466.91 (C–Hδ), 1650.12 (C=Cν) 1690.21 (C–CO–Cν), 1742.02 (C=Oν), 2856.74 (C–Hν), 2921.22 (C–Hν), and 3010.12 (aromatic C–Hν); 1H NMR (500 MHz, d-chloroform, δ in ppm) δ 7.77–7.69 (m, 2H), 7.55 (dq, J = 6.8 Hz, 3H), 5.15 (m, 1H), 4.22 (t, J = 6.8 Hz, 2H), 4.08 (d, 2H), 3.78 (m, 1H), 2.30 (m, 1H), 2.18 (m, 2H), 2.02 (m, 1H), 1.72 (m, 2H), 1.63 (m, 2H) 1.44 (t, 2H), 1.39 (m,2H), 1.36 (m, 1H), 1.26 (m, 2H), 0.98 (m, 2H), 0.91 (t, 3H), and 0.81 (t, 3H); 13C NMR (125 MHz, d-chloroform, δ in ppm) δ 167.72 (C-7), 132.5 (C-1), 132.31 (C-6), 130.91 (C-2), 130.76 (C-12), 128.86 (C-3), 128.84 (C-5), 128.71 (C-4), 126 (C-11), 72.41 (C-13), 71.79 (C-18), 65.58 (C-8), 45.02 (C-16), 40.97 (C-14), 38.05 (C-9), 37.88 (C-15), 34.62 (C-17), 30.57 (C-10), 29.9 (C-21), 29.73 (C-22), 27.73 (C-23), 19.76 (C-19), 13.74 (C-20), and 18.65 (C-24); 1H–1H COSY, and HMBC data, refer to Table 2; HRMS (ESI) m/e calcd for C24H35O3 371.2587, found 371.2678 [M + H]+.
Antimicrobial assay
The crude solvent extract (10 μL in MeOH) and the title compounds (100 μg in MeOH) purified from seaweed and associated bacteria were subjected to antibacterial assays by disk diffusion method on Mueller Hinton agar (Difco) dressed with overnight cultures of the test pathogenic microbial suspensions (cell density of 105 CFU/mL) as described previously (Chakraborty et al. 2014). The inhibitory activity was expressed in terms of inhibitory zone diameter recorded after an incubation period of 18 h at 37 °C. The results were compared with antibiotic disk for Gram negatives (HiMedia, India).
Results
Isolation and antibacterial activities of B. subtilis MTCC 10407 associated with intertidal seaweed S. myriocystum
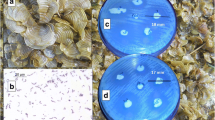
The bacterial isolate, B. subtilis with antibacterial activity was identified and submitted at the Institute of Microbial Technology (Chandigarh, India) as MTCC10407 (Fig. 1a). It was recognized during fatty acid methyl ester analysis (data not shown), biochemical, 16S rRNA gene sequencing, etc., deposited under the accession number JF834075, and the strain was found to be polyketide synthase gene (pks) positive with the KS domain sequences obtained under accession number KC589398. Although it is difficult to predict the pks products based upon the derived sequences due to the presence of smaller gene fragments, an attempt was made to correlate the existence of pks gene with the structural characteristics of the secondary metabolites. The strain possessed an extensive spectrum of antibacterial activity against human food pathogenic bacteria {A. hydrophilla MTCC 646 (11 mm), V. parahaemolyticus MTCC 451 (11 mm), V. parahaemolyticus ATCC®17802™ (17 mm), V. harveyi MTCC 3438 (16 mm), V. alginolyticus MTCC 4439 (11 mm), and V. vulnificus MTCC 1145 (17 mm)}.

a Brown seaweed S. myriocystum. The inhibitory activities of the B. subtilis MTCC 10407 associated with S. myriocystum against V. vulnificus MTCC 1145 on Mueller Hinton agar plates and the seaweed are shown as insets. b The Gram-stained B. subtilis MTCC 10407. The antibacterial activities of ethyl acetate extract of B. subtilis MTCC 10407 and the compounds purified (1–2) against A. hydrophilla on Mueller Hinton agar plates were shown as inset. EA ethyl acetate extract of B. subtilis MTCC 10407. The bactericidal inhibition zones are visualized as clearance around the disk impregnated with the test materials. c Antibacterial activities of isolated compounds (1–4) against A. hydrophilla on Mueller Hinton agar plates. d Inhibitory activities of (1–4) against V. alginolyticus MTCC 4439 as visualized on MTT-sprayed Mueller Hinton agar plates. The live cells were changed to blue color. The bactericidal zones of the active compounds were visualized as yellow color
Antibacterial activities of chromatographic fractions of S. myriocystum and associated B. subtilis MTCC 10407 by agar diffusion method
The ethyl acetate extract (10 μg on disk) of B. subtilis MTCC 10407 exhibited an inhibitory zone diameter of >20 mm against the experimental pathogens (Fig. 1b; Table 3). The compounds 1 and 2 isolated from B. subtilis MTCC 10407 demonstrated significant antibacterial activity (inhibitory zone diameter greater than 15 mm against A. hydrophilla, 10 μg on disk) against the same pathogens (Fig. 1c; Table 3). The seaweed-derived compounds (3 and 4) showed a negligible inhibition zone (<10 mm) (Fig. 1c; Table 3). These results were further validated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) spray on the Mueller Hinton agar plates (Fig. 1d).
Structural characterization of antibacterial O-heterocycle pyrans from B. subtilis MTCC 10407
Two novel O-heterocycle pyrans, 1 and 2 (as stated in the materials section) were isolated upon repeated chromatography in which compound 1, a new substituted O-heterocycle pyran, was isolated as yellowish oil. IR bending vibration bands of compound 1 at 1738 cm−1 is attributed to ester carbonyl absorption. The IR spectrum explained a broad absorption band at ν max of 1648 cm−1, symbolizing the olefinic system. Its molecular ion peak at m/e 386 (HRESIMS m/e 387.2315 [M + H]+) along with the 1H and 13C NMR experiments (Table 1) implied the elemental composition of C23H30O5 as having nine degrees of hydrogen deficiencies (Fig. S1). The 13C NMR spectrum in conjunction with DEPT experiments indicated 23 carbons in 1, together with carbonyl carbons (two in numbers) at δ 179.99 (C-11) and δ 167.72 (C-7); olefinic protons at δ 138.77 (C-15) and 123.12 (C-16) (Table 1). 1H–1H COSY couplings between the protons at δ 0.92 (H-25)/δ 1.26 (H-24)/δ 1.44 (H-20)/δ 1.01 (H-21)/δ 0.88 (H-22) in the spectrum supported the presence of 3-ethyl butane moiety (Fig. S2). Two methylene groups were attributed to C8–9 positions, and the one with δ 4.31 exhibited downfield shift due to the extended conjugation probably linked to an aromatic moiety. The aromatic protons showed their characteristic signals at δ 7.78–7.58. 1H–1H COSY experiments revealed that the protons at δ 4.31 (t) correlate with those (−CH2− protons) at δ 1.72 (assigned to be as H-9) and that at δ 2.38, the latter is assigned to be attached to a strongly electronegative group. HMBC correlations were apparent between H-9 (δ 1.72) with that of a carboxyl carbon at δ 179.99. The >C=O group at the C-11 position of 1 ensued intense deshielding of the methine proton at δ 2.38, and likewise, was accounted for the C-10 position. Proton chemical shifts at δ 1.53, 2.04, 5.37, and 4.09 along with detailed 2D NMR experiments established the presence of O-heterocyclic pyran network. The –CH− proton (t) recorded at δ 4.23 (C-14) is distinctive of the juncture of the bicyclic system. The low field quaternary signal (13C NMR) was in accordance with the presence of C-7 carbonyl group of 1, which was corroborated by the comparatively downfield shift of H-8 proton (δ 4.31), and likewise, referred to a possible oxygenation. The HMBC correlations from H-13 (δ 1.68) to C-14 (δ 72.41); H-14 (δ 4.23) to C-15 (δ 138.77), C-18 (δ 71.79); H-16 (δ 5.34) to C-17 (δ 38.05); H-17 (δ 2.04) to C-18 (δ 71.79) together with 1H–1H COSY relations, such as H-13 (δ 1.68)/H-14 (δ 4.23); H-16 (δ 5.34)/H-17 (δ 2.04)/H-18 (δ 4.09) established the pyrano-pyranone framework. The C–H and H–H connectivities in the HMBC and 1H–1H COSY spectra, respectively, implied that the nine degrees of hydrogen deficiencies were due to the three rings and six double bonds. NOE couplings were realized between Hα-14/Hα-10, which indicates that these groups must be disposed on the same side of the molecule. Therefore, the C-11 carboxyl group was attributed to be oriented at the opposite side (β-disposed) of the protons designated as Hα-14/Hα-10. The methine proton attributed to C-16 did not exhibit NOE relationship with α-oriented H-14 and H-18 protons, and was therefore, disposed at the opposite face of the protons H-14/H-18. The molecular ion peak at m/e 386 eliminated benzoic acid (m/e 122) to yield 3-ethyl-7-(2-ethylbutyl)-tetrahydropyrano-[3,2b]-pyran-2(3H)-one at m/e 266. The occurrence of the fragment ion at m/e 154 designated the presence of tetrahydropyrano-[3,2b]-pyran-2 (3H)-one moiety, resulted from the side chain elimination in 3-ethylpentane (Fig. S1). Intramolecular rearrangement of tetrahydropyrano-[3,2b]-pyran-2-(3H)-one resulted in the formation of heptenoic acid (m/e 128) via the intermediate (E)-6-ethylidene-tetrahydropyran-2-one (m/e 126). The presence of tropylium ion (m/e 91) supports the presence of aryl ring system in 1.
The IR absorption band of 2, yellowish oil, exhibited close resemblance with that of 1, except the presence of greater olefinic signals near 1653 cm−1, which apparently indicated that 1 and 2, shared close structural similarities. The molecular ion peak was recorded at m/e 399 (HRESIMS m/e 399.2268 [M + H]+), which along with the 1H/13C NMR data (Table 1) designated the elemental composition of 2 as C24H30O5 with ten degrees of unsaturation. A significant similarity in the NMR spectral data between 2 and 1 indicated their structural similarity. However, unlike the compound 1, no DEPT signal was apparent for the C-16 for 2, which indicated the carbon as quaternary (δ 142.78). Additional olefinic signals were apparent at δ 130.14, 123.78, and 114.63. The 1H–1H COSY correlation with H-18 (δ 5.27 dd, 1H)/H-19 (δ 1.94–1.97, m, 2H)/H-20 (δ 5.80)/H-21 (δ 4.97, dd, 1H)/H-22 (δ 0.87, d, 3H) supported the presence of the six member hexadiene moiety (Fig. S3). The attachment of olefinic proton at H-18 (δ 5.27) to the oxopyrano pyran at C-12 (δ 53.61) was confirmed from the detailed HMBC analyses. (Figs. S3 and S4; Table 1). The 1H NMR in conjugation with 13C NMR recorded the signals at δ 2.38, 1.62, 3.91, and 3.51, and the 1H–1H COSY correlations were evident between these protons assigned to be as H-10/H-14/H-13/H-12 supported the presence of six member tetrahydropyranone ring systems. The proton at δ 4.32 along with the quaternary carbon atom at δ 132.96, which were attributed to the junction point (at the C-7 position of 2) between the aromatic ring and carboxyl ester group, experienced intense deshielding. 1H–1H COSY correlations and detailed HMBC experiments revealed the presence of methine proton at δ 2.38 between the tetrahydropyranone ring and ethyl benzoate moiety (Table 1). The cyclization point of the substituted lactone skeleton was determined by the downfield shift of H-10 at δ 2.38, which also gives clear 1H–1H COSY correlation with H-10/H-14/H-13, thus supporting the presence of the six-member lactone moiety. The relative stereochemistry of C-10, C-12, and C-17 of the hexahydropyrano-[3,2b]-pyranone framework was inferred by detailed NOESY experiments. NOE correlations were apparent between Hα-13 (δ 3.91)/Hα-10 (δ 2.38) thus indicating that these groups must be on the equal plane of the molecule (Fig. S4). The C-12 methine proton did not display NOE interactions with α-oriented H-13 and H-10, thus indicating that H-12 is disposed at the opposite plane of the compound 2.
Structural characterization of secondary metabolites from seaweed S. myriocystum
Two compounds 3 and 4 (as stated in the materials section) were isolated as white crystalline solid and amorphous yellow powder, respectively. The structural details of the compounds were as follows.
The IR bending bands of compound 3 at 1736.96 cm−1 attributed to the ester carbonyl absorption. The mass spectra of 3 accounted for the molecular ion peak at m/e 416 (HRESIMS m/e 417.2768 [M + H]+) enclosing eight degrees of unsaturation, and the elemental composition of C27H36O3 based upon combined 1H and 13C NMR data (Table 2). The comparison of 13C NMR between the compounds 1 isolated from B. subtilis MTCC 10407 and 3 from seaweed S. myriocystum was illustrated in Fig. 2a, b. The 13C NMR/DEPT spectra of 3 connoted the presence of 25 carbons, along with the carbonyl carbons (at δ 173.30 and δ 167.70) and olefinic carbons at δ 132.46 and 114.05 (Table 2). The –CH2- protons appeared at δ 4.11 was on account of the existence of dihydropyran framework, and was attributed to C-18 position of the heterocyclic skeleton. 1H–1H COSY experiments revealed that the protons at δ 4.33 (d) correlated with the methylene protons at δ 1.72 (attributed to H-9) and the –CH proton at δ 2.34, the latter was assigned to be attached to a strongly electronegative group (Fig. S5). A typical signal characteristic of –OCH3 group was apparent at δ 3.69, which enabled to deduce the presence of methoxy carbonyl moiety in the compound. It was further confirmed from the HMBCs between H-12 (δ 3.69) and carbonyl carbon, C-11 (δ 173.30). The methine proton (t) at δ 4.24 has been attributed to the junction of 3, 6-dihydro-2H-pyran moiety with that of the side chain. Detailed 1H–1H COSY, HMBC and HSQC experiments affirmed the proton and carbon connectivity of the 3-ethyl butane moiety connected with the dihydro-2H-pyran framework at the C-17 position of the ring structure (S5-S6). Intense 1H–1H COSY correlations were evident between δ 2.02 (−CH, assigned to be as C-17)/δ 1.52 (−CH2, C-19)/δ 1.44 (−CH2, C-20)/δ 1.02 (−CH2, C-21)/δ 0.88 (−CH3, C-22), which along with the results detailed above support the presence of 3-butyl-3,6-dihydro-5-methyl-2H-pyran moiety. The other HMBC relations from H-13 (δ 1.61) to C-14 (δ 68.16); H-17 (δ 2.02) to C-18 (δ 71.79), C-20 (δ 22.63); H-18 (δ 4.11) to C-16 (δ 129.93); H-20 (δ 1.44) to C-25 (δ 13.96) were further confirming the structure of title compound. The combined spectral analyses unequivocally explained the presence of the relative stereochemistry (NOESY spectrum and the J-values) of the chiral centers, especially that of C-10 (with δH 2.34 of proton at C-10, HSQC) bearing the –COOMe moiety, and that of C-14 (bearing the –CH group proton at δ 4.24) and C-17 (bearing the –CH group proton at δ 2.02). NOE couplings were observed between Hα-10 (δ 2.34)/ Hα-17 (δ 2.02), which indicate that these protons were disposed on the equal plane of the molecule (Fig. S6).

Comparison of 13C NMR between the compounds a 1 isolated from B. subtilis MTCC 10407 and b 3 from seaweed S. myriocystum
The IR bending vibration bands of compound 4 at 1742 cm−1 attributed to the ester carbonyl absorption. The mass spectra of the title compound 4 accounted for the molecular ion peak at m/e 370 [HRESIMS m/e 371.2678 (M + H)+] bearing eight degrees of unsaturation, and the molecular formula as C24H34O3 based upon combined 1H and 13C NMR data (Table 2). Two –CH2− groups were attributed to occur at the C8–9 positions, and the one with δ 4.22 showed a downfield shift as a result of an extended conjugation, which was probably linked to an aromatic moiety. The 13C/DEPT spectra designated the existence of 24 carbons along with one carbonyl at δ 167.70 and olefinic carbons at δ 130 and 125.98 (Table 2). The HMBC correlation of the proton at δ 4.22 with the carbon atom at δ 167.72 apparently indicated the presence of –C=O(O) moiety. 1H–1H COSY experiments revealed that the protons at δ 4.22 (t) correlate with the methylene protons (δ 1.72, attributed to H-9) and HMBC correlations were apparent between H-9 (δ 1.72) with that of a carboxyl carbon at δ 167.72, which apparently indicated the presence of ethyl benzoate moiety. The C-18 position of 4 appeared on account of intense deshielding (δ 4.08) of –CH2− proton probably linked to oxygen. The chemical shift of the protons at δ 4.08, 2.02, 1.57, 2.18, and 3.78 ppm along with detailed 2D NMR experiments established the presence of pyranose moiety. This detailed spectral experiments established that the C4 skeleton was attached to the C-12 position of the hexahydro-2H-chromene framework (Figs. S7 and S8). The HMBC spectroscopic values further confirmed the structure, which were H-13 (δ 3.78) to C-14 (δ 40.97) and C-9 (δ 38.05); H-14 (δ 2.30) to C-13 (δ 72.41), C-18 (δ 71.79), and C-17 (δ 34.62); H-18 (δ 4.08) to C-20 (δ 13.74) and C-14 (δ 40.97); and H-19 (δ 0.98) to C-18 (δ 71.79) and C-17 (δ 34.62). The relative stereochemistry of C-10 chiral center (with δH 1.36, HSQC) carrying the –C=C− group and that of C-7 (bearing the –CH2 group proton at δ 4.29) and C-18 (bearing the –CH2 group proton at δ 4.08), was deduced by extensive NOESY experiments (Fig. S8). NOE couplings were observed between Hα-10 (δ 1.36)/Hα-18 (δ 4.08), which indicate that these protons were disposed on the equal plane of the molecule. NOESY correlation between 10-H (δ 1.36) and 11-H (δ 5.17) designated that they were on the same plane of 4. The mass fragmentation pattern of compounds 3 and 4 from seaweed S. myriocystum are shown in Figs. S9 and S10.
Discussion
The seaweed-associated bacterial flora was reported as symbiotic organisms on seaweeds and is potential sources of antibacterial metabolites. Aeromonas hydrophila, V. vulnificus, and V. parahaemolyticus, which were reported to be as human pathogens, and cause gastroenteritis and lethal instances of septicemia in immunocompromised individuals, were used to understand the antagonistic properties of the seaweed-associated bacteria. The study of the chemical ecology of living surfaces of marine organisms and the symbiotic relationships between seaweeds and their microbial flora can provide important biotechnological information with significance for the production of valuable antimicrobial metabolites. Bacillus species of marine origin was found to produce bioactive compounds belonging to macrolactones and polyketide analogs (Armstrong et al. 2001). Kanagasabhapathy et al. (2008) investigated the highest activity of Bacillus species in particular Bacillus cereus and Bacillus pumilus from some red seaweed and which was in good agreement with present studies since the major populations of our isolates were Bacillus comprising B. subtilis, Bacillus amyloliquefaciens, and B. cereus.
In our survey, the antagonistic bacteria associated with seaweed S. myriocystum predominantly represented by the phylum Firmicutes (Bacillus spp.) which was reported to dominate among the strains with polyketide synthase (pks) genes (Chakraborty et al. 2014). The bacterial strain MTCC10407 (NCBI accession number-JF834075), used in the current study, showed identity to B. subtilis strain jinfen 1 (JX960641). The pks gene constitutes essential molecular machines that take part in the biosynthesis of various natural products bearing polyketide chemistry (Donadio et al. 2007). B. subtilis MTCC 10407 under the present study is found to be positive for pks gene with an NCBI accession number KC607823. The phylogenetic study has shown that the amplified gene products were of bacterial type I pks (results not shown). Earlier studies showed that bacterial type I pks has potential to produce a broad spectrum of biomedically important secondary metabolites (Piel et al. 2004). During our study of biologically active natural products from seaweeds and its associated bacteria, we could notice that the antibacterial activities of seaweed-derived compounds were lesser than those of the associated bacterial communities (Chakraborty et al. 2014, 2010). This led us to investigate whether bacteria or the seaweed is the original source of antibacterial compounds so that the present study dealt with the B. subtilis strain MTCC10407 associated to seaweed host S. myriocystum as a representative organism to explain the chemical interactions of seaweed and its affiliated bacterial flora. The guiding principles to determine the bioactivity of the secondary metabolites from bacteria vis-à-vis seaweed was designed by utilizing electronic, hydrophobic, and steric parameters (Chakraborty et al. 2014). The descriptors, polarisability (Pl), molecular polar surface area (tPSA), hydrophobic parameter (logarithmic scale of the octanol/water partition coefficient, log Pow), molar refractivity (MR), molar volume (MV), and parachor (P) (Table 4) were experimentally determined or adopted from ChemDraw Ultra 8.0/ACD Chemsketch (version 8.0) databases.
Using bioassay-guided fractionation, two antibacterial compounds 1 (2-(7-(2-ethylbutyl)-2,3,4,4a,6,7-hexahydro-2-oxopyrano-[3, 2b]-pyran-3-yl)-ethyl benzoate) and 2 [2-((3Z)-2-ethyl-octahydro-6-oxo-3-((E)-pent-3-enylidene)-pyrano-[3, 2b] pyran-7-yl)-ethyl benzoate] bearing the typical polyketide backbone, with potential antibacterial activity against human food pathogenic bacteria, have been isolated from the ethyl acetate extract of from B. subtilis MTCC 10407. The multifactorial polyketide structures are endued with supplementary O-heterocyclic moieties that contribute rigor to polyketide structure. Two homologous compounds 3 (3-(methoxycarbonyl)-4-(5-(2-ethylbutyl)-dihydro-3-methyl-2H-pyran-2-yl)-butyl benzoate) and 4 [2-(8-butyl-3-ethyl-hexahydro-2H-chromen-6-yl)-ethyl benzoate] also have been isolated from the ethyl acetate extract of host seaweed S. myriocystum. It is interesting to note that the tetrahydropyran-2-one moiety of the tetrahydropyrano-[3, 2b]-pyran-2(3H)-one system of 1 might be cleaved by the metabolic pool of seaweeds to afford methyl 3-(dihydro-3-methyl-2H-pyranyl)-propanoate moiety of 3, which was found to have no significant antibacterial activity (Fig. 3). It is therefore imperative that the presence of dihydro-methyl-2H-pyran-2-yl propanoate system is essentially required to impart the greater activity (IZD 17 mm, 10 μg on disk). It is significant to note that seaweed might metabolically engineer the parent compound 1 of bacterial origin in its metabolic pool for some other reasons probably related to the structural integrity and rigidity of the seaweed cell wall. This hypothesis can further be supported by the fact that transformed product 3 has greater values of steric descriptors (P 982.5 cm3, MR 118.59 cm3/mol, MV 402.4 cm3) than the parent compound 1 (P 872.9 cm3, MR 107.71 cm3/mol, MV 338.0 cm3) purified from B. subtilis MTCC 10407. The biotransformation of the bacterial metabolite to 3 in seaweed might thus contribute to the adaptive mechanism of the seaweed to form a tougher cell wall to resist the pathogenic bacterial flora in the oceanic ecosystem. Thus, there exists an interesting chemical ecological interaction between the secondary metabolites produced by the seaweed bacteria and the seaweed host organism.

Hypothetical biosynthetic transformation of 1 in the seaweed metabolic pool by methyltransferase reactions that utilized S-adenosyl methionine type of compound. The oxygen atom of 1 engages in SN2 nucleophilic attack on the electrophilic methyl carbon of S-adenosyl methionine to afford 3-(methoxycarbonyl)-4-(5-(2-ethylbutyl)-3,6-dihydro-2H-pyran-2-yl)-butyl benzoate. This followed the electrophilic addition reactions to asymmetrical alkenes, where the nucleophile is a π-bond on 4-(benzoyloxy)-2-((5-(2-ethylbutyl)-5,6-dihydro-2H-pyran-2-yl)methyl)-butanoic acid, proceed through the stabilization of the carbocation intermediate to yield 3-(methoxycarbonyl)-4-(3,6-dihydro-3-methyl-5-(2-methylbutyl)-2H-pyran-2-yl)-butyl benzoate. This was followed by the double-bond shifting in the pyran ring system of 3,6-dihydro-3-methyl-5-(2-methylbutyl)-2H-pyran-2-yl to 5,6-dihydro-3-methyl-5-(2-methylbutyl)-2H-pyran-2-yl
The antibacterial compound 2 isolated from the ethyl acetate extract of from B. subtilis MTCC 10407 and 4 from seaweed S. myriocystum shared similar structures, and therefore, might be the result of the identical metabolic pool of seaweed and bacteria. In particular, the presence of ethyl benzoate moiety in 2 and 4 from seaweed-associated bacteria and seaweed strongly suggested the ecological and metabolic relationship between these compounds. The seaweed derived metabolite 4 did not show an appealing antibacterial property, and therefore, it can be concluded that the compound has a different role, probably related to structural functionality of seaweed. It is interesting to note the greater hydrophobic coefficient of 4 (log P ow 5.68) than that recorded in 2 (log P ow 4.28), which apparently support its role in cell wall structure and its integrity. Polyketides were found to occupy an emerging pertinence as potential pharmacological leads in drugs and nutraceutical products for utilization against disease causing microorganisms and different immune-compromising ailments. Among different bacterial genera, Bacillus spp. were perceived to possess different pks gene groups and bioactive compounds with polyketide moiety. The β-ketoacyl synthase (KS) domains are imparted in pks gene groups, and hence, the KS domains are valuable to screen for microbial pks genes. The present study demonstrated that the antibacterial polyketide metabolites of B. subtilis MTCC 10407 was recognized to represent the platform of pks-1 gene encoded products.
It is of note that the hydrophobic (log P ow) and steric descriptors (P, MR, and MV) had a major role to describe the bioactivity of compound 1 isolated from B. subtilis MTCC 10407 and compound 3 from the host seaweed. Although the tPSA depicting the electronic descriptor is identical (61.83) in 1 and 3, the activity of the latter was lesser (IZD 8 mm; 10 μg on disk) than of the former (IZD 17 mm; 10 μ on disk), apparently due to the greater steric values (P = 982.5 cm3) of 3 than that recorded in 1 (P = 872.9 cm3) (Table 4). The compound 2 isolated from B. subtilis MTCC 10407 also demonstrated to possess greater antibacterial activities against pathogenic organisms than 4 purified from S. myriocystum. It is of note that unlike compound 1, the compound 2 with 6-oxo-3-(pentenylidene) pyrano-[3,2b] pyranyl moiety showed greater polarisability (44.8 × 10−24 cm3), and therefore, the electronic descriptors might play a predominant role in determining the antibacterial activity. The electronic factor such as tPSA was found to significantly contribute towards the greater antibacterial activity of 2. In particular, the tPSA of 2 was recorded to be significantly greater (61.8) than the related seaweed metabolite 4, which recorded a lesser tPSA value (35.5). This might be due to the absence of the –O–C=O group from 4, possibly due to the biochemical transformation of 2 in the seaweed metabolic pool. The electronic descriptor, Pl was found to be directly involved with the antibacterial properties of 2, which signified that electronic (inductive/field) rather than bulk factors (parachor) play predominant roles to determine the antibacterial properties of the title compound.
The polyketide gene network of type I pks shelters the acyl carrier protein (ACP) and KS domains, whereas the former was found to collegially catalyze decarboxylative type of Claisen condensation (Chakraborty et al. 2014) comprising different starter/extender entities on ACP and KS subunits of pks. This result in the polyketide chain extension resulting in the formation of ACP-S-7-(2-ethylbutyl)-hexahydro-3-(2-hydroxyethyl)-2-mercaptopyrano-[3,2b]-pyran-2-ol as an intermediate (Fig. 4a). A sequel of reactions comprising condensation, dehydration and ketoreduction lead to the formation of 2-(7-(2-ethylbutyl)-hexahydro-2-oxopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate. Notably, the polyketide biosynthetic avenue resulting in the formation of 2-(octahydro-2-oxo-7-(pent-3-enylidene) pyrano-[3,2b]-pyran-3-yl)-ethyl benzoate is a pks-derived product (Fig. 4b). A putitive biosynthetic route leading to the formation of the pks gene product can be proposed (Fig. 4c, d), which accounted S-3-(1-hydroxypropyl)-4,7-dioxooctanethioate as the starter elementary unit rather than acetate/malonate. Earlier reports indicated the concerted zipper-type cyclization reaction of polyepoxides by pks-1 system through a semiacetal hydroxy group (Bhatt et al. 2005; Gallimore et al. 2006). Similar biosynthetic routes have been postulated with regard to the biosynthesis of maitotoxin polyethers and heterocyclic pyran-derived tetronomycin of marine origin bearing the polyketide frame of reference (Gallimore et al. 2006). The connections of pks metabolite gene with auxiliary metabolites fitting in with polyketide analogs, and their biosynthetic pathways in relevant microbes were accounted for in prior reports (Moldenhauer et al. 2007). The pks gene uses malonyl/methylmalonyl/ethylmalonyl acyl coenzyme A starter units and amino/hydroxyl/alkoxy malonyl-ACP extenders to produce distinct groups of polyketide compounds (Zhao et al. 2008). It is of note that during the course of pks-catalyzed biosynthesis of 2-(7-(2-ethylbutyl)-hexahydro-2-oxopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate, the ACP and KS subunits of the enzyme build their products from the starter unit of 2-(7-(2-ethylbutyl)-hexahydro-2-oxopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate, whereas malonate has been used as an extender unit. The biosynthetic route of 2-(7-(2-ethylbutyl)-hexahydro-2-oxopyrano-[3, 2b]-pyran-3-yl)-ethyl benzoate starts from thioacetate unit attached to ACP involving a sequel of ketoreduction and decarboxylative Claisen condensation. The biosynthetic route of pyran ring system features the extraordinary blueprint of nature to yield complex molecules from simpler units by pks enzymatic cascade.


a Hypothetical pks-1 assisted biosynthetic pathway of 1. The intermediate products were built upon 5-ethyl-3-(hydroxymethyl) heptanethioate starter and malonate extenders. ACP-S-7-ethyl-5-(hydroxymethyl)-3-oxononanethioate was shown to form by the process of decarboxylation of malonyl-ACP unit on the KS domain. The 7-ethyl-5-(hydroxymethyl)-3-oxononanethioate group is then moved to the KS domain from ACP. KS-catalyzed decarboxylative Calisen condensation leads to the formation of ACP-S-9-ethyl-2-(2-hydroxyethyl)-7-(hydroxymethyl)-3,5-dioxoundecanethioate. The latter undergoes ketoreduction followed by dehydration to afford ACP-S-9-ethyl-3,5-dihydroxy-2-(2-hydroxyethyl)-7-(hydroxymethyl) undecanethioate. The mercaptyl sulfur of the resulting product experiences a nucleophilic attack by the 5OH in substituted undecanethioate yielding ACP-S-6-(4-ethyl-2-(hydroxymethyl)hexyl)-3,6-dihydro-3-(2-hydroxyethyl)-2-mercapto-2H-pyran-2-ol. C5 olefinic carbon atom of the 2H-pyran ring system undergoes intramolecular nucleophilic attack by the 2′-OH group of 4-ethyl-2-(hydroxymethyl)hexyl moiety to afford ACP-S-7-(2-ethylbutyl)-octahydro-3-(2-hydroxyethyl)-2-mercaptopyrano-[3,2b]-pyran-2-ol, which undergoes dehydrogenation yielding ACP-S-7-(2-ethylbutyl)-hexahydro-3-(2-hydroxyethyl)-2-mercaptopyrano-[3,2b]-pyran-2-ol. b The biosynthetic route of 1 starts from thioacetate unit enclosed with the ACP domain, and was followed by sequential decarboxylative Claisen condensations/ketoreduction. DH catalyzed the formation of KR product benzothioic S-ACP by the removal of a molecule of water from the intermediate 5,7-dihydroxy-3-oxooctanethioic S-ACP. The nucleophilic attack of 2OH group (in 2-hydroxyethyl group) of 7-(2-ethylbutyl)-hexahydro-3-(2-hydroxyethyl)-2-mercaptopyrano-[3,2b]-pyran-2-ol-S-ACP on benzothioic S-ACP result in the formation of 2-(7-(2-ethylbutyl)-hexahydro-2-hydroxy-2-mercaptopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate-S-ACP. A successive removal of ACP-SH resulted in the formation of 2-(7-(2-ethylbutyl)-hexahydro-2-oxopyrano-[3,2b]-pyran-3-yl)-ethyl benzoate. c The array of events in putitive biosynthetic pathway of pks product as compound 2, displaying the pks-1 catalyzed steps of decarboxylation and elongation. The intermediate products were built upon S-3-(1-hydroxypropyl)-4,7-dioxooctanethioate starter and malonate extenders. ACP-S-5-(1-hydroxypropyl)-3,6,9-trioxodecanethioate was shown to form by the process of decarboxylation of malonyl-ACP unit on the KS domain. The latter undergoes ketoreduction and dehydration to afford ACP-S-5-(1-hydroxypropyl)-3-oxodeca-5,8-dienethioate. The ACP-S-5-(1-hydroxypropyl)-3-oxodeca-5,8-dienethioate group is then moved to the KS domain from ACP. KS-catalyzed decarboxylative Calisen condensation of ACP-S-7-ethyl-5-(hydroxymethyl)-3-oxononanethioate resulting in the formation of ACP-S-2-(2-hydroxyethyl)-7-(1-hydroxypropyl)-3,5-dioxododeca-7,10-dienethioate. Two alternate steps of ketoreduction of the latter yielded ACP-S-3-hydroxy-2-(2-hydroxyethyl)-7-(1-hydroxypropyl)-5-oxododeca-7,10-dienethioate. d DH catalyzed the formation of KR product ACP-S-5-hydroxy-2-(2-hydroxyethyl)-7-(1-hydroxypropyl) dodeca-3,7,10-trienethioate by the removal of one molecule of water from the intermediate ACP-S-3-hydroxy-2-(2-hydroxyethyl)-7-(1-hydroxypropyl)-5-oxododeca-7,10-dienethioate. The nucleophilic attack of 5OH group {in 5-hydroxy-2-(2-hydroxyethyl) moiety} on ACP-S-5-hydroxy-2-(2-hydroxyethyl)-7-(1-hydroxypropyl)dodeca-3,7,10-trienethioate result in the formation of 2H-pyranyl analog, 3,6-dihydro-3-(2-hydroxyethyl)-6-(2-(1-hydroxypropyl)hepta-2,5-dienyl)-2-mercapto-2H-pyran-2-ol. A consecutive intramolecular nucleophilic attack by 1-hydroxypropyl moiety in the 2-(1-hydroxypropyl)hepta-2,5-dien-1-yl) system on the C-3 of the 2H-pyran ring system yielded 6-ethyl-3-(2-hydroxyethyl)-2-mercapto-7-(pent-3-en-1-ylidene)-octahydropyrano-[3,2b]-pyran-2-ol-S-ACP. Benzothioic S-ACP experiences nucleophilic attack by the 3OH group in 3-(2-hydroxyethyl)-system of 6-ethyl-3-(2-hydroxyethyl)-2-mercapto-7-(pent-3-en-1-ylidene)octahydropyrano-[3,2b]-pyran-2-ol-S-ACP yielding pyrano [3,2b]pyran-3-yl) ring system of ACP-S-2-(6-ethyl-octahydro-2-hydroxy-2-mercapto-7-(pent-3-enylidene)pyrano-[3,2b]-pyran-3-yl)-ethyl benzoate. A subsequent elimination of ACP-SH afforded the final PKS product as 2-(octahydro-2-oxo-7-(pent-3-enylidene)-pyrano-[3,2b]-pyran-3-yl)-ethyl benzoate
The previous decade saw the disclosure of different bacterial polyketide leads with colossal significance as novel alternatives to the existing antibiotics for use against multi-drug resistant human opportunistic food pathogens. In this study, we depicted two new variants of antibacterial O-heterocyclic pyran derivatives of polyketide origin. These novel polyketide products may function as promising new-generation drug templates to counter the multi-resistant human food pathogenic bacteria. These polyketides are unprecedented from seaweeds, and the structural similarity of the seaweed-derived homologous compounds to the microbial secondary metabolites proposes that these might be the products of symbiotic bacteria dwelling on host seaweeds and is biotransformed by the seaweed metabolic pool. We have likewise given confirmation to a putitive biosynthetic course of these compounds, and this may prompt to identify novel drug target. The present work may have a footprint on the use of O-heterocyclic polyketide products for biotechnological, food, and pharmaceutical applications mainly as novel antimicrobial secondary metabolites.
References
Ali AIB, Bour ME, Ktari L, Bolhuis H, Ahmed M, Boudabbous A, Stal LJ (2012) Jania rubens Associated bacteria: molecular identification and antimicrobial activity. J Appl Phycol 24:525–534
Armstrong E, Yan L, Boyd KG, Wright CP, Burgess JG (2001) The symbiotic role of marine microbes on living surfaces. Hydrobiologia 461:37–40
Bhatt A, Stark CBW, Harvey BM, Gallimore AR, Demydchuk YA, Spencer JB, Staunton J, Leadlay PF (2005) Accumulation of an E,E,E-triene by the monensin producing polyketide synthase when oxidative cyclization is blocked. Angew Chem 117:7237–7240
Chakraborty K, Lipton AP, Paulraj R, Chakraborty RD (2010) Guaiane sesquiterpenes from seaweed Ulva fasciata Delile and their antibacterial properties. Eur J Med Chem 45:2237–2244
Chakraborty K, Thilakan B, Raola VK (2014) Polyketide family of novel antibacterial 7-O-methyl-5′-hydroxy-3′-heptenoate macrolactin from seaweed-associated Bacillus subtilis MTCC 10403. J Agric Food Chem 62:12194–12208
Donadio S, Monciardini P, Sosio M (2007) Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat Prod Rep 24:1073–1109
Gallimore AR, Stark CB, Bhatt A, Harvey BM, Demydchuck Y, Bolanos-Garcia V, Fowler DJ, Staunton J, Leadlay PF, Spenceer JB (2006) Evidence for the role of the monB genes in polyether ring formation during monensin biosynthesis. Chem Biol 13:453–460
Goecke F, Labes A, Wiese J, Imhoff JF (2010) Chemical interactions between marine macroalgae and bacteria. Mar Ecol Prog Ser 409:267–300
Hollants J, Leliaert F, Clerck OD, Willems A (2012) What we can learn from sushi: a review on seaweed-bacterial associations. FEMS Microbiol Ecol 83:1–16
Kanagasabhapathy M, Hideaki S, Shinichi N (2008) Phylogenetic identification of epibiotic bacteria possessing antimicrobial activities isolated from red algal species of Japan. World J Microbiol Biotechnol 24:2315–2321
Kubanek J, Jensen PR, Keifer PA, Sullards MC, Collins DO, Fenical W (2003) Seaweed resistance to microbial attack: a targeted chemical defense against marine fungi. Proc Natl Acad Sci U S A 100:6916–6921
Kumar V, Rao D, Thomas T, Kjelleberg S, Egan S (2011) Antidiatom and antibacterial activity of epiphytic bacteria isolated from Ulva lactuca in tropical waters. World J Microbiol Biotechnol 27:1543–1549
Moldenhauer J, Chen XH, Borriss R, Piel J (2007) Biosynthesis of the antibiotic bacillaene, the product of a giant polyketide synthase complex of the trans-AT family. Angew Chem 119:1–4
Penesyan A, Marshall-Jones Z, Holmstrom C, Kjelleberg S, Egan S (2009) Antimicrobial activity observed among cultured marine epiphytic bacteria reflects their potential as a source of new drugs. FEMS Microbiol Ecol 69:113–124
Piel J, Hui D, Fusetani N, Matsunaga S (2004) Targeting modular polyketide synthases with iteratively acting acyltransferases from metagenomes of uncultured bacterial consortia. Environ Microbiol 6:921–927
Quevrain E, Roue M, Bourguet-Kondracki IM (2014) Assessing the potential bacterial origin of the chemical diversity in calcareous sponges. J Mar Sci Technol 22:36–49
Raola VK, Chakraborty K (2016) Two rare antioxidative prenylated terpenoids from loop-root Asiatic mangrove Rhizophora mucronata (family Rhizophoraceae) and their activity against pro-inflammatory cyclooxygenases and lipoxidase. Nat Prod Res. doi:10.1080/14786419.2016.1174232
Zhang W, Zhang Li F, Miao X, Meng Q, Zang X (2009) Investigation of bacteria with polyketide synthase genes and antimicrobial activity isolated from South China Sea sponges. J Appl Microbiol 107:1364–5072
Zhao J, Yang N, Zeng R (2008) Phylogenetic analysis of type I polyketide synthase and nonribosomal peptide synthetase genes in Antarctic sediment. Extremophiles 12:97–105
Zhao K, Zheng Y, Penttinen P, Guan T, Xiao J, Chen Q, Xu J, Lindstrom K, Zhang L, Zhang X, Strobel GA (2011) The diversity and antimicrobial activity of endophytic actinomycetes from medicinal plants in Panxi Plateau, China. Curr Microbiol 62:182–190
Acknowledgments
This work was supported by funding under Indian Council of Agricultural Research Network Project High Value Compounds (grant no. HVC/ICAR 2012-2017). The authors are thankful to Indian Council of Agricultural Research (ICAR), New Delhi for providing facilities to carry out the work. The authors thank the Director, Central Marine Fisheries Research Institute for support. Thanks are due to the Head, Marine Biotechnology Division, Central Marine Fisheries Research Institute for facilitating the research activity. B.T., V.K.R., and M.J. acknowledge ICAR, for the award of scholarships.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare no competing financial interest.
Electronic supplementary material
ESM 1
(PDF 867 kb)
Rights and permissions
About this article

Cite this article
Chakraborty, K., Thilakan, B., Chakraborty, R.D. et al. O-heterocyclic derivatives with antibacterial properties from marine bacterium Bacillus subtilis associated with seaweed, Sargassum myriocystum . Appl Microbiol Biotechnol 101, 569–583 (2017). https://doi.org/10.1007/s00253-016-7810-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7810-3