
Abstract
Metagenomic libraries from diverse environments have been extensive sources of many lipases and esterases; nevertheless, most of these enzymes remain biochemically uncharacterized. We previously built a metagenomic fosmid library from a microbial consortium specialized for diesel oil degradation and tested it for lipolytic activity. In the present study, we identified the PL14.H10 clone that was subcloned and sequenced, which enabled the identification of the EST3 protein. This enzyme exhibited 74 % amino acid identity with the uncharacterized alpha/beta hydrolase from Parvibaculum lavamentivorans [GenBank: WP012110575.1] and was classified into lipolytic enzyme family IV. Biochemical characterization revealed that EST3 presents high activity in a wide range of temperature with highest activity from 41 to 45 °C. Also, this thermostable esterase acts from mild acidic to alkaline conditions with an optimum pH of 6.0. The enzyme exhibited activity against p-nitrophenyl esters of different chain lengths and highest catalytic efficiency against p-nitrophenyl caprylate. The activity of the protein was increased in the presence of 0.5 mM of Mn+2, Li+, EDTA, and 1 % of CTAB and exhibited half of the activity in the presence of 10 % methanol and ethanol. Moreover, the homology model of EST3 was built and compared to other esterases, revealing a substrate channel that should fit a wide range of substrates. Taken together, the data presented in this work reveal the unique and interesting characteristics of EST3 that might be explored for further use in biotechnological applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The replacement of chemical catalysts with enzymatic processes enables reductions in time, energy, and industrial wastes that might compromise the environment. Thereby, enzymes could be used to either replace or even complement conventional methodology resulting in cleaner production processes (Lorenz et al. 2002). Recently, enzymatic catalysis has been implemented in a broad range of industries (Jegannathan and Nielsen 2013) and it is expected that biotechnological applications must stimulate the industrial enzyme market (Li et al. 2012).
Metagenomics is a useful approach that enables access to the collective genomes of environmental samples without the need for isolation and cultivation of microorganisms (Handelsman et al. 1998), representing a method with high potential in the search for new molecules. Various industrial sectors are interested in exploring unculturable microorganisms (Lorenz and Eck 2005) to identify new enzymes for industrial catalysis. Many lipolytic enzymes have been identified via metagenomic studies from different environments that include the following: marine sediment (Hårdeman and Sjöling 2007; Chu et al. 2008; Hu et al. 2010; Jeon et al. 2011), marine polluted sediments (Elsaied et al. 2011), glaciers (Cieśliński et al. 2009; Fu et al. 2013), ponds, lakes, (Rees et al. 2003; Wu and Tsai 2004; Ranjan et al. 2005) activated sludge (Roh and Villatte 2008; Liaw et al. 2010), forest soil (Nacke et al. 2011; Faoro et al. 2012), mangrove soil (Couto et al. 2010), and bovine rumen (Privé et al. 2015). However, many of these enzymes have not been fully biochemically characterized (Steele et al. 2009).
Lipases (EC 3.1.1.3) and esterases (EC 3.1.1.1) act on long-chain acylglycerols that have very low solubilities in water (carbon chain lengths >10) and short-chain water-soluble acylglycerols (carbon chain lengths ≤10), respectively (Arpigny and Jaeger 1999). They possess the following characteristics that make them a group of enzymes with many industrial applications: stereoselectivity, no requirements for cofactors, stability in organic solvents, and broad substrate specificity (Bornscheuer 2002; Jaeger and Eggert 2002). These properties allow them to be used in the synthesis of biopolymers, in the production of enantiopure pharmaceuticals and agrochemicals, and in the paper and food industries (Jaeger and Eggert 2002). Also, their potentials in the synthesis of biodiesel and in the bioremediation of industrial wastes of lipid origin have been highlighted (Hasan et al. 2006; Jeon et al. 2009). Lipases/esterases are the third leading group in the enzyme sales market after carbohydrases and proteases (Hasan et al. 2006; Selvin et al. 2012), and are expected to exhibit high growth potential in the World Industrial Enzymes Market since they can be used in many industrial bioconversions (López-López et al. 2014). At this moment, lipolytic enzymes that are extremely stable in non-aqueous media and exhibiting high levels of activity are desirable (Glogauer et al. 2011).
Based on conserved amino acid sequence motifs and biological properties, bacterial lipases and esterases were first classified into eight families (Arpigny and Jaeger 1999). Although this classification system has since been adopted, further studies resulted in the discovery of novel lipolytic enzymes that could not be grouped into any family. Consequently, novel families and subfamilies have been proposed (Lee et al. 2006; Kim et al. 2009). Lee et al. (2006) described the discovery of a gene that encodes a lipolytic enzyme and the creation of a new family referred to as LipG. The creation of a new subfamily within lipolytic enzymes family IV was proposed (Jeon et al. 2012) in a phylogenetic analysis from metagenomic library from tidal flat sediment. In another study, of 12 clones that encoded lipolytic enzymes found in a metagenomic library from activated sludge, four of the enzymes clustered to form a new family (Liaw et al. 2010).
In order to identify enzymes for different applications, a fosmid metagenomic library consisting of 4224 clones was previously constructed in the Laboratory of Biochemistry of Microorganisms and Plants at UNESP/Jaboticabal-Brazil. The soil was collected in a region contaminated with petroleum hydrocarbons from a former automotive lubricant factory in the city of Ribeirão Preto, São Paulo State, Brazil (Accorsini et al. 2012), and was used to develop a microbial consortium that was specialized for diesel oil degradation (Paixão et al. 2010) and for the subsequent construction of the metagenomic library. Recently, we have performed a screening for clones exhibiting lipolytic activity on agar containing tributyrin as the indicator substrate, which resulted in the identification of 30 positive clones. From one of the positive clones, a new esterase member of family V of bacterial lipolytic enzymes, designated as Est16, was cloned, overexpressed and characterized (Pereira et al. 2015).
In this research, the consistent interest in lipolytic enzymes with properties suitable for industrial purposes prompted us to investigate another positive clone from this metagenomic library to identify the gene responsible for its lipolytic activity. The assembled contig of this clone exhibited one open-reading frame (ORF) identified as a putative gene encoding the lipolytic enzyme EST3, which was further cloned and overexpressed. Additionally, structural analyses based on molecular modeling produced information concerning further site-directed mutagenesis with the goal of increasing the enzyme’s efficiencies on desired substrates. The functional and structural characterization of this novel esterase revealed its promising potential that can be explored for biotechnological applications.
Materials and methods
Subcloning and sequence analysis
To identify the gene responsible for the lipolytic activity of the PL14.H10 clone, its DNA was subcloned and sequenced. DNA was extracted with Wizard® Plus SV Minipreps DNA Purification System (Promega, Madison, WI, USA) according to the manufacturer’s instructions, and randomly sheared by nebulization into fragments of 1–3 kb. These fragments were recovered from a 0.8 % low melting point agarose gel. The blunt-ended DNA fragments were ligated into a SmaI-digested and dephosphorylated pUC19 vector (Thermo Scientific, Waltham, MA, USA).
The ligation product was used for the transformation of Escherichia coli DH5α-competent cells, and the transformants were selected. The resulting subcloned library was fully sequenced from both ends using M13-forward and M13-reverse primers with the automated sequencer ABI3100 (Applied Biosystems, Foster City, USA). Sequence assembly and contig editing were performed with the Phred/Phrap (Ewing et al. 1998) and Consed (Gordon et al. 1998) programs. To identify the ORFs, the ORF Finder tool provided by the National Center for Biotechnology Information (NCBI: http://www.ncbi.nlm.nih.gov) was used. The translated ORFs were compared to known sequences deposited in the non-redundant protein databases using the local alignment tool BLASTX (Altschul et al. 1990).
To analyze the phylogenetic relationship among EST3 and other lipolytic enzymes, a phylogenetic tree was constructed with neighbor-joining method (Saitou and Nei 1987) by means of Molecular Evolutionary Genetics Analysis software (MEGA 6) (Tamura et al. 2011) using 34 bacterial lipolytic enzymes selected from the first esterase/lipase classification proposed by Arpigny and Jaeger (1999), and the metagenome-derived lipolytic enzymes from two new families, LipG (Lee et al. 2006) and LipEH166 (Kim et al. 2009). Bootstrapping based on 1000 resamplings was used to estimate the robustness of the tree (Felsenstein 1985). Multiple alignments between protein sequences were performed with ClustalW (Thompson et al. 1994). Signal sequence searches were performed with the SignalP 3.0 program (Bendtsen et al. 2004).
Molecular Modeling
A search for proteins that are structurally similar to EST3 was undertaken using BLASTP against the Protein Data Bank (PDB). A three-dimensional structural model of EST3 was built based on the structural coordinates of the carboxylesterase Est2 from Alicyclobacillus acidocaldarius [PDB code: 1EVQ] (De Simone et al. 2000) and the esterase from Sulfolobus tokodai [PDB code: 3AIK] (Angkawidjaja et al. 2012). The models were built using Modeller 9.10 program (Sali and Blundell 1993) with the default parameters. Multiple-sequence alignments of EST3 and other esterases were performed with ClustalW (Thompson et al. 1994). Structural superposition of the EST3 model with the esterases structures was performed using the Secondary Structure Matching program inside COOT (Emsley et al. 2010). PyMOL software (PyMOL molecular graphics system, Schrödinger, LLC) was used to analyze the three-dimensional structure model and residues from active site and to prepare the figures.
Heterologous expression and purification of the recombinant protein
The est3 gene was amplified using the following synthetic oligonucleotides (the respective EcoRI and HindIII restriction sites are underlined): forward 5′-GAAAAGAATTCCAGATGGCACTCGATCC-3′ and reverse 5′-ACCGGAAGCTTCCTCATGCCAGCG-3′. The PCR product was digested with EcoRI and HindIII and then ligated into a pET28a(+) vector (Novagen, Madison, WI, USA) that had been digested with the same restriction enzymes.
The recombinant plasmids were transformed into E. coli BL21(DE3) cells by heat shock to express the recombinant enzyme. For protein expression, a 1 % pre-inoculum was seeded in Luria-Bertani (LB) liquid medium containing 50 μg mL−1 kanamycin and agitated at 200 rpm at 37 °C. When the optical density (OD600) of the culture reached an absorbance of 0.4–0.6, 0.1 mM isopropyl-b-d-thiogalactopyranoside (IPTG) was added to induce protein expression at 30 °C for 6 h. After this period, the cells were centrifuged and the pellet homogenized in lysis buffer (50 mM Tris-HCl, pH 8.0; 100 mM NaCl). Lysozyme was added at a final concentration of 4 μg ml−1 and the material was incubated at an ice bath for 1 h. The cells were disrupted by sonication using the Branson Sonifier equipment (Branson, CT, USA) with 6 cycles of 10 pulses at 30 % amplitude with 20-s intervals. The disrupted cells were centrifuged at 38,724g for 20 min at 4 °C. The obtained extract was incubated for 1 h under agitation with Ni-NTA resin previously equilibrated with the lysis buffer containing 20 mM imidazole and applied to a gravity flow column (Qiagen, Venlo, Netherlands). The target protein was eluted with a binding buffer containing 80 mM imidazole and then concentrated with Vivaspin 20 30,000 MWCO (Sartorius Stedim, Aubagne, France) under centrifugation at 200g. The final protein concentration was determined using a Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). The equipment was calibrated with the extinction coefficient of the purified protein.
Electrophoresis and zymogram
The protein samples were analyzed on 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the gel was stained with Coomassie Brilliant Blue R-250 and destained with 10 % (v/v) acetic acid (Laemmli 1970). The lipolytic activities of the bands (zymogram) were detected using tributyrin as the substrate according to Oh et al. (1999) with some modifications: the sample was not previously heated, and the SDS was removed from gel after electrophoresis by washing it with gentle agitation for 10 min in 50 mM Tris-HCl buffer at pH 8.0 containing 1 % Triton X-100 and twice in the same buffer free of detergent. Subsequently, the gel was placed in an emulsion of tributyrin agar and incubated at 30 °C until the appearance of clear halos (Glogauer et al. 2011).
Enzyme assays
EST3 activity was measured by spectrophotometric detection against p-nitrophenyl (pNP) esters. Unless otherwise indicated, p-nitrophenyl butyrate was used as the substrate for the standard assay method. The reaction mixture contained 1 mM pNP-butyrate in isopropanol/acetonitrile (4:1 v/v) (Faoro et al. 2012) and 50 mM Tris-HCl buffer (pH 8.0) with 0.3 % (v/v) Triton X-100. The reaction was initiated by the addition of 54.21 nM of the purified enzyme in solution (10 mM Tris-HCl, pH 8.0, and 50 mM NaCl), and the final volume of the reaction mixture was 100 μL. The enzyme activity was determined by continuously monitoring the product, p-nitrophenol, at 348 nm, which is the pH-independent isosbestic point of p-nitrophenol and the p-nitrophenoxide ion (Hriscu et al. 2013), with a SpectraMax M2e spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The molar extinction coefficients of p-nitrophenol were corrected for pH variation (Brod et al. 2010).
All reactions were performed in triplicate, and control reactions were performed without the enzymes for every measurement under the different conditions to subtract the values for the nonenzymatic hydrolysis of the substrates. To determine the initial reaction velocities, the linear regressions and the mean standard deviations were calculated.
The effect of pH on enzyme activity was determined at 30 °C with 50 mM with each of following buffers under standard assay conditions: citrate (pH 3.0 to 6.0), sodium phosphate (pH 6.0 to 8.0), Tris-HCl (pH 7.5 to 9.0), and CAPS (pH 9.5 to 11). The optimal temperature was determined in a range of 18 to 75 °C. Then, the released p-nitrophenol was measured. After these preliminary experiments, standard enzyme assays were developed on 50 mM sodium phosphate buffer pH 6.0 at 41 °C. A thermostability assay was performed over the temperature range of 30 to 80 °C by pre-incubating the purified enzyme for 15, 30, and 60 min at each given temperature. The residual esterase activity was determined after each incubation time. The non-incubated enzyme was taken as the control.
The EST3 substrate preference was measured for p-nitrophenyl esters consisting of the following aliphatic side chain lengths: pNP-acetate (C2), pNP-butyrate (C4), pNP-valerate (C5), pNP-caprylate (C8), pNP-caprate (C10), pNP-laurate (C12), pNP-myristate (C14), and pNP-palmitate (C16) (Sigma, St. Louis, MO, USA).
The effects of various reagents on enzyme activity were investigated by measuring the residual activity under standard conditions following pre-incubation of the enzyme solution with each reagent for 5 min at 4 °C. The activity without additives was defined as 100 %. To investigate the effect of salts and metal ions on EST3 activity, 0.5 mM of Na+, K+, Mg2+, Ca2+, Mn2+, Co2+, Cu+2 and Fe+2, Ni+2, Al+3, or Li+ was added to the enzyme solution. The effects of commonly used detergents, such as Tween 20, Tween 80, and Triton X-100, were measured by including 1 and 5 % (v/v) concentrations of these compounds in the reaction mixtures. Sodium dodecyl sulfate (SDS), hexadecyltrimethylammonium bromide (CTAB), and ethylenediaminetetraacetic acid (EDTA) were tested at the concentrations of 0.5 mM and 1 mM (w/v). The stability of EST3 in the presence of organic solvents and glycerol was assessed by measuring the residual activity after incubating the enzyme solution with 10 % (v/v) concentrations of methanol, ethanol, 2-propanol, dimethyl sulfoxide (DMSO), or glycerol at 4 °C for 5 min and subsequently measuring the residual activity. The control without added solvent was used to calculate the residual activity.
All data obtained were analyzed using the R software. ANOVA and Tukey’s test at 5 % probability were used to compare the treatment methods.
The kinetic parameters K m and K cat were calculated from activity measurements with pNP esters in the concentration range of 0.04 to 3 mM under standard assay conditions. The initial reaction velocities were measured using 5.42 nM of the enzyme for substrate C2 and 54.2 nM for substrates C4–C8. The kinetic parameters for each substrate were obtained by non-linear regression of the data on Michaelis-Menten equation using the software GraphPad Prism version 5.0.
Circular Dichroism
For this analysis, an additional purification step was performed. Concentrated protein from the affinity chromatography was loaded onto a gel filtration column (Superdex 75 16/60 GL; GE Healthcare Bio-Sciences, Uppsala, Sweden), equilibrated with 20 mM Tris-HCl pH 8.0, 50 mM NaCl, and 5 % glycerol, and separation was conducted at a flow rate of 0.5 mL min−1 on a Akta System (GE Healthcare).
To determine the thermal stability of the enzyme at four different pHs, the following buffers were tested: 2.5 mM sodium citrate pH 5.6, 2.5 mM sodium phosphate pHs 7.0 and 8.0, and 2.5 mM CHES pH 9.0. The changes in ellipticity at 222 nm of 5.4 μmol of the protein were monitored using a Jasco-815 spectropolarimeter (Jasco, Tokyo, Japan) in a 1-cm cuvette, wherein the temperature was increased by 1 °C min−1 in the range of 20 to 110 °C. The signal was averaged over five measurements at each temperature. The melting point (T m ) represents the temperature at the midpoint of the unfolding transition.
Nucleotide sequence accession number
The DNA sequence of est3 gene was deposited at GenBank with the reference code KP692205.
Results
Sequence analysis of the lipolytic clone
From the 30 positive clones previously identified (Pereira et al. 2015), the clone PL14.H10 was selected for further investigation since it showed strong hydrolytic activity as evidenced by a clear halo around the colony. The sublibrary of the clone PL14.H10, which contained 480 clones with insert fragments of 1–3 kb, was sequenced and its complete nucleotide sequence was obtained with 32,000 base pairs. Sequence annotations from the ORF Finder program and subsequent BLAST analysis revealed the presence of 29 ORFs that exhibited similarities to genes that have been annotated with predicted functions. Among these ORFs, four proteins with potential lipolytic activity were identified (Fig. S1) and only one selected to be characterized in this work while the other three will be further investigated. An additional file shows the prediction of the ORFs encoding lipolytic enzymes identified in the clone PL14.H10 (Table S1).
The est3 gene [GenBank: KP692205] encodes the protein named EST3. The sequence composition of est3 consists of 939 bp, which the G + C content is 67 %, and no signal peptide cleavage site was identified, as revealed by the SignalP program. Amino acid sequence alignments revealed sequence similarities ranging from 65 to 74 % and query coverage around 94 % with an alpha/beta hydrolase protein from Parvibaculum lavamentivorans [GenBank: WP012110575.1], a hypothetical protein from an uncultured microorganism [GenBank: AEM45144.1], and the carbohydrate esterase CE10 from the uncultured bacterium Ec32 [GenBank: CDO59205.1]. To date, only the hypothetical protein AEM45144.1 from an uncultured microorganism has been partially characterized as an esterase (Nacke et al. 2011), but the biochemical properties of this enzyme have not been analyzed.
Family identification and corresponding motifs of the lipolytic enzyme
To analyze the phylogenetic relationship of EST3 with representative members of bacterial lipolytic enzymes, a phylogenetic tree was built. EST3 clustered with lipolytic enzymes from family IV (Fig. 1a), being closely related to a lipase from Pseudomonas sp. (accession number AAC38151.1), a lipase-like enzyme from Alcaligenes euthophus (accession number AAC41424.1), and an acetyl esterase from E. coli (accession number AAC73578.1). These enzymes show activities against p-nitrophenyl esters of fatty acids with short to medium chains (Choo et al., 1998), and they are used for purposes like degradation of hemicelluloses (Wang et al. 2011; Till et al. 2013).

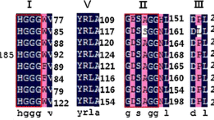
Phylogenetic relationship of lipolytic enzymes. a Unrooted phylogenetic tree of EST3 in this study (filled circle), Est16 (empty circle) (Pereira et al. 2015), and representative members of other previously identified families. The tree was constructed using the MEGA 6 program with the neighbor-joining algorithm. The analyses were bootstrapped (1000 replications), and only values greater than 50 % are shown. The scale bar indicates the number of amino acid substitutions per site. b Multiple sequence alignments of the partial amino acid sequences of EST3 with representative members of family IV. The numbers beside each sequence indicate the amino acid positions. The conserved motifs are indicated with a box. The amino acid residues belonging to the catalytic triad are marked with a filled circle
Multiple-sequence alignment corroborated that EST3 belongs to the family IV of lipolytic enzymes and the presence of HSL family conserved motifs (Fig. 1b). The catalytic triad residues (Ser, Asp, and His) were identified. Additionally, we evidenced the highly conserved HGGG motif in the EST3 sequence upstream of the serine motif; according to some authors, this motif is involved in hydrogen-bonding interactions that stabilize the oxyanion hole and plays a role in catalysis (Hårdeman and Sjöling 2007; Bunterngsook et al. 2010). Interestingly, Est 16, a previously isolated esterase from the same library (Pereira et al. 2015) that shares 30 % of identity with EST3, was clustered with bacterial lipolytic family V, evidencing the diversity of enzymes that can be isolated from similar sources.
Recombinant EST3 is stable and active in a broad range of pH and temperature
Following the structural and phylogenetic analyses, the protein was expressed in E. coli BL21(DE3) in the soluble form after optimization tests at different temperatures (data not shown). Significant amounts of active protein was achieved when E. coli BL21(DE3) cells were induced with 0.1 mM IPTG at 30 °C for 4 h. SDS-PAGE analysis of the purified protein revealed the expected molecular weight of 36 kDa as predicted by ProtParam tool (http://us.expasy.org/tools/protparam.html) (Fig. 2a). In addition, the zymogram exhibited a clear band in the region corresponding to the enzyme indicating that the purified EST3 was active (Fig. 2b).

Expression, purification, and lipolytic activity of recombinant EST3. a SDS-PAGE (10 %) of the expression and Ni-NTA affinity chromatography purification fractions of EST3 from E. coli BL21(DE3) cells carrying the pET28a-est3 vector. M: molecular weight standard (Thermo Scientific). Lane 1: crude extract of the non-induced cells. Lane 2: crude extract of the induced cells. Lane 3: soluble extract. Lane 4: flow-through fraction from affinity chromatography. Lanes 5 and 6: eluted fractions with 20 and 80 mM imidazole, respectively. b Zymogram of EST3. M: molecular weight standards (Bio-Rad, Hercules, USA). Lane 1: purified protein. Lane 2: lipolytic activity of purified EST3 band using tributyrin as substrate
The effects of pH and temperature on the enzymatic activity of EST3 were assayed spectrophotometrically using pNP-butyrate as substrate. The enzyme displayed activity over a broad pH range of 5.5 to 8.5 with optimal activity at pH 6.0, which is near the theoretical isoelectric point of 5.78 according the ProtParam prediction (Fig. 3a, gray shadow).

Effects of pH and temperature on EST3 activity. The production of p-nitrophenol from p-nitrophenyl butyrate was monitored at 348 nm in 50 mM phosphate, pH 6.0, containing 0.3 % Triton X-100. The activity of a non-incubated sample was defined as 100 %. a Effect of pH on EST3. The enzymatic assay was performed at pHs ranging from 4.5 to 11.0. The buffers used were 50 mM sodium citrate (pH 3.5 to 6.0) (black circle), 50 mM sodium phosphate (pH 6.0 to 8.0) (black square), 50 mM Tris-HCl (pH 7.5 to 9.0) (black triangle), and 50-mM CAPS (pH 9.5 to 11) (black diamond). b Effect of temperature on EST3. The activity of the enzyme was measured from 18 to 75 °C. c Analysis of EST3 thermostability after incubation of the enzyme for different periods at 30 °C (black circle), 40 °C (black square), 50 °C (black triangle), 60 °C (black diamond), and 70 °C (+). d Thermal denaturation profile of EST3. The changes in molar ellipticity at 222 nm at a scan rate of 1 °C min−1 in the temperature range of 20 to 110 °C were measured. Tested pHs: 5.6 (dark grey diamond), 7.0, (black square) 8.0 (grey diamond), and (grey circle) 9.0. Letters (a) and (b), on the top in the graphics indicate the significant difference between each condition performed in the experiment, according to ANOVA and Tukey’s test at 5 % probability
The subsequent described results were obtained in 50 mM sodium phosphate buffer at pH 6.0. EST3 protein displayed relative activities above 50 % in the temperature range of 28–57 °C and exhibited the highest activity at 35–48 °C (Fig. 3b, gray shadow). Interestingly, even in temperatures above 50 °C, EST3 still exhibited around 40 % of activity indicating the enzyme is thermostable. In fact, analysis of EST3 activity measured after longer incubation times at different temperatures corroborated the previous results and revealed the enzyme maintains its relative activity around 60 % (Fig. 3c). Moreover, circular dichroism assays at pH 7.0 showed that EST3 presented two steps of unfolding, with T m values of 48 and 68 °C, respectively (Fig. 3d).
Substrate specificity and effect of additives on EST3 activity
The substrate specificity of the enzyme investigated against pNP-esters revealed its ability to hydrolyze substrates up to 12C in the acyl-chain (C4–C12) and higher values of relative activity for pNP-butyrate and pNP-valerate (Fig. 4a). However, the analysis of the kinetic parameters of the enzyme revealed that its catalytic efficiency (K cat/K m ) is two-fold increased for pNP-caprylate (Table 1). In the presence of Mn2+ and Li+ ions, the EST3 activity increased 24 and 28 %, while Co+2, Ni+2, and Al+3 had an inhibitory effect decreasing the EST3 activity more than 40 %. The chelating agent EDTA exerted an inhibitory effect on the enzyme activity at 1 mM but exerted a stimulatory effect at a lower concentration (Fig. 4b, gray and black bars, respectively).

Activity of EST3 against different substrates and influence of ions and solvents. a Determination of the substrate specificity of the purified recombinant EST3. p-nitrophenyl esters with various fatty acid residue chain lengths were used as substrates. The maximal activity was defined as 100 %, and the relative activities are shown as percentages of the maximal activity. b Effect of different metal ions, salts and EDTA [0.5 mM (black square) and 1 mM (grey square)] on EST3 activity. c Effects of organic solvents on the enzyme activity. The activities were measured with p-nitrophenyl butyrate as the substrate in 50 mM phosphate at pH 6.0. One hundred percent indicates the activity measured under standard conditions without metal or solvents (b and c). No significant standard deviation values for the triplicates were identified when no error bar is present. In the graphics, letters indicate the significant difference between each condition performed in the experiment, according to ANOVA and Tukey’s test at 5 % probability
Since organic solvents are used in many chemical applications (Hårdeman and Sjöling 2007; Mei et al. 2012), the activity of EST3 was also monitored in the presence of 10 % concentration of these substances. The results revealed that the enzyme activity was decreased in the presence of DMSO, ethanol, methanol, and isopropanol but still maintained a residual activity of 68, 46.8, 47.7, and 38.2 %, respectively. In addition, 10 % of glycerol had an opposite effect increasing the enzyme activity by approximately 16 % when compared to the control (Fig. 4c).
The effect of detergents on EST3 is summarized in Table 2. Non-ionic detergents, such as Triton X-100 and Tween 20, decreased the enzyme activity, while the ionic detergent (CTAB) increased its activity up to ~180 %. On the other hand, EST3 activity was completely inhibited by Tween 80 and SDS.
Three-dimensional features of the EST3 model
The amino acid sequence of EST3 was submitted to BLASTP against PDB and exhibited 45 and 42 % identity with a carboxylesterase from A. acidocaldarius [PDB code: 1EVQ] and a carboxylesterase from the archaeon S. tokodaii [PDB code: 3AIK], respectively. The structural coordinates of both proteins were used for EST3 model building based on the high amino acid sequence identity and similar function. The structural model of EST3 fitted very well in the three-dimensional structures of lipases/esterases, consisting of two domains: the alpha/beta and the helical (Fig. 5a, respectively shown in blue and pink). Residues from the catalytic triad, formed by the Ser198, His322, and Asp292, are located in the interface between both domains (Fig. 5a, shown in yellow sticks). The Ser198 is located in the conserved sequence 196GDSAG200, which is situated in the nucleophilic elbow (in green). Similar to a typical member of the hormone sensitive family, the formation of the oxyanion hole of EST3 is likely mediated by Gly125–127, which are located in the 124HGGG127 motif (salmon stick).

Three-dimensional structure model of EST3 and comparison with other esterases. Homology model of EST3 produced with the MODELLER 9.10 program based on the X-ray structure of the carboxylesterases from Alicyclobacillus acidocaldarius [PDB code: 1EVQ] and Sulfolobus tokodaii [PDB code: 3AIK]. a Structural representation of the EST3 model with the helical (pink) and alpha/beta domains (blue) evidenced in cartoon. The residues from the catalytic triad (in yellow stick) are evidenced in the structure and in a detailed view. The HGGG and GDSAG motifs also are evidenced, respectively, in salmon stick and green cartoon. b Structural superimposition of EST3 (blue), A. fulgidus carboxylesterase (yellow), and S. tokodaii carboxyesterase (pink) showing the fitting of the regions. The presence of the cap-domain in the structures of EST3 and A. fulgidus structures are evidenced with a circle. The perfect fitting of the residues from the active site from the three proteins is also shown in stick. The numbers are presented in the following order: EST3, A. acidocaldarius, and S. tokodaii carboxylesterases. c Surface view of the three structures evidencing the differences in the enzyme pockets that may affect substrate range and passage. The domains of the proteins are delimited, and the residues from the catalytic triad are shown as yellow spheres. d Structural superimposition of EST3 and Est16 models for comparison. R.m.s.d. values were taken for both domains separately to evidence the main differences in the helical domain. e Comparison of the pockets of both proteins and relationship with substrate preference
As expected, the structural model of EST3 superposed nicely with the A. acidocaldarius and S. tokodaii carboxylesterases structures, and revealed main differences in the helical domain (Fig. 5b). Interestingly, only EST3 and the carboxylesterase of A. acidocaldarius were identified as having a cap-domain (Fig. 5b, blue and yellow cartoon, respectively), which is a common characteristic present in the other esterases/lipases of family IV. Indeed, the residues that form the catalytic triad in the three structures were nicely superimposed (Fig. 5b, details in sticks). Despite these similarities, the proteins exhibited quite significant differences in the pocket that can affect the type of substrate that can be cleaved (Fig. 5c). EST3 and S. tokodaii proteins showed a bigger channel crossing both sides of the proteins, differently of the enzyme from A. acidocaldarius. The channel is in accordance with the substrates cleaved by EST3, which exhibited preference for the substrate with longer acyl-chain pNP-caprylate. On the other hand, despite the presence of a channel in the pocket from the carboxylesterase of S. tokodaii, this enzyme revealed preference for pNP-butyrate (Suzuki et al. 2004).
In addition to these carboxylesterases, we performed a comparison of EST3 with Est16, another esterase isolated from the same metagenomic source, which biochemical and biophysical properties were previously characterized. Est16 belongs to family V of lipases and esterases and the comparison revealed that both proteins showed significant differences, mainly evidenced in the helical domain as evidenced by the values of r.m.s.d. (Fig. 5d). The secondary structures (α-helices) from the helical domain are oriented in distinct conformations that affect the pocket (Fig. 5e) and that are directly related to the preference for substrates. As shown, EST3 that has a long channel has preference to cleave substrates with up to 8C in the acyl-chain, while Est16 only cleaved substrates with C4 and C5 acyl-chains, in accordance with its pocket that is limited.
Discussion
Metagenomic libraries from contaminated soils have proven to be great sources of many lipolytic enzymes (Elend et al. 2006; Glogauer et al. 2011; Garcia et al. 2015; Kim et al. 2015; Pereira et al. 2015). In the present work, we characterized the esterase EST3 isolated from a metagenomic library derived from soil contaminated with petroleum hydrocarbons, which belongs to family IV of the lipolytic enzymes.
Enzymatic characterization of EST3 indicated that this enzyme exhibited a mildly acidic pH optimum, in contrast to the majority of esterases in the literature, which exhibit alkaline pH optimums (Wu and Sun 2009; Hu et al. 2010; Tao et al. 2011; Wang et al. 2011; Jeon et al. 2012) including Est16, which we have recently characterized, and showed maximal activity at pH 8 and 9. Lipolytic enzymes that act under mild acidic conditions could be useful to the dairy industry, to develop flavors, to accelerate cheese ripening, in animal feed supplements (Bunterngsook et al. 2010) and in the treatment of waste oil (Morohoshi et al. 2011). Site environmental conditions are important to consider for bioremediation, and generally enzymes that act from pH 6 to 8, as EST3, are desirable (Adams et al. 2015).
Chu et al. (2008) characterized EstA and EstB, esterases from marine metagenome that presented highest activity in the pH range from 6.5 to 8.0. Also, both showed their highest activity at 45 °C but lower than 20 % activities above 60 °C, in contrast to EST3, that presented high activity (more than 40 %) from 28 up to 75 °C. Lipolytic enzymes that display high levels of activity in mild conditions are desirable for some purposes, as the bioremediation of low-temperature soils or waters. Moreover, mild temperatures may reduce energy consumption and also make possible to heat-inactivate the enzyme during biotransformation processes (Luo et al. 2006; Hårdeman and Sjöling 2007). By comparison with our previous work, similar behavior under temperature variation was found for Est16 (Pereira et al., 2015). On the other hand, besides the mild optimum temperature, the denaturation curve of the EST3 showed that the protein is fully unfolded in two steps and maintained 50 % of its activity even after incubation in high temperatures as 60 °C, which suggested that this enzyme also displays potential applicability as a thermal enzyme. Interestingly, the stability assay at different temperatures revealed that although EST3 lost its activity after 15 min of incubation at 30 and 40 °C, the activity was completely restored over time in these temperatures. Further analyses are required to investigate if the enzyme will show higher activity upon longer incubation times to confirm that these results are due to the phenomenon called thermal activation (Faoro et al. 2012). It is interesting to note that the thermal activation is not a common feature of esterases.
The protein exhibited relative resistance to organic solvents, detergents, and a set of tested ions. EST3 was relatively stable in the presence of 1 % Triton X-100 and Tween 20. The presence of surfactants reduces the interfacial tension of the aqueous phase, which typically leads to protein unfolding that decreases the interaction of the enzyme with its substrate (Salameh and Wiegel 2010). CTAB significantly enhances EST3 activity probably stabilizing the interfacial area and facilitating the access of the substrate to the catalytic site (Saranya et al. 2014), which is considered a desirable characteristic for enzymes that act in bioremediation (Li and Chen 2009) and is not common among esterases (Hu et al. 2010; Fu et al. 2011; Wemheuer et al. 2013).
In relation to the structural model of EST3 in comparison with the protein structures used as a model, it showed that residues from the catalytic triad are structurally placed in the same position. Moreover, the main differences among the proteins that shared the same family are located in the helical domain, which forms a long tunnel in EST3 that crossed the protein from the front to the back part (Fig. 5c) and likely favored the access of the tested substrates to the catalytic triad and the subsequent hydrolysis of pNP-esters with acyl-chain length up to C12, as shown in the functional characterization (Fig. 4a). A channel is also present in the S. tokodaii carboxylesterase but it does not cross the protein, limiting the length of the acyl-chains as previously observed in the enzymatic characterization of this protein (Suzuki et al. 2004). Again, the comparison with Est16 also revealed significant differences in this domain that might explain the distinct range of substrates presented by both enzymes.
EST3 showed the highest catalytic efficiency for C8 when compared with other esterases described in literature, as EstDL30 (Tao et al. 2011) and EstOF4 (Rao et al. 2013) (Table 3). For all evaluated substrates in this work, EST3 exhibited higher kinetic parameter values compared to RmEstA (Liu et al. 2013), a family IV esterase from the thermophilic zygomycete Rhizomucor miehei. In comparison with the esterase from our previous work, Est16 showed higher catalytic efficiency for pNP-butyrate, while EST3 is more efficient in hydrolyzing pNP-caprylate, but still with K cat/K m ratio 1.39 times smaller than the value achieved by Est16. This substrate hydrolysis profile is characteristic of esterases, which are enzymes typically active only on short-chain fatty acid esters that are at least partially soluble in water (Arpigny and Jaeger 1999).
In conclusion, this study described the successful isolation and characterization of a novel esterase from a metagenomic library of a consortium that was specialized for diesel oil degradation. EST3 was classified into family IV of the bacterial lipolytic enzymes that presents a long channel for substrates that might explain its capability to cleave substrates consisting of different acyl-chain length. The enzyme was active under mild acidic to alkaline conditions, exhibited high activity in a wide range of temperatures. Altogether, these results have raised a set of characteristics from EST3 that reveal its potential for biotechnological applications.
References
Accorsini FR, Mutton MJR, Lemos EGM, Benincasa M (2012) Biosurfactantes production by yeasts using soybean oil and glycerol as low cost substrate. Brazilian J Microbiol 55:116–125. doi:10.1590/S1517-83822012000100013
Adams GO, Fufeyin PT, Okoro SE, Ehinomen I (2015) Bioremediation, Biostimulation and Bioaugmention: A Review. Int J Environ Bioremediation Biodegrad 3:28–39. doi:10.12691/ijebb-3-1-5
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic Local Alignment Search Tool. J Mol Biol 215:403–410
Angkawidjaja C, Koga Y, Takano K, Kanaya S (2012) Structure and stability of a thermostable carboxylesterase from the thermoacidophilic archaeon Sulfolobus tokodaii. FEBS J 279:3071–3084. doi:10.1111/j.1742-4658.2012.08687.x
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343(Pt 1):177–183
Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340:783–795. doi:10.1016/j.jmb.2004.05.028
Bornscheuer UT (2002) Microbial carboxyl esterases : classification, properties and application in biocatalysis. FEMS Microbiol Rev 26:73–81. doi:10.1111/j.1574-6976.2002.tb00599.x
Brod FCA, Vernal J, Bertoldo JB, Terenzi H, Arisi ACM (2010) Cloning, expression, purification, and characterization of a novel esterase from Lactobacillus plantarum. Mol Biotechnol 44:242–249. doi:10.1007/s12033-009-9232-2
Bunterngsook B, Kanokratana P, Thongaram T, Tanapongpipat S, Uengwetwanit T, Rachdawong S, Vichitsoonthonkul T, Eurwilaichitr L (2010) Identification and Characterization of Lipolytic Enzymes from a Peat-Swamp Forest Soil Metagenome. Biosci Biotechnol Biochem 74:1848–1854. doi:10.1271/bbb.100249
Choo D, Kurihara T, Suzuki T, Soda K, Esaki N (1998) A Cold-Adapted lipase of an Alaskan gene cloning and enzyme purification and characterization. Appl Environ Microbiol 64:486–491.
Chu X, He H, Guo C, Sun B (2008) Identification of two novel esterases from a marine metagenomic library derived from South China Sea. Appl Microbiol Biotechnol 80:615–625. doi:10.1007/s00253-008-1566-3
Cieśliński H, Białkowskaa A, Tkaczuk K, Długołecka A, Kur J, Turkiewicz M (2009) Identification and molecular modeling of a novel lipase from an Antarctic soil metagenomic library. Pol J Microbiol 58:199–204
Couto GH, Glogauer A, Faoro H, Chubatsu LS, Souza EM, Pedrosa FO (2010) Isolation of a novel lipase from a metagenomic library derived from mangrove sediment from the south Brazilian coast. Genet Mol Res 9:514–523. doi:10.4238/vol9-1gmr738
De Simone G, Galdiero S, Manco G, Lang D, Rossi M, Pedone C (2000) A snapshot of a transition state analogue of a novel thermophilic esterase belonging to the subfamily of mammalian hormone-sensitive lipase. J Mol Biol 303:761–771. doi:10.1006/jmbi.2000.4195
Elend C, Schmeisser C, Leggewie C, Babiak P, Carballeira JD, Steele HL, Reymond J, Jaeger K, Streit WR (2006) Isolation and Biochemical Characterization of Two Novel Metagenome-Derived Esterases. Appl Environ Microbiol 72:3637–3645. doi:10.1128/AEM.72.5.3637
Elsaied H, Stokes HW, Kitamura K, Kurusu Y, Kamagata Y, Maruyama A (2011) Marine integrons containing novel integrase genes, attachment sites, attI, and associated gene cassettes in polluted sediments from Suez and Tokyo Bays. ISME J 5:1162–1177. doi:10.1038/ismej.2010.208
Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi:10.1107/S0907444910007493
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185. doi:10.1101/gr.8.3.175
Faoro H, Glogauer A, Couto GH, de Souza EM, Rigo LU, Cruz LM, Monteiro RA, Pedrosa FDO (2012) Characterization of a new Acidobacteria-derived moderately thermostable lipase from a Brazilian Atlantic Forest soil metagenome. FEMS Microbiol Ecol 81:386–394. doi:10.1111/j.1574-6941.2012.01361.x
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the Bootstrap. Evolution (N Y) 39:783–791
Fu C, Hu Y, Xie F, Guo H, Ashforth EJ, Polyak SW, Zhu B, Zhang L (2011) Molecular cloning and characterization of a new cold-active esterase from a deep-sea metagenomic library. Appl Microbiol Biotechnol 90:961–970. doi:10.1007/s00253-010-3079-0
Fu J, Leiros H-KS, de Pascale D, Johnson KA, Blencke H-M, Landfald B (2013) Functional and structural studies of a novel cold-adapted esterase from an Arctic intertidal metagenomic library. Appl Microbiol Biotechnol 97:3965–3978. doi:10.1007/s00253-012-4276-9
Garcia RAM, Pereira MR, Maester TC, de Macedo Lemos EG (2015) Investigation, expression, and molecular modeling of ORF2, a metagenomic lipolytic enzyme. Appl Biochem Biotechnol 175:3875–3887. doi:10.1007/s12010-015-1556-8
Glogauer A, Martini VP, Faoro H, Couto GH, Müller-Santos M, Monteiro RA, Mitchell DA, de Souza EM, Pedrosa FO, Krieger N (2011) Identification and characterization of a new true lipase isolated through metagenomic approach. Microb Cell Factories 54:1–15. doi:10.1186/1475-2859-10-54
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202. doi:10.1101/gr.8.3.195
Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM (1998) Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5:R245–R249
Hårdeman F, Sjöling S (2007) Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol Ecol 59:524–534. doi:10.1111/j.1574-6941.2006.00206.x
Hasan F, Shah AA, Hameed A (2006) Industrial applications of microbial lipases. Enzym Microb Technol 39:235–251. doi:10.1016/j.enzmictec.2005.10.016
Hriscu M, Chiş L, Toşa M, Irimie FD (2013) pH-Profiling of thermoactive lipases and esterases: caveats and further notes. Eur J Lipid Sci Technol 115:571–575. doi:10.1002/ejlt.201200305
Hu Y, Fu C, Huang Y, Yin Y, Cheng G, Lei F, Lu N, Li J, Ashforth EJ, Zhang L, Zhu B (2010) Novel lipolytic genes from the microbial metagenomic library of the South China Sea marine sediment. FEMS Microbiol Ecol 72:228–237. doi:10.1111/j.1574-6941.2010.00851.x
Jaeger K-E, Eggert T (2002) Lipases for biotechnology. Curr Opin Biotechnol 13:390–397
Jegannathan KR, Nielsen PH (2013) Environmental assessment of enzyme use in industrial production—a literature review. J Clean Prod 42:228–240. doi:10.1016/j.jclepro.2012.11.005
Jeon JH, Kim J-T, Kim YJ, Kim H-K, Lee HS, Kang SG, Kim S-J, Lee J-H (2009) Cloning and characterization of a new cold-active lipase from a deep-sea sediment metagenome. Appl Microbiol Biotechnol 81:865–874. doi:10.1007/s00253-008-1656-2
Jeon JH, Kim JT, Lee HS, Kim S-J, Kang SG, Choi SH, Lee J-H (2011) Novel lipolytic enzymes identified from metagenomic library of deep-sea sediment. Evid Based Complement Alternat Med 2011:1–9. doi:10.1155/2011/271419
Jeon JH, Lee HS, Kim JT, Kim S-J, Choi SH, Kang SG, Lee J-H (2012) Identification of a new subfamily of salt-tolerant esterases from a metagenomic library of tidal flat sediment. Appl Microbiol Biotechnol 93:623–631. doi:10.1007/s00253-011-3433-x
Kim E-Y, Oh K-H, Lee M-H, Kang C-H, Oh T-K, Yoon J-H (2009) Novel cold-adapted alkaline lipase from an intertidal flat metagenome and proposal for a new family of bacterial lipases. Appl Environ Microbiol 75:257–260. doi:10.1128/AEM.01400-08
Kim HJ, Jeong YS, Jung WK, Kim SK, Lee HW, Kahng H-Y, Kim J, Kim H (2015) Characterization of novel family IV esterase and family I.3 lipase from an oil-polluted mud flat metagenome. Mol Biotechnol 57:781–792. doi:10.1007/s12033-015-9871-4
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lee M-H, Lee C-H, Oh T-K, Song JK, Yoon J-H (2006) Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: evidence for a new family of bacterial lipases. Appl Environ Microbiol 72:7406–7409. doi:10.1128/AEM.01157-06
Li J-L, Chen B-H (2009) Surfactant-mediated biodegradation of polycyclic aromatic hydrocarbons. Materials (Basel) 2:76–94. doi:10.3390/ma2010076
Li S, Yang X, Yang S, Zhu M, Wang X (2012) Technology prospecting on enzymes: application, marketing and engineering. Comput Struct Biotechnol J 2:1–11. doi:10.5936/csbj.201209017
Liaw R-B, Cheng M-P, Wu M-C, Lee C-Y (2010) Use of metagenomic approaches to isolate lipolytic genes from activated sludge. Bioresour Technol 101:8323–8329. doi:10.1016/j.biortech.2010.05.091
Liu Y, Xu H, Yan Q, Yang S, Duan X, Jiang Z (2013) Biochemical characterization of a first fungal esterase from Rhizomucor miehei showing high efficiency of ester synthesis. PLoS One 8:e77856. doi:10.1371/journal.pone.0077856
López-López O, Cerdán ME, Gonzalez-Siso MI (2014) New extremophilic lipases and esterases from metagenomics. Curr Protein Pept Sci 15:445–455
Lorenz P, Eck J (2005) Metagenomics and industrial applications. Nat Rev Microbiol 3:510–516
Lorenz P, Liebeton K, Niehaus F, Eck J (2002) Screening for novel enzymes for biocatalytic processes: accessing the metagenome as a resource of novel functional sequence space. Curr Opin Biotechnol 13:572–577. doi:10.1016/S0958-1669(02)00345-2
Luo Y, Wang G, Ma Y, Wei D (2006) Application of a silica gel prolonged-release system for methanol in the production of biodiesel. J Chem Technol Biotechnol 81:1846–1848. doi:10.1002/jctb.1602
Mei Y, Peng N, Zhao S, Hu Y, Wang H, Liang Y, She Q (2012) Exceptional thermal stability and organic solvent tolerance of an esterase expressed from a thermophilic host. Appl Microbiol Biotechnol 93:1965–1974. doi:10.1007/s00253-011-3504-z
Morohoshi T, Oikawa M, Sato S, Kikuchi N, Kato N, Ikeda T (2011) Isolation and characterization of novel lipases from a metagenomic library of the microbial community in the pitcher fluid of the carnivorous plant Nepenthes hybrida. J Biosci Bioeng 112:315–320. doi:10.1016/j.jbiosc.2011.06.010
Nacke H, Will C, Herzog S, Nowka B, Engelhaupt M, Daniel R (2011) Identification of novel lipolytic genes and gene families by screening of metagenomic libraries derived from soil samples of the German Biodiversity Exploratories. FEMS Microbiol Ecol 78:188–201. doi:10.1111/j.1574-6941.2011.01088.x
Oh B, Kim H, Lee J, Kang S, Oh T-K (1999) Staphylococcus haemolyticus lipase : biochemical properties, substrate specificity and gene cloning. FEMS Microbiol Ecol 179:385–392
Paixão DAA, Dimitrov MR, Pereira RM, Accorsini FR, Vidotti MB, Lemos EG de M (2010) Molecular analysis of the bacterial diversity in a specialized consortium for diesel oil degradation. Rev Bras Ciência do Solo 34:773–781.
Pereira MR, Mercaldi GF, Maester TC, Balan A, Lemos EG de M (2015) Est16, a New Esterase Isolated from a Metagenomic Library of a Microbial Consortium Specializing in Diesel Oil Degradation. PLoS One 10:1–16. doi:10.1371/journal.pone.0133723
Privé F, Newbold CJ, Kaderbhai NN, Girdwood SG, Golyshina OV, Golyshin PN, Scollan ND, Huws SA (2015) Isolation and characterization of novel lipases/esterases from a bovine rumen metagenome. Appl Microbiol Biotechnol 99:5475–5485. doi:10.1007/s00253-014-6355-6
Ranjan R, Grover A, Kapardar RK, Sharma R (2005) Isolation of novel lipolytic genes from uncultured bacteria of pond water. Biochem Biophys Res Commun 335:57–65. doi:10.1016/j.bbrc.2005.07.046
Rao L, Xue Y, Zheng Y, Lu JR, Ma Y (2013) A novel alkaliphilic bacillus esterase belongs to the 13(th) bacterial lipolytic enzyme family. PLoS One 8:1–10. doi:10.1371/journal.pone.0060645
Rees HC, Grant S, Jones B, Grant WD, Heaphy S (2003) Detecting cellulase and esterase enzyme activities encoded by novel genes present in environmental DNA libraries. Extremophiles 7:415–421. doi:10.1007/s00792-003-0339-2
Roh C, Villatte F (2008) Isolation of a low-temperature adapted lipolytic enzyme from uncultivated micro-organism. J Appl Microbiol 105:116–123. doi:10.1111/j.1365-2672.2007.03717.x
Saitou N, Nei M (1987) The neighbor-joining method : a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Salameh M a, Wiegel J (2010) Effects of detergents on activity, thermostability and aggregation of two alkalithermophilic lipases from Thermosyntropha lipolytica. Open Biochem J 4:22–28. doi:10.2174/1874091X01004010022
Sali A, Blundell TL (1993) Comparative Protein Modelling by Satisfaction of Spatial Testraints. J Mol Biol 234:779–815
Saranya P, Sukanya Kumari H, Prasad Rao B, Sekaran G (2014) Lipase production from a novel thermo-tolerant and extreme acidophile Bacillus pumilus using palm oil as the substrate and treatment of palm oil-containing wastewater. Environ Sci Pollut Res Int 21:3907–3919. doi:10.1007/s11356-013-2354-x
Selvin J, Kennedy J, Lejon DPH, Kiran GS, Dobson ADW (2012) Isolation identification and biochemical characterization of a novel halo-tolerant lipase from the metagenome of the marine sponge Haliclona simulans. Microb Cell Factories 11:72. doi:10.1186/1475-2859-11-72
Steele HL, Jaeger K-E, Daniel R, Streit WR (2009) Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol 16:25–37. doi:10.1159/000142892
Suzuki Y, Miyamoto K, Ohta H (2004) A novel thermostable esterase from the thermoacidophilic archaeon Sulfolobus tokodaii strain 7. FEMS Microbiol Lett 236:97–102. doi:10.1016/j.femsle.2004.05.026
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi:10.1093/molbev/msr121
Tao W, Lee MH, Wu J, Kim NH, Lee S-W (2011) Isolation and characterization of a family VII esterase derived from alluvial soil metagenomic library. J Microbiol 49:178–185. doi:10.1007/s12275-011-1102-5
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi:10.1093/nar/22.22.4673
Till M, Goldstone DC, Attwood GT, Moon CD, Kelly WJ, Arcus VL (2013) Structure and function of an acetyl xylan esterase (Est2A) from the rumen bacterium Butyrivibrio proteoclasticus. Proteins 81:911–917. doi:10.1002/prot.24254
Wang G, Meng K, Luo H, Wang Y, Huang H, Shi P, Pan X, Yang P, Yao B (2011) Molecular cloning and characterization of a novel SGNH arylesterase from the goat rumen contents. Appl Microbiol Biotechnol 91:1561–1570. doi:10.1007/s00253-011-3289-0
Wemheuer B, Taube R, Akyol P, Wemheuer F, Daniel R (2013) Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula. Archaea 2013:1–13. doi:10.1155/2013/136714
Wu C, Sun B (2009) Identification of Novel Esterase from Metagenomic Library of Yangtze River. J Microbiol Biotechnol 19:187–193. doi:10.4014/jmb.0804.292
Wu H-S, Tsai M-J (2004) Kinetics of tributyrin hydrolysis by lipase. Enzym Microb Technol 35:488–493. doi:10.1016/j.enzmictec.2004.08.002
Acknowledgments
We would like to thank Prof. João Martins Pizauro Junior (São Paulo State University, UNESP, Campus of Jaboticabal, Brazil) for providing the kinetics equipment and the Spectroscopy and Calorimetry Facility (Brazilian Biosciences National Laboratory-LNBio-Campinas, Brazil) for enabling the CD measurements.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This work was supported by the São Paulo Research Foundation–FAPESP (grant numbers 2011/09064-6 and 2013/03568-8).
Conflict of Interest
The authors declare that they have no competing interests.
Electronic supplementary material
ESM 1
(PDF 215 kb)
Rights and permissions
About this article

Cite this article
Maester, T.C., Pereira, M.R., Machado Sierra, E.G. et al. Characterization of EST3: a metagenome-derived esterase with suitable properties for biotechnological applications. Appl Microbiol Biotechnol 100, 5815–5827 (2016). https://doi.org/10.1007/s00253-016-7385-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7385-z