
Abstract
Eighteen strains of Escherichia coli were compared for maximum specific growth rate (μ MAX) on 85 mM acetate as the sole carbon source. The C strain ATCC8739 had the greatest growth rate (0.41 h−1) while SCS-1 had the slowest growth rate (0.15 h−1). Transcriptional analysis of three of the strains (ATCC8739, BL21, SMS-3-5) was conducted to elucidate why ATCC8739 had the greatest maximum growth rate. Seventy-one genes were upregulated 2-fold or greater in ATCC8739, while 128 genes were downregulated 2-fold or greater in ATCC8739 compared to BL21 and SMS-3-5. To generate a strain that could grow more quickly on acetate, ATCC8739 was cultured in a chemostat using a progressively increasing dilution rate. When the dilution rate reached 0.50 h−1, three isolated colonies each grew faster than ATCC8739 on 85 mM acetate, with MEC136 growing the fastest with a growth rate of 0.51 h−1, about 25 % greater than ATCC8739. Transcriptional analysis of MEC136 showed that eight genes were downregulated 2-fold or greater and one gene was upregulated 2-fold or greater compared to ATCC8739. Genomic sequencing revealed that MEC136 contained a single mutation, causing a serine to proline change in amino acid 266 of RpoA, the α subunit of the RNA polymerase core enzyme. The 260–270 amino acid region of RpoA has been shown to be a key region of the protein that affects the interaction of the α subunit of the RNA polymerase core enzyme with several global transcriptional activators, such as CRP and FNR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diminishing petroleum reserves, unsustainable net rates of greenhouse gas emissions, and technological advancements have revived interest in the microbial formation of fuels and biochemicals from renewable resources (Stephanopoulos 2007; Bartley and Ronald 2009; Liu and Khosla 2010). Abundant hardwood, softwood, grasses, and agricultural residues contain up to 70 % cellulose and hemicellulose that are closely associated with lignin (Klinke et al. 2004). Pretreatment methods required to remove lignin and make the hemicellulose and cellulose fractions accessible for fermentation inevitably generate inhibitors, which negatively impact the microorganisms that generate the desired biochemical or fuel product (Sun and Cheng 2002; Klinke et al. 2004; Parawira and Tekere 2011).
Acetic acid (acetate) is one of the most abundant inhibitors generated through lignocellulosic pretreatment processes (Pettersen 1984; Sakai et al. 2007), and this acid affects the specific growth rate and biomass yield of microbes utilizing glucose or xylose (Luli and Strohl 1990; Roe et al. 1998, 2002; Takahashi et al. 1999). For example, 8 mM acetate (<0.5 g/L) decreased the growth rate of Escherichia coli on glucose by 50 % (Roe et al. 1998), and an increase in acetate from 34 to 68 mM led to a 60 % decrease rate of CO2 formation by Saccharomyces cerevisiae (Maesen and Lako 1952). A recent review details the tolerance of bacteria to acetic acid (Trĉek et al. 2015).
Several approaches have been proposed to remove acetate prior to microbial conversion, including adsorption with activated charcoal (Carvalho et al. 2006), liquid-liquid extraction (Um et al. 2011), and electrodialysis (Wong et al. 2009, 2010). A microbial method to remove acetate selectively from a mixture has also recently been reported (Lakshmanaswamy et al. 2011; Xia et al. 2012). This method is based on knocking out various sugar uptake systems to yield a strain which will consume only acetate and leave the sugars generated in lignocellulosic hydrolysates unaffected. Such microbial detoxification of acetate-containing lignocellulosic hydrolysates converts a portion of the acetate carbon into cellular metabolites that can be used in subsequent microbial conversion processes. The rate of acetate removal depends on the specific growth rate of the microorganism selected for acetate detoxification. Acetate “tolerance” (i.e., growth rate of cells when exposed to an elevated concentration) may be less important since under various continuous or semi-continuous modes of operation the microorganism is exposed to a very low acetate concentration regardless of its continuous consumption. Despite the broad interest in acetate “detoxification,” little is known about what genetic factors elevate microbial acetate utilization.
Continuous culture systems, such as chemostats, auxostats, and turbidostats, are frequently used for studying evolution and selection (Dykhuizen and Hartl 1983; Sauer 2001). For example, de Crécy et al. (2007) described a small volume turbidostat to adapt a mutant to higher growth rates, and more recently demonstrated the evolution of E. coli for thermophilic growth (Blaby et al. 2012). Using continuous cultures to select for increased growth is straightforward because under various operational modes slow growing members of the population are “washed out” of the system, selectively leaving members with the desired high growth rate phenotype.
The goal of this study was to compare strains of E. coli using acetate as the sole carbon source. The transcriptome of selected strains was compared to elucidate which genes were differentially expressed during growth on acetate. Additionally, the strain with the greatest growth rate was grown in a progressively increasing pseudo-steady-state (chemostat) culture for adaptive evolution, and the fastest growing isolate was sequenced to determine what mutations had occurred.
Materials and methods
Bacterial strains
Eighteen strains of E. coli were used in this study (Table 1), including many commonly used wild-type E. coli strains.
Growth medium and conditions
A defined medium was used for all the shake flask (growth rate) experiments containing the following (per liter): 1.70 g citric acid, 13.30 g KH2PO4, 4.50 g (NH4)2HPO4, 1.2 g MgSO4·7H2O, 13 mg Zn(CH3COO)2·2H2O, 1.5 mg CuCl2·2H2O, 15 mg MnCl2·4H2O, 2.5 mg CoCl2·6H2O, 3.0 mg H3BO3, 2.5 mg Na2MoO4·2H2O, 100 mg Fe(III) citrate, 4.5 mg thiamine·HCl, 8.4 mg Na2(EDTA)·2H2O, and 85 mM acetate using Na(CH3COO)·3H2O. Additional experiments were conducted with the 85 mM lactic acid or 85 mM galactose instead of acetate. In each case, solutions were sterilized separately, combined, and the pH adjusted to 7.0 with a 20 % (w/v) NaOH solution. Cells were routinely cultured on Lysogeny Broth (LB) agar plates, transferred in 1:50 dilution to 50 mL defined medium in 250 mL shake flasks, grown to an optical density (OD) of 2.0, and then transferred again to two to three identical flasks used for growth rate determination and analysis. For each growth rate study, three to six flasks were incubated at 37 °C with an agitation of 350 RPM, and at least six samples examined for OD during the early exponential phase. Samples were stored at −20 °C for subsequent acetate analysis.
Adaptive evolution
Adaptive evolution using the fastest growing strain of E. coli was conducted in a chemostat with 800 mL defined medium in a 2.5-L bioreactor (Bioflo 2000, New Brunswick Scientific Co. Edison, NJ, USA). To prevent wall growth during the process, the glass vessel and the stainless steel parts of the system were coated with PEG-Silane. First, the fermenter was cleaned and hydroxyl groups were created on surfaces by exposure to hot piranha solution (7:3 1.2 M H2SO4:3.0 % H2O2). Silane was used to attach anti-biofouling polyethylene glycol (PEG) to the surface as previously described (Papra et al. 2001; Lan et al. 2005). The PEG-silane was coated onto the all surfaces by 3 days of exposure using 30 mM 2-[methoxy(polyethyleneoxy)propyl] trimethoxysilane (PEG-silane, Gelest SIM6492.7) in toluene. The coating on the stainless steel was confirmed by contact angle analysis, with an average contact angle of 34° indicating the surface presence of the hydrophilic PEG-silane. Air was sparged into the bioreactor at a flow rate of 1.0 L/min, the agitation was 500 rpm, the temperature was maintained at 37 °C, and the pH controlled at 7.0 using 20 % (w/v) NaOH and 20 % (v/v) H2SO4. The feed used for adaptive evolution contained 35 mM acetate, and all other nutrients at one quarter the concentrations used in the shake flask studies. When the dilution rate reached 0.50 h−1, the culture was plated onto LB plates, and three isolates designated MEC134, MEC135, MEC136 were selected for further study.
A spectinomycin-resistant mutant (rpsE) of MEC136 was obtained by plating 0.2 mL of a culture grown overnight on LB onto multiple LB agar plates containing 100 μg/mL spectinomycin. The rpsE mutant was obtained at a frequency of 1 in 109 cells. A P1 lysate was made from MEC136 and used to transfer the rpoA mutation back into ATCC8739 by selecting for spectinomycin resistance.
Analytical methods
The optical density at 600 nm (OD) (UV-650 spectrophotometer, Beckman Instruments, San Jose, CA) was used to monitor cell growth. Liquid chromatography with a refractive index detector and a Coregel 64-H ion-exclusion column (Transgenomic Ltd., Glasgow, UK) using a mobile phase of 4 mN H2SO4 was used for acetate analysis as described previously (Eiteman and Chastain 1997). For dry cell weight measurement, samples at different ODs were centrifuged (8400×g for 10 min), the pellets washed twice with DI water and dried on an aluminum weighing boat at 60 °C for 24 h.
Transcriptome analysis
Samples having OD600 of 0.5–0.6 were mixed with the RNA protect-reagent Bacteria Reagent (Qiagen Sciences Inc., Germantown, MD, USA) in the volumetric ratio of 1:2, incubated for 5 min at room temperature (20–25 °C) and centrifuged (5000×g for 10 min). Supernatant was discarded, and the pellet was stored at −20 °C. Two microliters of TE buffer containing 15 mg/mL lysozyme and 15 μL proteinase K was used as the lysis reagent. Addition of lysis reagent was followed by incubation in a water bath at 37 °C for 10 min and three freeze-thaw cycles (Lahtvee et al. 2009). Total RNA was extracted using QIAGEN RNeasy Mini kit and genomic DNA was removed by on-column DNase digestion using QIAGEN RNase-free DNase set. Single-stranded cDNA was synthesized from 5 μg total RNA in a 40 μL total reaction volume using superscript III first strand synthesis system (Invitrogen, Carlsbad, CA) following manufacturer’s instructions, including RNase H treatment. cDNA cleanup was carried out using QIAGEN Qiaquick PCR purification kit.
E. coli K-12 (4 × 72K format) microarrays were used in this study (Roche NimbleGen catalog #A6697-00-01). The labeling, hybridization, washing, and image scanning steps were performed at a core facility in Florida State University. The samples were labeled with cy3 random nonamers on a hybed on a 4 bay NimbleGen hybridization (Applied Biosystems, Foster City, CA). Scanning of the array was performed using a NimbleGen MS200 2 μm scanner (Roche NimbleGen, Madison, Wisconsin). Raw data extraction, image analysis, and normalization steps were performed using NimbleGen’s DEVA software. ArrayStar software was used for data analysis (DNASTAR, Madison, WI). For each strain, transcriptome analyses were performed in technical duplicates and on three independently grown shake flask cultures (n = 6).
Next generation sequencing (NGS) data analysis
Illumina paired end reads for the parent (ATCC8739) and the evolved strain (MEC136) were generated using the MiSeq platform by Research and Testing Laboratory (Lubbock, TX). The next generation sequencing data from this project has been made available through the NCBI BioProject/Sequence Read Archive data repository and can be accessed via the NCBI BioProject Accession ID: PRJNA314220 or SRA Accession ID: SRP071112. Individual raw data reads from this project can be accessed using SRA Accession ID: SRR3202030 (ATCC 8739) and SRA Accession ID SRR3190692 (MEC136). The parent strain library yielded a total of 2,373,109 sequences with 628,873,885 bases and the evolved strain library yielded a total of 2,373,109 sequences with 635,993,212 bases.
The sequencing reads were checked for quality using FastQC (Andrews 2014) and trimmed of adapters using trimmomatic (Bolger et al. 2014). The trimmed data for each strain was aligned to the E. coli ATCC8739 reference genome (NC_010468.1) using bowtie2 (Langmead and Salzberg 2012). Samtools was used to convert the resulting .sam files to .bam files, index the .bam data files, and generate variant statistics for all nucleotide positions based on an indexed genome file, which was also generated using samtools (Li et al. 2009). Bcftools was then used to generate the variant call files based on the variant statistics (Li et al. 2009). The indexed genome sequence file, gene annotation file for E. coli, indexed.bam file, and the variant call file were co-visualized using the Integrated Genome Viewer (Thorvaldsdóttir et al. 2013) to inspect the depth of coverage at variant call positions for each strain. Depth of coverage, which was evaluated using IGV, ranged from just over 100 to nearly 1900 for both libraries. The confirmed variant positions were then compared for the two strains and a single variant position that was present only in the evolved strain was identified.
Results
Growth rate on acetate
Eighteen strains were compared (three to six replicates each) for growth in defined medium containing acetate as the sole carbon source at a final concentration of 85 mM (Table 2). The C strain ATCC8739 had the greatest specific growth rate of 0.41 h−1, and SCS-1 had the lowest specific growth rate of 0.15 h−1. The five strains with the highest growth rate were all wild-type strains. A wide distribution of growth rates was observed, although 15 of the 18 showed growth rates between 0.26 and 0.37 h−1. Observed biomass yield for the strains was also calculated as the ratio of biomass generated to the acetate consumed, and no correlation was found between the biomass yield and specific growth rate (data not shown).
Transcriptional analysis
Three strains (ATCC8739, SMS-3-5, and BL21) were selected for transcriptional analysis using microarrays to understand why ATCC8739 grows fastest on acetate. These three strains differ in their maximum specific growth rate by approximately 0.05 h−1 from each other in the decreasing order ATCC8739, SMS-3-5, BL21. Seventy-one genes were 2-fold or more upregulated in ATCC8739 compared to SMS-3-5 and BL21 (Supplement, Table S1). The list includes prophage-related genes, phenylacetate metabolic genes, pseudo-genes, and transcriptional regulators. Ten genes (yqe and yge) belong to the type 3 secretion system pathogenicity island ETT2. The ETT2 gene cluster is present in a majority of E. coli strains (pathogenic or commensal) and has been subjected to varying degree of mutational attrition that render it unable to encode a functioning secretion system (Ren et al. 2004). Furthermore, over 90 % of these upregulated genes (66 genes) showed a greater expression level in SMS-3-5 compared to BL21. Thus for these genes, the expression level correlated with the maximum specific growth rate. One hundred twenty-eight genes were 2-fold or more downregulated in ATCC8739 compared to SMS-3-5 and BL21 (Supplement, Table S2). Of these, more than half (61 %, 77 genes) showed a lower expression with SMS-3-5 compared to BL21, and were correlated with maximum specific growth rate.
Adaptive evolution
The strain with the greatest μ MAX, ATCC8739, was evolved for growth rate by exposure to a 35 mM acetate feed at progressively greater dilution rates in a pseudo-steady-state chemostat. Specifically, after achieving steady-state at a growth rate of 0.25 h−1, the dilution rate was increased step-wise by 0.01 h−1 every 6 h, with the culture OD consistently maintained between 1.0 and 1.3. When the dilution rate reached 0.35 h−1, this step-wise increase was made every 8–10 h. After 185 h with a dilution rate of 0.47 h−1, the culture OD had decreased to about 0.4, and the effluent acetate concentration was about 17 mM. In order to ensure that the culture did not wash out, the dilution rate was reduced to 0.45 h−1 for about 10 h, then the step-wise increase in dilution rate continued until the dilution rate attained 0.50 h−1, at which point a sample was plated onto a Petri dish to isolate individual colonies. The experiment corresponded to about 450 generations.
Three isolates designated MEC134, MEC135, and MEC136 from the chemostat operating at 0.50 h−1 were selected to calculate (in triplicate) the maximum specific growth rate on a defined medium containing 85 mM acetate. Whereas the growth rate of ATCC8739 on acetate was determined to be 0.41 ± 0.01 h−1, the μ MAX of all three isolates was greater: MEC134 (0.50 ± 0.01 h−1), MEC135 (0.43 ± 0.01 h−1), and MEC136 (0.51 ± 0.01 h−1).
Transcriptional analysis of the evolved strain MEC136
Transcriptional analysis was performed on the evolved strain with the greatest growth rate on acetate, MEC136, and compared with ATCC8739 to elucidate what genes were differentially expressed in the two strains and might play a role in the elevated growth rate of MEC136 on acetate. Only one gene (ylcG) was upregulated 2-fold or more in MEC136 compared to ATCC8739, while eight genes were downregulated 2-fold or more in MEC136 (Table 3). Interestingly, ylcG was found to be downregulated in ATCC8739 compared to SMS-3-5 and BL21 (Supplement, Table S2), and would thus appear not to correlate with growth rate. Three genes were differentially expressed consistently between the four strains (Supplement, Table S2): mcrB, yihO, yjdQ. Each of these genes was downregulated in MEC136 compared to ATCC8739, and each of these genes was also downregulated in ATCC8739 relative to SMS-3-5 and BL21.
Sequencing and reconstructing the evolved strain MEC136
Next generation sequencing as detailed in “Materials and methods” showed that MEC136 was caused by a single T to C transition mutation at base pair 454,696, which resulted in a S266P change in the α subunit of the RNA polymerase core enzyme coded by the rpoA gene and the presence of the mutation was confirmed by conventional sequencing. Interestingly, neither the evolved MEC134 nor MEC135 strain contained a mutation in rpoA. Because the rpoA gene is less than 4000 bp from the rpsE gene that codes for the 30S ribosomal subunit protein S5, which can be mutated to yield spectinomycin resistance, we expected that P1 lysates prepared from a spectinomycin-resistant mutant of MEC136 would allow us to transduce the rpoA mutation back into wild-type ATCC8739 thus recreating MEC136 at a frequency of about 90 %. The rpoA gene was sequenced in one such transductant (designated MEC537) and found to contain the S266P mutation. MEC537 was determined to have a specific growth rate of 0.52 ± 0.02 h−1 on a defined medium containing 85 mM acetate, statistically identical to the growth rate of MEC136 on the same medium.
Growth rates of the evolved MEC136 strain on other carbon sources
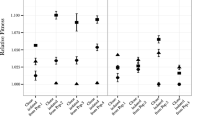
Because the altered RpoA protein could affect the utilization of other carbon sources besides acetate, we also determined the growth rates of MEC136 and MEC537, as well as the wild-type parental strain ATCC8739 and the wild-type strain MG1655 on another organic acid, lactate, and one carbohydrate, galactose (Fig. 1). For each of these three carbon sources, ATCC8739 and its derivatives, MEC136 and MEC537, attained a growth rate significantly greater than MG1655. MEC136 and MEC537 grew significantly faster than ATCC8739 on acetate. In contrast to growth on acetate, the growth rates observed for ATCC8739 were slightly greater than the growth rates observed for either MEC136 or MEC537 for both lactate and galactose. For all these carbon sources, the growth rate of MEC136 was indistinguishable from the growth rate of MEC537.

Comparison of the specific growth rate (h−1) of the four strains MG1655, ATCC8739, MEC136, and MEC537 on defined medium containing 85 mM acetate, lactate, or galactose as the sole carbon source. Data represent mean of three to six replicate experiments and standard error of the mean
Discussion
Growth rate affects the rate of transcription or mRNA turnover, leading to changes in gene expression levels and protein synthesis. Pedersen et al. (1978) studied the expression of 140 individual E. coli proteins as a function of growth rates in five different media. The expression of nearly three quarters of these proteins (102) increased monotonically with growth rate. Similarly, Pease et al. (2002) demonstrated that the expression of pyridoxal 5′-phosphate synthesis genes in E. coli K-12 increased with growth rates, measured as the amount of mRNA synthesized and the rate of protein accumulation. These proteins are examples of growth rate-dependent regulation (Pearse and Wolf 1994; Tao et al. 1999).
In our study, 18 strains of E. coli were compared for growth on acetate. The transcriptional analysis of three strains was compared when each was growing at its own maximum specific growth rate on acetate. Of the many genes differentially expressed between ATCC8739, SMS-3-5, and BL21, none have any known relationship with acetate. A few of the genes can be attributed to changes in growth rate more generally.
For example, the folK gene encoding 7,8-dihydro-6-hydroxymethylpterin pyrophosphokinase (HPPK) was upregulated more than 2-fold in ATCC8739 compared to both SMS-3-5 and BL21. HPPK catalyzes the transfer of pyrophosphate from ATP to 7,8-dihydro-6-hydroxymethylpterin, leading to the biosynthesis of folate cofactors required for the syntheses of many essential nutrients including thymidylate, methionine, purines, serine, glycine, and pantothenate. Harvey (1973) observed that 0.1 μg/mL trimethoprim, an inhibitor of dihydrofolate reductase, reduced the growth rate of E. coli by 50 %. The highest intracellular folate levels in S. cerevisiae were observed during the highest growth rates in either batch or steady-state cultivation (Hjortmo et al. 2008), suggesting that greater demand for nucleotides and amino acids leads to a elevated folate pool. The increased expression of folK in ATCC8739 compared to SMS-3-5 and BL21 may thus merely reflect the increased pool for biosynthetic precursors in the strain growing at the highest growth rate.
Similarly, yafP was more than 10-fold downregulated in ATCC8739 compared to SMS-3-5 or BL21. YafP and PolIV proteins are hypothesized to form a toxin-antitoxin pair: in the absence of PolIV, YafP increased the cytotoxicity of 4-nitroquinoline-1-oxide (NQO) and nitrofurazone (Gutierrez et al. 2011). Lower expression of the yafP gene in ATCC8739 might thus merely indicate reduced cytotoxicity leading to an improved growth compared to the other strains. A few of the differentially expressed genes are related to transport functions. The glutamate, aspartate ABC transporter permease subunit (gltJ), the fructuronate transporter (gntP) and the L-idonate/D-gluconate transporter (idnT), a predicted N-acetylneuraminic acid transporter (yjhB), an uncharacterized sodium-dependent transporter (yidK), and an uncharacterized galactose-pentose-hexuronide transporter (yihO) were each downregulated in ATCC8739 compared with BL21 and SMS-3-5. Only yihO was also downregulated in MEC136. Although the function of YihO is not known, it is proposed to be involved in an ion-driven metabolite uptake system (Poolman et al. 1996). However, it is not clear how these genes might play a role in growth on acetate.
No difference was observed in the steady-state transcription of any metabolic genes associated with acetate consumption. Indeed, the absence of differentially expressed genes which can be specifically attributed to acetate suggests that high growth rate of ATCC8739 growing on acetate may simply be a general characteristic of this strain rather than particular to acetate as the sole carbon source. For example, neither genes for acetyl-CoA synthetase (acs) nor acetate kinase/phosphotransacetylase (ackA/pta) mediating the conversion of acetate to acetyl-CoA were differentially expressed. Genes expressing ICL (aceA), ICDH (icd), or ICDH kinase/phosphatase (aceK) were also not overexpressed. Similarly, no genes associated with acid stress response (Foster 2004) were differentially expressed, a result which may simply be a consequence of all three strains been exposed identically to acetate. The transcription profiles provide little insight as to why ATCC8739, BL21, and SMS-3-5 have different growth rates on acetate.
Using a chemostat, ATCC8739, the fastest growing strain on acetate that we tested, was adapted to yield MEC136, which had a growth rate of 0.51 h−1 on defined medium containing 85 mM acetate. ATCC8739 had a growth rate of 0.41 h−1 on the same media. While transcriptional analysis did not reveal any significant differences between MEC136 and ATCC8739, whole genomic sequencing showed that the MEC136 strain contained a single mutation, which resulted in a S266P change in RpoA, the α subunit of RNA polymerase.
RpoA facilitates the action of upstream-promoter (UP) elements as well as the activators and repressors that interact with these elements, including the well-characterized global transcriptional regulators, cAMP receptor protein (CRP), which regulates transcription in response to carbon availability, and fumarate and nitrate reductase (FNR), which regulates the transcriptional response to oxygen limitation (for a general review, see Grainger and Busby 2008). The C-terminal domain (αCTD) of RpoA is responsible for making contact with the UP elements and their regulators and several researchers have shown that amino acids 260–270 of RpoA are crucial for this interaction (Tao et al. 1995; Murakami et al. 1996; Yang et al. 1997; Savery et al. 1998, 2002).
The location of the single mutation that we observed at amino acid 266 of RpoA is within the key 260–270 amino acid region and suggests that this mutation affects the transcription of a gene or genes that increase the ability of MEC136 to grow on acetate as a sole carbon source. Interestingly, other studies have shown that RpoA is involved in the ability of E. coli to grow on acetate. Nègre et al. (1997) showed that the αCTD region of RpoA binds to the UP region of the promoter for the aceBAK operon, which encodes isocitrate lyase (aceB), malate synthase (aceA), and isocitrate dehydrogenase kinase/phosphorylase (aceK). Arnold et al. (2001) demonstrated that rpoA expression was repressed by 100 mM acetate. Beatty et al. (2003) found that acs, which codes for acetyl-coenzyme A synthetase, the high-affinity acetate-scavenging enzyme that converts acetate to acetyl-coenzyme, is activated by CRP via the aCTD region of RpoA. Collectively, these results support the likelihood that the rpoA S266P mutation that we observed in MEC136 is responsible for transcriptional changes in a gene or genes that enable E. coli to grow faster on acetate as a sole carbon source.
References
Andrews S (2014) FastQC. A quality control tool for high throughput sequence data http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ Accessed June–December 2014
Arnold CN, Mcelhanon J, Lee A, Leonhart R, Siegele DA (2001) Global analysis of Escherichia coli gene expression during the acetate-induced acid tolerance response. J Bacteriol 183:2178–2186
Bartley LE, Ronald PC (2009) Plant and microbial research seeks biofuel production from lignocellulose. Calif Agric 63:178–184
Beatty CM, Browning DF, Busby SJW, Wolfe AJ (2003) Cyclic AMP receptor protein-dependent activation of the Escherichia coli acsP2 promoter by a synergistic class III mechanism. J Bacteriol 185:5148–5157
Blaby IK, Lyons BJ, Wroclawska-Hughes E, Phillips GCF, Pyle TP, Chamberlin SG, Benner SA, Lyons TJ, de Crécy-Lagard V, de Crécy E (2012) Experimental evolution of a facultative thermophile from a mesophilic ancestor. Appl Environ Microbiol 78:144–155
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Carvalho GBM, Mussatto SI, Candido EJ, Silva J (2006) Comparison of different procedures for the detoxification of eucalyptus hemicellulosic hydrolysate for use in fermentative processes. J Chem Technol Biotechnol 81:152–157
de Crécy E, Metzgar D, Allen C, Penicaud M, Lyons B, Hansen CJ, de Crécy-Lagard V (2007) Development of a novel continuous culture device for experimental evolution of bacterial populations. Appl Microbiol Biotechnol 77:489–496
Dykhuizen DE, Hartl DL (1983) Selection in chemostats. Microbiol Rev 47:150–168
Eiteman MA, Chastain MJ (1997) Optimization of the ion-exchange analysis of organic acids from fermentation. Anal Chim Acta 338:69–75
Foster JW (2004) Escherichia coli acid tolerance: tales of an amateur acidophile. Nat Rev Microbiol 2:898–907
Grainger DC, Busby SJW (2008) Global regulators of transcription in Escherichia coli: mechanisms of action and methods for study. Adv Appl Microbiol 65:93–113
Gutierrez A, Elez M, Clermont O, Denamur E, Matic I (2011) Escherichia coli YafP protein modulates DNA damaging property of the nitroaromatic compounds. Nucleic Acids Res 39:4192–4201
Harvey RJ (1973) Growth and initiation of protein-synthesis in Escherichia coli in presence of trimethoprim. J Bacteriol 114:309–322
Hjortmo S, Patring J, Andlid T (2008) Growth rate and medium composition strongly affect folate content in Saccharomyces cerevisiae. Int J Food Microbiol 123:93–100
Klinke HB, Thomsen AB, Ahring BK (2004) Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl Microbiol Biotechnol 66:10–26
Lahtvee PJ, Valgepea K, Nahku R, Abner K, Adamberg K, Vilu R (2009) Steady state growth space study of Lactococcus lactis in A-stat cultures. Ant Van Leeuw 96:487–496
Lakshmanaswamy A, Rajaraman E, Eiteman MA, Altman E (2011) Microbial removal of acetate selectively from sugar mixtures. J Ind Microbiol Biotechnol 38:1477–1484
Lan S, Veiseh M, Zhang M (2005) Surface modification of silicon and gold-patterned silicon surfaces for improved biocompatibility and cell patterning selectivity. Biosens Bioelect 20:1697–1708
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25:2078–2079
Liu TG, Khosla C (2010) Genetic engineering of Escherichia coli for biofuel production. Annu Rev Genet 44:53–69
Luli GW, Strohl WR (1990) Comparison of growth, acetate production, and acetate inhibition of Escherichia coli strains in batch and fed-batch fermentations. Appl Environ Microbiol 56:1004–1011
Maesen TJM, Lako E (1952) The influence of acetate on the fermentation of Baker’s yeast. Biochim Biophys Acta 9:106–107
Murakami K, Fujita N, Ishihama A (1996) Transcription factor recognition surface on the RNA polymerase α subunit is involved in contact with the DNA enhancer element. EMBO J 15:4358–4367
Nègre D, Bonod-Bidaud C, Oudot C, Prost J-F, Kolbl A, Ishihama A, Cozzone AJ, Cortay J-C (1997) DNA flexibility of the UP element is a major determinant for transcriptional activation at the Escherichia coli acetate promoter. Nucleic Acids Res 25:713–718
Papra A, Gadegaard N, Larsen NB (2001) Characterization of ultrathin poly(ethylene glycol) monolayers on silicon substrates. Langmuir 17:1457–1460
Parawira W, Tekere M (2011) Biotechnological strategies to overcome inhibitors in lignocellulose hydrolysates for ethanol production: review. Crit Rev Biotechnol 31:20–31
Pearse AJ, Wolf RE Jr (1994) Determination of the growth rate-regulated steps in expression of the Escherichia coli K-12 gnd gene. J Bacteriol 176:115–122
Pease AJ, Roa BR, Luo W, Winkler ME (2002) Positive growth rate-dependent regulation of the pdxA, ksgA, and pdxB genes of Escherichia coli K-12. J Bacteriol 184:1359–1369
Pedersen S, Bloch PL, Reeh S, Neidhardt FC (1978) Patterns of protein-synthesis in Escherichia coli—catalog of amount of 140 individual proteins at different growth-rates. Cell 14:179–190
Pettersen RC (1984) The chemical composition of wood. In: Roswell R (ed) The chemistry of solid wood. American Chemical Society, Washington, DC, pp 57–126
Poolman B, Knolm J, van der Does C, Henderson PJF, Liang WJ, Leblanc G, Pourcher T, Musveteau I (1996) Cation and sugar selectivity determinants in a novel family of transport proteins. Mol Microbiol 19:911–922
Ren CP, Chaudhuri RR, Fivian A, Bailey CM, Antonio M, Barnes WA, Pallen MJ (2004) The ETT2 gene cluster, encoding a second type III secretion system from Escherichia coli, is present in the majority of strains but has undergone widespread mutational attrition. J Bacteriol 186:3547–3560
Roe AJ, Mclaggan D, Davidson I, O’Byrne C, Booth IR (1998) Perturbation of anion balance during inhibition of growth of Escherichia coli by weak acids. J Bacteriol 180:767–772
Roe AJ, O’Byrne C, Mclaggan D, Booth IR (2002) Inhibition of Escherichia coli growth by acetic acid: a problem with methionine biosynthesis and homocysteine toxicity. Microbiol 148:2215–2222
Sakai S, Tsuchida Y, Nakamoto H, Okino S, Ichihashi O, Kawaguchi H, Watanabe T, Inui M, Yukawa H (2007) Effect of lignocellulose-derived inhibitors on growth of and ethanol production by growth-arrested Corynebacterium glutamicum R. Appl Environ Microbiol 73:6349–6353
Sauer U (2001) Evolutionary engineering of industrially important microbial phenotypes. Adv Biochem Eng Biotechnol 73:129–169
Savery NJ, Lloyd GS, Kainz M, Gaal T, Ross W, Ebright RH, Gourse RL, Busby SJW (1998) Transcription activation at Class II CRP-dependent promoters: identification of determinants in the C-terminal domain of the RNA polymerase α subunit. EMBO J 17:3439–3447
Savery NJ, Lloyd GS, Busby SJW, Thomas MS, Ebright RH, Gourse RL (2002) Determinants of the C-terminal domain of the Escherichia coli RNA polymerase α subunit important for transcription at Class I Cyclic AMP Receptor Protein-dependent promoters. J Bacteriol 184:2273–2280
Stephanopoulos G (2007) Challenges in engineering microbes for biofuels production. Science 315:801–804
Sun Y, Cheng JY (2002) Hydrolysis of lignocellulosic materials for ethanol production: a review. Biores Technol 83:1–11
Takahashi CM, Takahashi DF, Carvalhal MLC, Alterthum F (1999) Effects of acetate on the growth and fermentation performance of Escherichia coli KO11. Appl Biochem Biotechnol 81:193–203
Tao K, Zou C, Fujita N, Ishihama A (1995) Mapping of the OxyR protein contact site in the C-terminal region of RNA polymerase α subunit. J Bacteriol 177:6740–6744
Tao H, Bausch C, Richmond C, Blattner FR, Conway T (1999) Functional genomics: expression analysis of Escherichia coli growing on minimal and rich media. J Bacteriol 181:6425–6440
Thorvaldsdóttir H, Robinson JT, Mesirov JP (2013) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14:178–192
Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, Bidet P, Bingen E, Bonacorsi S, Bouchier C, Bouvet O, Calteau A, Chiapello H, Clermont O, Cruveiller S, Danchin A, Diard M, Dossat C, El Karoui M, Frapy E, Garry L, Ghigo JM, Gilles AM, Johnson J, Le Bouguénec C, Lescat M, Mangenot S, Martinez-Jéhanne V, Matic I, Nassif X, Oztas S, Petit MA, Pichon C, Rouy Z, Saint Ruf C, Schneider D, Tourret J, Vacherie B, Vallenet D, Médigue C, Rocha EPC, Denamu E (2009) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5:e1000344
Trĉek J, Mira NP, Jarboe LR (2015) Adaptation and tolerance to bacteria against acetic acid. Appl Microbiol Biotechnol 99:6215–6229
Um BH, Friedman B, van Walsum GP (2011) Conditioning hardwood-derived pre-pulping extracts for use in fermentation through removal and recovery of acetic acid using trioctylphosphine oxide (TOPO). Holzforschung 65:51–58
Wong M, Wright M, Woodley JM, Lye GJ (2009) Enhanced recombinant protein synthesis in batch and fed-batch Escherichia coli fermentation based on removal of inhibitory acetate by electrodialysis. J Chem Technol Biotechnol 84:1284–1291
Wong M, Woodley JM, Lye GJ (2010) Application of bipolar electrodialysis to E. coli fermentation for simultaneous acetate removal and pH control. Biotechnol Lett 32:1053–1057
Xia T, Eiteman MA, Altman E (2012) Simultaneous utilization of glucose, xylose and arabinonse in the presence of acetate by a consortium of Escherichia coli strains. Microb. Cell Fact. 11:77 doi:10.1186/1475-2859-11-77
Yang J, Murakami K, Camakaris H, Fujita N, Ishihama A, Pittard AJ (1997) Amino acid residues in the α-subunit C-terminal domain of Escherichia coli RNA polymerase involved in activation of transcription from the mtr promoter. J Bacteriol 179:6187–6191
Acknowledgments
MAE and EA acknowledge the Consortium for Biotechnology Research (DOE Prime Agreement DE-FG36-02GO12026), Southeastern Sun Grant Center under Prime Award No. DTOS59-07-G-00050 and NSF (CBET-0929893) for financial support of portions of this research. The authors also acknowledge Sarah A. Lee and Afaq M. M. Niyas for assistance with growth studies and Ronni Altman for strain construction.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human or animal subjects.
Additional information
Ankit Agarwal and Jacob Crigler contributed equally to this work.
An erratum to this article is available at http://dx.doi.org/10.1007/s00253-017-8271-z.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
Table S1: Genes which are two-fold or more upregulated in ATCC8739 compared to SMS-3-5 and BL21. Table S2: Genes which are two-fold or more downregulated in ATCC8739 compared to SMS-3-5 and BL21. (PDF 95 kb)
Rights and permissions
About this article

Cite this article
Rajaraman, E., Agarwal, A., Crigler, J. et al. Transcriptional analysis and adaptive evolution of Escherichia coli strains growing on acetate. Appl Microbiol Biotechnol 100, 7777–7785 (2016). https://doi.org/10.1007/s00253-016-7724-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7724-0